
Digital Alchemy: The Rise of Machine and Deep Learning in Small-Molecule Drug Discovery



Abstract

1. Introduction and Scope
1.1. Traditional Drug Discovery Challenges
1.2. ML and DL: A Paradigm Shift in Drug Discovery
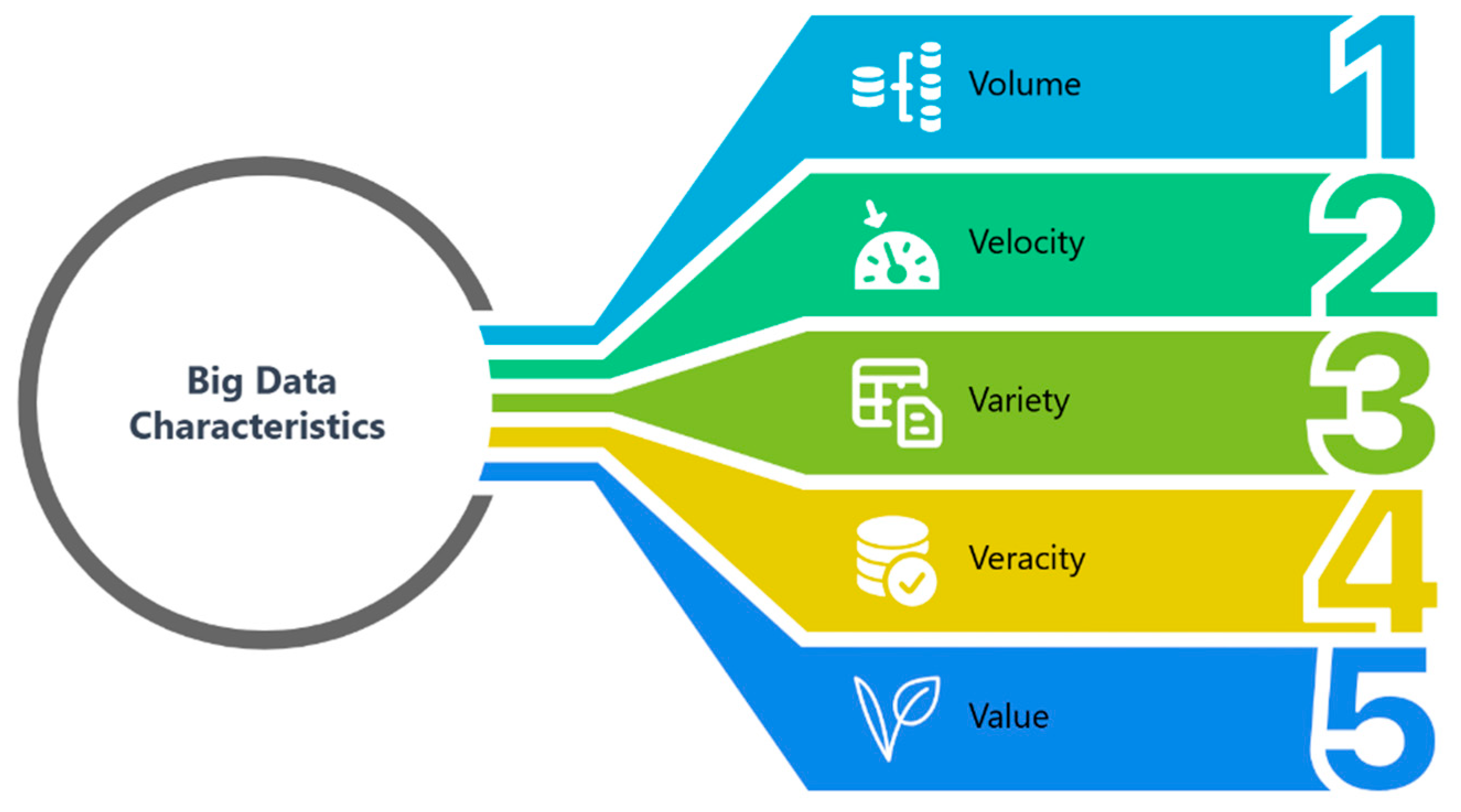
1.3. AI Drug Discovery: From Big Data to Smart Data
1.4. Molecular Representations
2. Big Data Sources in Small-Molecule Drug Discovery
2.1. Public Chemical Databases
2.2. Public Biological and Omics Repositories
2.3. Scientific Literature and Electronic Health Records (EHRs)
2.4. Proprietary and In-House Data
2.5. High-Throughput Screening (HTS) and Novel Data
2.6. Generated Data from Virtual Screening (VS)
3. Classical ML Models
4. Deep Learning Models
4.1. Artificial Neural Networks (ANNs)
4.2. Deep Neural Networks (DNNs)
4.3. Convolutional Neural Networks (CNNs)
4.4. Recurrent Neural Networks (RNNs)
4.5. Graph Neural Networks (GNNs)
4.6. Generative Models for De Novo Drug Design
4.7. Transformer-Based Encoders
5. AI-Driven Applications Across the Drug Discovery Pipeline
5.1. Target Identification
5.2. Hit Discovery and Virtual Screening
5.2.1. Structure-Based Virtual Screening (SBVS)
5.2.2. Ligand-Based Virtual Screening (LBVS)
5.2.3. Generative Virtual Screening (GVS)
5.3. Lead Optimization
5.4. ADMET Prediction
5.5. Drug Repurposing
5.6. Clinical Trial Design and Optimization
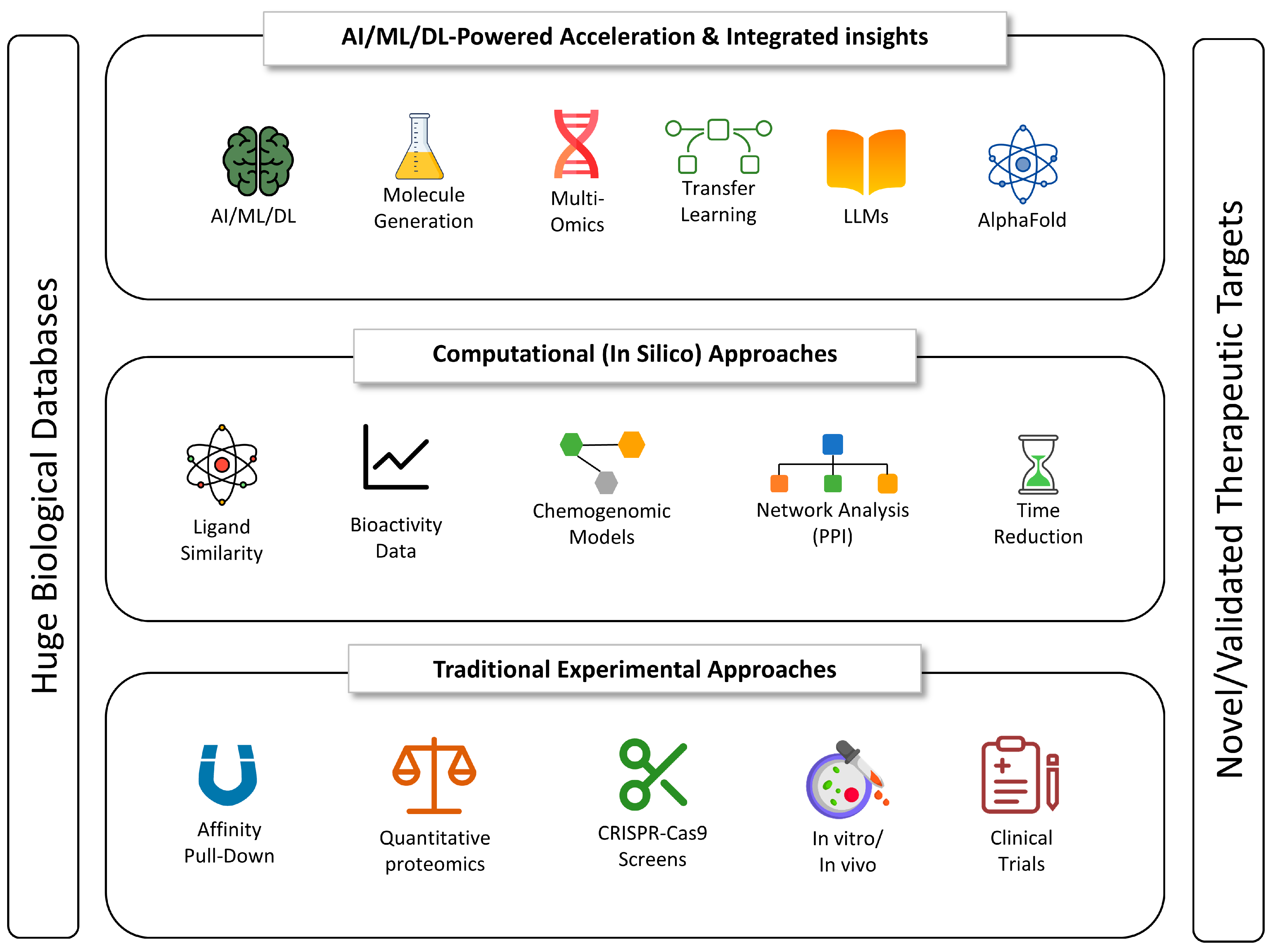
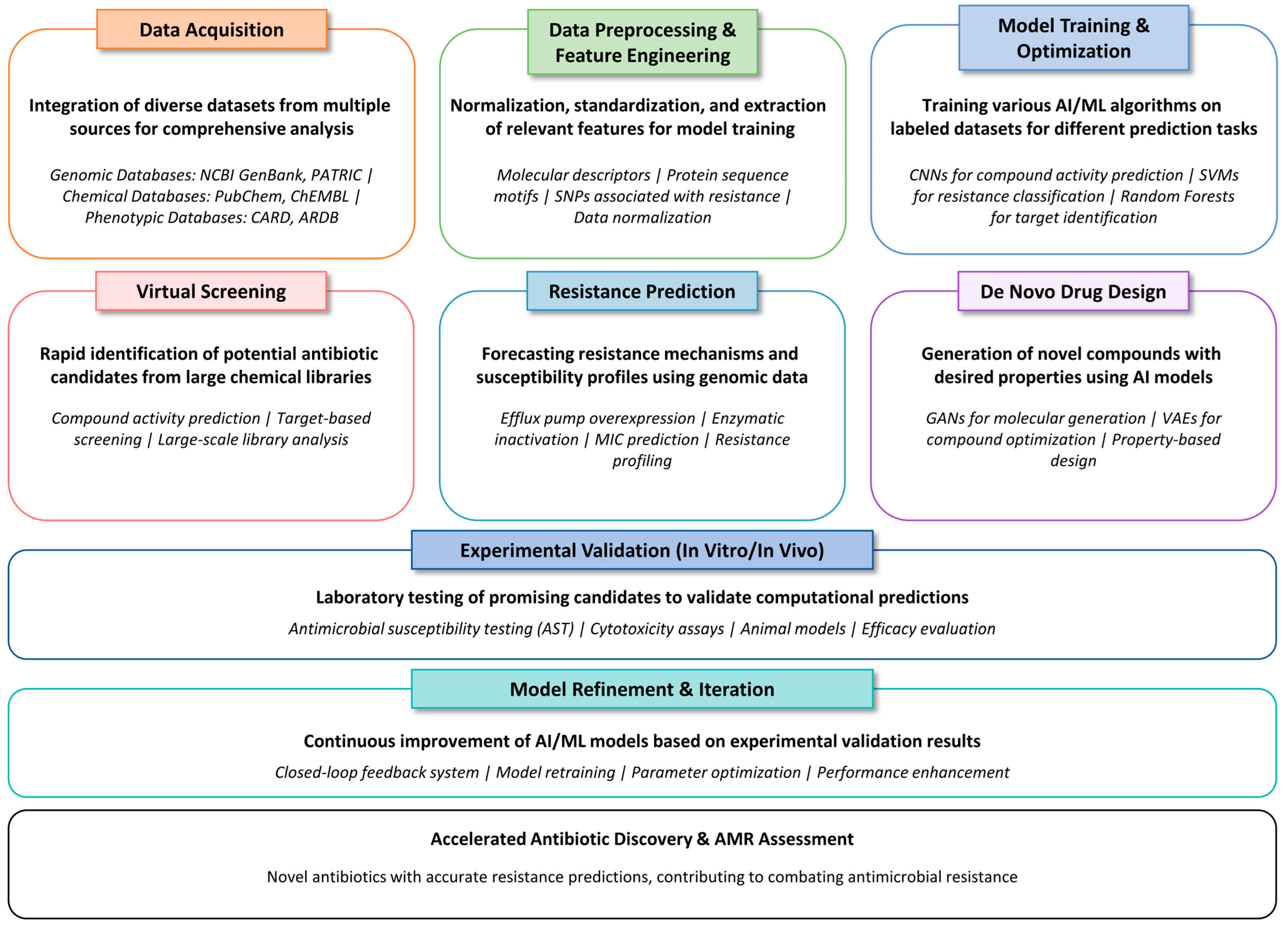
5.7. AI in Antibiotic Discovery and Resistance Prediction
6. Real-World Evidence (RWE) and Case Studies
6.1. Accelerated Timelines and Improved Success Rates
6.2. Case Studies: Notable AI-Designed Drugs in Clinical Development
6.3. Collaborative Ecosystem
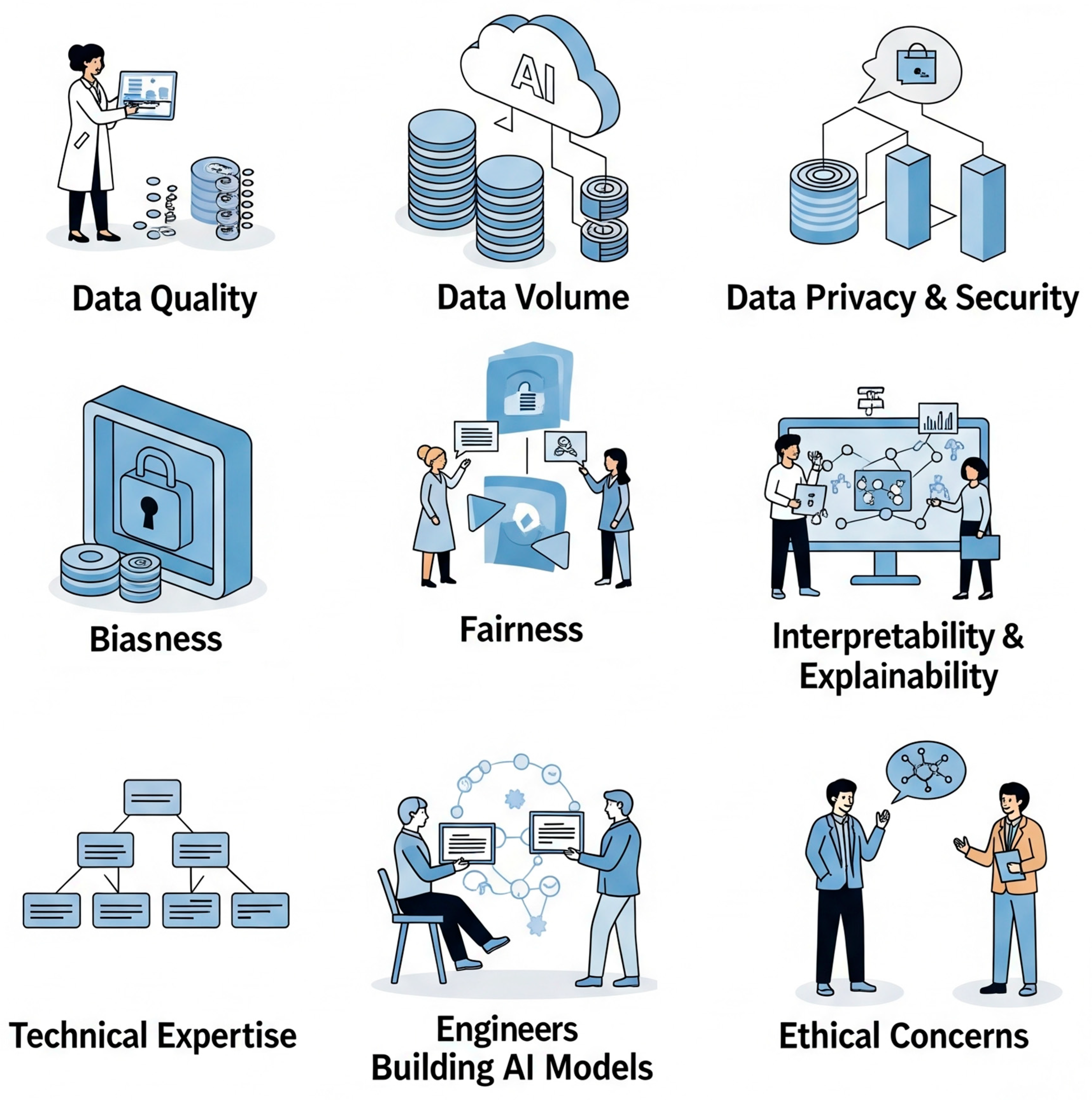
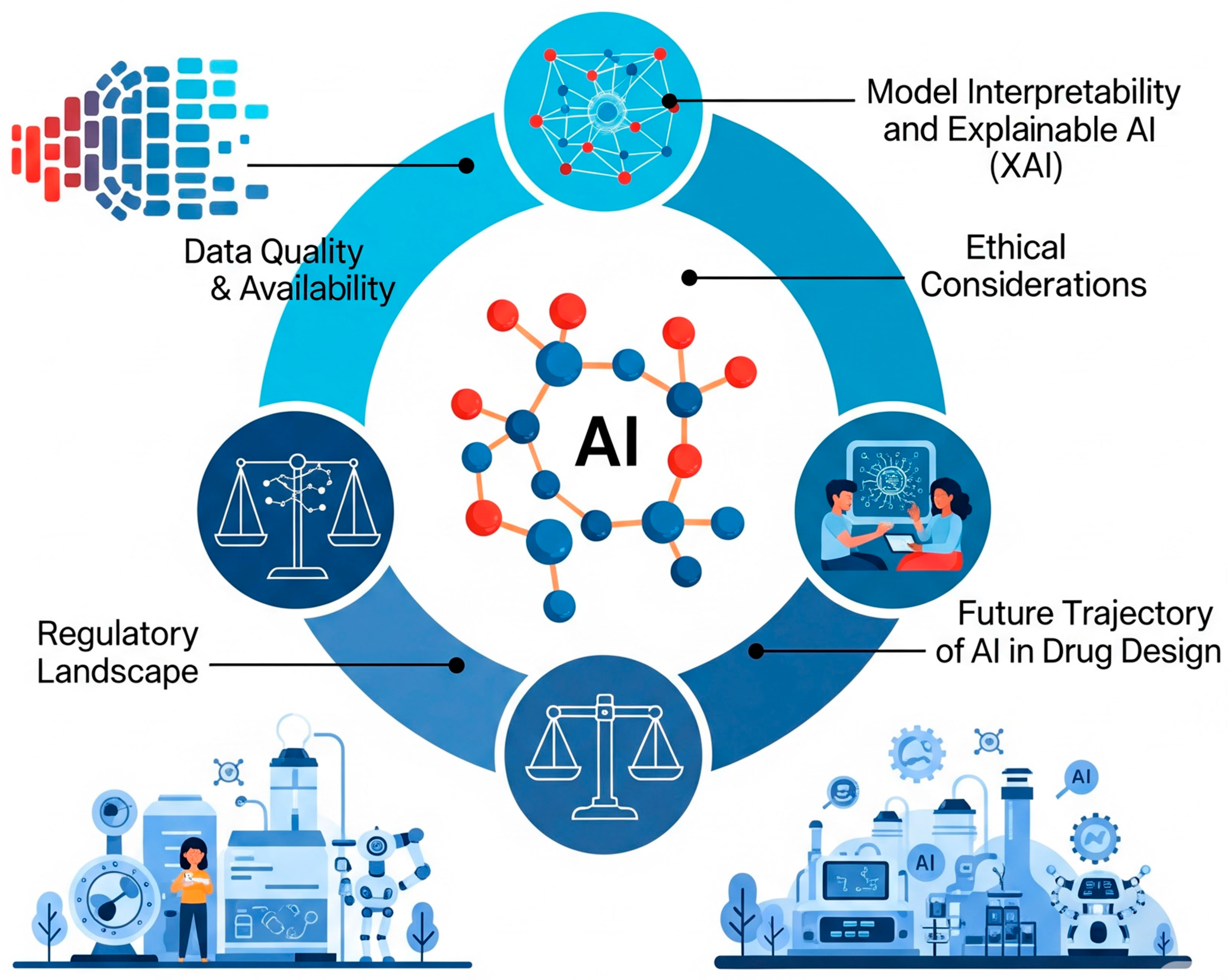
7. Challenges and Future Perspectives
7.1. Data Quality and Availability
7.2. Model Interpretability and Explainable AI (XAI)
7.3. Ethical Considerations
7.4. Regulatory Landscape
7.5. Future Trajectory of AI in Drug Design
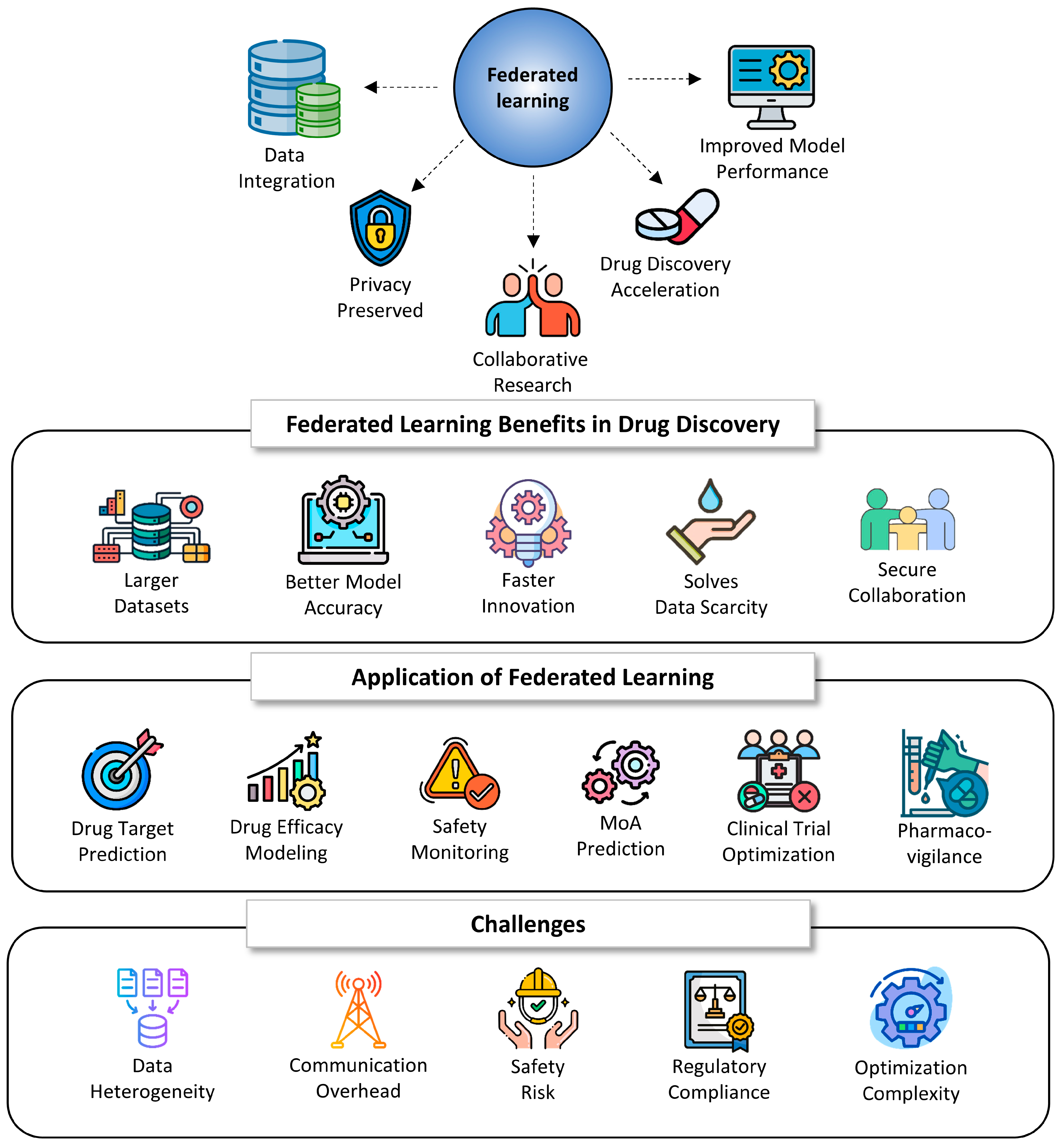
7.6. Federated Learning (FL)
8. Critical Perspective on the Application of AI in Small Molecule Design
8.1. Method Comparisons and Contextual Performance
8.2. Limitations of Current Benchmarks and Datasets
8.3. Overemphasis on Accuracy: The Blind Spot of Explainability
8.4. Reproducibility and Generalizability Concerns
8.5. Disconnect from Real-World Drug Discovery
8.6. Toward Trustworthy and Sustainable AI in Molecular Design
9. Conclusions and Future Vision
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAE | Adversarial Autoencoder |
| AMR | Antimicrobial Resistance |
| ANN | Artificial Neural Network |
| ARDB | Antibiotic Resistance Genes Database |
| BN | Bayesian Network |
| CARD | Comprehensive Antibiotic Resistance Database |
| CPTAC | Clinical Proteomic Tumor Analysis Consortium |
| CRISPR-Cas9 | Clustered Regularly Interspaced Short Palindromic Repeat |
| cryo-EM | Cryo-Electron Microscopy |
| CSM | Computed Structure Models |
| DNN | Deep Neural Network |
| DT | Decision Tree |
| DTI | Drug–Target Interaction |
| ECFP | Extended Connectivity Fingerprints |
| EHR | Electronic Health Record |
| EST | Expressed Sequence Tag |
| GAN | Generative Adversarial Network |
| GAT | Graph Attention Networks |
| GBM | Gradient Boosting Algorithm |
| GCN | Graph Convolutional Network |
| GCPNet | Generic Crystal Pattern Graph Neural Network |
| GNN | Graph Neural Network |
| GPCRs | G Protein-Coupled Receptors |
| GPR | Gaussian Process Regression |
| GRU | Gated Recurrent Unit |
| GVS | Generative Virtual Screening |
| GWAS | Genome-Wide Association Study |
| HTS | High-Throughput Screening |
| IND | Investigational New Drug |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| k-NN | k-Nearest Neighbor |
| KRR | Kernel Ridge Regression |
| LBVS | Ligand-Based Virtual Screening |
| LLM | Large Language Models |
| LR | Logistic Regression |
| LSTM | Long Short-Term Memory |
| MGC | Molecular Graph Convolution |
| MIPD | Model-Informed Precision Dosing |
| MLP | Multilayer Perceptron |
| MPNN | Message Passing Neural Network |
| MS | Mass Spectrometry |
| NB | Naïve Bayes |
| NCIGDC | National Cancer Institute Genomic Data Commons |
| NGS | Next-Generation Sequencing |
| NMR | Nuclear Magnetic Resonance |
| PCA | Principal Component Analysis |
| PDB | Protein Data Bank |
| PLS-DA | Partial Least Squares Discriminant Analysis |
| PPI | Protein–Protein Interaction |
| PTM | Post Translational Modification |
| QED | Quantitative Estimate of Drug Likeness |
| QSAR/QSPR | Quantitative Structure–Activity/Property Relationship |
| RF | Random Forest |
| RGNs | Recurrent Geometric Networks |
| RL | Reinforcement Learning |
| RNN | Recurrent Neural Network |
| SBVS | Structure-Based Virtual Screening |
| SE (3) | Special Euclidean |
| SELFIES | Self-Referencing Embedded Strings |
| SILAC | Stable Isotope Labeling by Amino Acids in Cell Culture |
| SMILES | Simplified Molecular Input Line Entry System |
| SNP | Single-Nucleotide Polymorphism |
| SOM | Self-Organizing Map |
| SRA | Sequence Read Archive |
| SVR | Support Vector Regression |
| TFN | Tensor Field Network |
| VAE | Variational Autoencoder |
| VEGFR2 | Vascular Endothelial Growth Factor Receptor 2 |
References
- Visan, A.I.; Negut, I. Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life 2024, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of Clinical Drug Development Fails and How to Improve It? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Obrezanova, O.; Martinsson, A.; Whitehead, T.; Mahmoud, S.; Bender, A.; Miljković, F.; Grabowski, P.; Irwin, B.; Oprisiu, I.; Conduit, G.; et al. Prediction of in Vivo Pharmacokinetic Parameters and Time-Exposure Curves in Rats Using Machine Learning from the Chemical Structure. Mol. Pharm. 2022, 19, 1488–1504. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Das, B.; Rawat, V.S.; Wahlang, J.B.; Nongpiur, A.; Tiewsoh, I.; Lyngdoh, N.M.; Das, D.; Bidarolli, M.; Sony, H.T. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. Int. J. Mol. Sci. 2023, 24, 2026. [Google Scholar] [CrossRef] [PubMed]
- Kattuparambil, A.A.; Chaurasia, D.K.; Shekhar, S.; Srinivasan, A.; Mondal, S.; Aduri, R.; Jayaram, B. Exploring Chemical Space for “Druglike” Small Molecules in the Age of AI. Front. Mol. Biosci. 2025, 12, 1553667. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, N.; Zhang, J.; Yang, B. Advances in Microfluidic Single-Cell RNA Sequencing and Spatial Transcriptomics. Micromachines 2025, 16, 426. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.; Moayedpour, S.; Li, R.; Corrochano-Navarro, A.; Kötter, A.; Kogler-Anele, L.; Riahi, S.; Grebner, C.; Hessler, G.; Matter, H.; et al. Deep Batch Active Learning for Drug Discovery. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ahmad, W.; Simon, E.; Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa-2: Towards Chemical Foundation Models. In Proceedings of the ELLIS Machine Learning for Molecule Discovery Workshop. arXiv 2022. [Google Scholar] [CrossRef]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. In Proceedings of the Machine Learning for Molecules Workshop at NeurIPS. arXiv 2020. [Google Scholar] [CrossRef]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.J.; Tkatchenko, A.; Müller, K.R. SchNet—A Deep Learning Architecture for Molecules and Materials. J. Chem. Phys. 2018, 148, 241722. [Google Scholar] [CrossRef] [PubMed]
- Gallego, V.; Naveiro, R.; Roca, C.; Ríos Insua, D.; Campillo, N.E. AI in Drug Development: A Multidisciplinary Perspective. Mol. Divers. 2021, 25, 1461–1479. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, N.W.A.; Gastegger, M.; Schütt, K.T. Symmetry-Adapted Generation of 3d Point Sets for the Targeted Discovery of Molecules. Adv. Neural Inf. Process Syst. 2020, 32, 680. [Google Scholar]
- Kong, W.; Hu, Y.; Zhang, J.; Tan, Q. Application of SMILES-Based Molecular Generative Model in New Drug Design. Front. Pharmacol. 2022, 13, 1046524. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Müller, A.T.; Huisman, B.J.H.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative Recurrent Networks for De Novo Drug Design. Mol. Inform. 2018, 37, 1700111. [Google Scholar] [CrossRef] [PubMed]
- Flores-Hernandez, H.; Martinez-Ledesma, E. A Systematic Review of Deep Learning Chemical Language Models in Recent Era. J. Cheminform. 2024, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Dalke, A. DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures. ChemRxiv 2018. [Google Scholar] [CrossRef]
- Scalia, G.; Grambow, C.A.; Pernici, B.; Li, Y.-P.; Green, W.H. Evaluating Scalable Uncertainty Estimation Methods for Deep Learning-Based Molecular Property Prediction. J. Chem. Inf. Model. 2020, 60, 2697–2717. [Google Scholar] [CrossRef] [PubMed]
- Reiser, P.; Neubert, M.; Eberhard, A.; Torresi, L.; Zhou, C.; Shao, C.; Metni, H.; van Hoesel, C.; Schopmans, H.; Sommer, T.; et al. Graph Neural Networks for Materials Science and Chemistry. Commun. Mater. 2022, 3, 93. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, M.; Wang, S.; Zhang, S. Deep Learning Methods for Molecular Representation and Property Prediction. Drug Discov. Today 2022, 27, 103373. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Kwon, J.J.; Liu, C.; Margiotta, E.; Shekhar, M.; Gould, A.E. Target-Driven Machine Learning-Enabled Virtual Screening (TAME-VS) Platform for Early-Stage Hit Identification. Front. Mol. Biosci. 2023, 10, 1163536. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Chen, Y.; Cheng, Y.; Wei, Y.; Li, Y.; Wang, J.; Wei, Y.; Chan, T.-F.; Li, Y. Deep Autoencoder for Interpretable Tissue-Adaptive Deconvolution and Cell-Type-Specific Gene Analysis. Nat. Commun. 2022, 13, 6735. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ma, L.-H.; Maletic-Savatic, M.; Liu, Z. NMRQNet: A Deep Learning Approach for Automatic Identification and Quantification of Metabolites Using Nuclear Magnetic Resonance (NMR) in Human Plasma Samples. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Gomari, D.P.; Schweickart, A.; Cerchietti, L.; Paietta, E.; Fernandez, H.; Al-Amin, H.; Suhre, K.; Krumsiek, J. Variational Autoencoders Learn Transferrable Representations of Metabolomics Data. Commun. Biol. 2022, 5, 645. [Google Scholar] [CrossRef] [PubMed]
- Galal, A.; Talal, M.; Moustafa, A. Applications of Machine Learning in Metabolomics: Disease Modeling and Classification. Front. Genet. 2022, 13, 1017340. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, A.; Gelzo, M.; Caterino, M.; Costanzo, M.; Ruoppolo, M.; Castaldo, G. Challenges in Metabolomics-Based Tests, Biomarkers Revealed by Metabolomic Analysis, and the Promise of the Application of Metabolomics in Precision Medicine. Int. J. Mol. Sci. 2022, 23, 5213. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial Chemistry in Drug Discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Adaptation of High-Throughput Screening in Drug Discovery—Toxicological Screening Tests. Int. J. Mol. Sci. 2011, 13, 427–452. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef] [PubMed]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of Machine Learning in Drug Discovery and Development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Kapsiani, S.; Howlin, B.J. Random Forest Classification for Predicting Lifespan-Extending Chemical Compounds. Sci. Rep. 2021, 11, 13812. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, M.; Kim, D. Utilizing Random Forest QSAR Models with Optimized Parameters for Target Identification and Its Application to Target-Fishing Server. BMC Bioinform. 2017, 18, 567. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Bae, H.; Jo, J.; Yoon, S. Comprehensive Ensemble in QSAR Prediction for Drug Discovery. BMC Bioinform. 2019, 20, 521. [Google Scholar] [CrossRef] [PubMed]
- Olier, I.; Sadawi, N.; Bickerton, G.R.; Vanschoren, J.; Grosan, C.; Soldatova, L.; King, R.D. Meta-QSAR: A Large-Scale Application of Meta-Learning to Drug Design and Discovery. Mach. Learn. 2018, 107, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.P.; Babu, M.S.; Garg, R.; Sowmya, S. Quantitative Structure-Activity Relationship Studies on Cyclic Urea-Based HIV Protease Inhibitors. J. Enzyme Inhib. 1998, 13, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Support Vector Regression-Based QSAR Models for Prediction of Antioxidant Activity of Phenolic Compounds. Sci. Rep. 2021, 11, 8806. [Google Scholar] [CrossRef] [PubMed]
- Guha, R. On Exploring Structure–Activity Relationships. Methods Mol. Biol. 2013, 993, 81–94. [Google Scholar] [PubMed]
- Jolliffe, I. A 50-Year Personal Journey through Time with Principal Component Analysis. J. Multivar. Anal. 2022, 188. [Google Scholar] [CrossRef]
- Mughal, H.; Bell, E.C.; Mughal, K.; Derbyshire, E.R.; Freundlich, J.S. Random Forest Model Predictions Afford Dual-Stage Antimalarial Agents. ACS Infect. Dis. 2022, 8, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Nedunchezhian, D.; Langeswaran, K.; Santhoshkumar, S. Identification of Novel Inhibitor Targeting Fyn Kinase Using Molecular Docking Analysis. Bioinformation 2019, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pérez, R.; Bajorath, J. Evolution of Support Vector Machine and Regression Modeling in Chemoinformatics and Drug Discovery. J. Comput. Aided Mol. Des. 2022, 36, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chen, J.; Cai, J.; Cao, M.; Yin, S.; Ji, M. Simultaneously Optimized Support Vector Regression Combined with Genetic Algorithm for Qsar Analysis of Kdr/Vegfr-2 Inhibitors. Chem. Biol. Drug Des. 2010, 75, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Y.; Wu, J.; Zhao, D.; Huang, J.; Lin, M.J.; Wang, L. Large-Scale Comparison of Machine Learning Methods for Profiling Prediction of Kinase Inhibitors. J. Cheminform. 2024, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Raymer, M.L.; Doom, T.E.; Kuhn, L.A.; Punch, W.F. Knowledge Discovery in Medical and Biological Datasets Using a Hybrid Bayes Classifier/Evolutionary Algorithm. IEEE Trans. Syst. Man Cybern. Part B (Cybern.) 2003, 33, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xiong, W.; Jia, L.; Xu, L.; Cai, Y.; Chen, Y.; Jin, J.; Gao, M.; Zhu, J. Developing a Naïve Bayesian Classification Model with PI3Kγ Structural Features for Virtual Screening against PI3Kγ: Combining Molecular Docking and Pharmacophore Based on Multiple PI3Kγ Conformations. Eur. J. Med. Chem. 2022, 244, 114824. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tropsha, A. Novel Variable Selection Quantitative Structure−Property Relationship Approach Based on the k-Nearest-Neighbor Principle. J. Chem. Inf. Comput. Sci. 2000, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction Using Deep Learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Tiwari, S.; Jain, G.; Shetty, D.K.; Sudhi, M.; Balakrishnan, J.M.; Bhatta, S.R. A Comprehensive Review on the Application of 3D Convolutional Neural Networks in Medical Imaging. Eng. Proc. 2023, 59, 3. [Google Scholar] [CrossRef]
- Francoeur, P.; Masuda, T.; Koes, D.R. 3D Convolutional Neural Networks and a CrossDocked Dataset for Structure-Based Drug Design. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De Novo Design of Protein Structure and Function with RFdiffusion. Nature 2023, 620, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Besharatifard, M.; Vafaee, F. A Review on Graph Neural Networks for Predicting Synergistic Drug Combinations. Artif. Intell. Rev. 2024, 57, 49. [Google Scholar] [CrossRef]
- Wang, C.; Kumar, G.A.; Rajapakse, J.C. Drug Discovery and Mechanism Prediction with Explainable Graph Neural Networks. Sci. Rep. 2025, 15, 179. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, G.L.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, Y.; Li, H.; Liu, X. Drug-Protein Interaction Prediction via Variational Autoencoders and Attention Mechanisms. Front. Genet. 2022, 13, 1032779. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Fan, M.; Cui, H.; Zhang, T.; Nakaguchi, T. GVDTI: Graph Convolutional and Variational Autoencoders with Attribute-Level Attention for Drug-Protein Interaction Prediction. Brief. Bioinform. 2022, 23, bbab453. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular De-Novo Design through Deep Reinforcement Learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; Isayev, O.; Tropsha, A. Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gao, K.; Nguyen, D.D.; Chen, X.; Jiang, Y.; Wei, G.W.; Pan, F. Algebraic Graph-Assisted Bidirectional Transformers for Molecular Property Prediction. Nat. Commun. 2021, 12, 3521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zheng, L.; Wang, S.; Lin, M.; Wang, Z.; Kong, A.W.K.; Mu, Y.; Wei, Y.; Li, W. A Fully Differentiable Ligand Pose Optimization Framework Guided by Deep Learning and a Traditional Scoring Function. Brief. Bioinform. 2023, 24, bbac520. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. DeepDR: A Network-Based Deep Learning Approach to in Silico Drug Repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A New Open-Source Player in the Drug Discovery Field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, L. Defining Dynamic Protein Interactions Using SILAC-Based Quantitative Mass Spectrometry. Methods Mol. Biol. 2014, 1188, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Hawkins, B.A.; Du, J.J.; Groundwater, P.W.; Hibbs, D.E.; Lai, F. A Guide to In Silico Drug Design. Pharmaceutics 2022, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-Y.; Lee, C.-Y.; Kim, C.-E. Predicting Activatory and Inhibitory Drug–Target Interactions Based on Structural Compound Representations and Genetically Perturbed Transcriptomes. PLoS ONE 2023, 18, e0282042. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Kantliwala, S.V.; Vybhavi, J.; Ravi, R.; Patel, H.; Patel, J. Review of AlphaFold 3: Transformative Advances in Drug Design and Therapeutics. Cureus 2024, 16, e63646. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold Accelerates Artificial Intelligence Powered Drug Discovery: Efficient Discovery of a Novel CDK20 Small Molecule Inhibitor. Chem. Sci. 2023, 14, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Sun, L.; Xia, Y.; Qin, T.; Zhang, S.; Poon, H.; Liu, T.-Y. BioGPT: Generative Pre-Trained Transformer for Biomedical Text Generation and Mining. Brief. Bioinform. 2022, 23, bbac409. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 117793221989905. [Google Scholar] [CrossRef] [PubMed]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s Biochemical and Cell-Based Approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.N.; Blackwell, J.H.; Holdgate, G.A.; Lucas, S.C.C.; Solovyeva, A.; Storer, R.I.; Whitehurst, B.C. Hit Me with Your Best Shot: Integrated Hit Discovery for the next Generation of Drug Targets. Drug Discov. Today 2024, 29, 104143. [Google Scholar] [CrossRef] [PubMed]
- Nada, H.; Meanwell, N.A.; Gabr, M.T. Virtual Screening: Hope, Hype, and the Fine Line in Between. Expert. Opin. Drug Discov. 2025, 20, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.N.; Sastry, G.N. Virtual Screening in Drug Discovery—A Computational Perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Lopez, J.; Aguila, S.A.; Gilson, M.K.; Brizuela, C.A. Improving Structure-Based Virtual Screening with Ensemble Docking and Machine Learning. J. Chem. Inf. Model. 2021, 61, 5362–5376. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H. The Science and Art of Structure-Based Virtual Screening. ACS Med. Chem. Lett. 2024, 15, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Pirzada, R.H.; Yasmeen, F.; Haseeb, M.; Javaid, N.; Kim, E.; Choi, S. Small Molecule Inhibitors of IL-1R1/IL-1β Interaction Identified via Transfer Machine Learning QSAR Modelling. Int. J. Biol. Macromol. 2024, 282, 137295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, Y.; Li, W.; Huang, N. A Comprehensive Survey of Prospective Structure-Based Virtual Screening for Early Drug Discovery in the Past Fifteen Years. Int. J. Mol. Sci. 2022, 23, 15961. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cui, D.; Jerome, S.V.; Michino, M.; Lenselink, E.B.; Huggins, D.J.; Beautrait, A.; Vendome, J.; Abel, R.; Friesner, R.A.; et al. Enhancing Hit Discovery in Virtual Screening through Absolute Protein–Ligand Binding Free-Energy Calculations. J. Chem. Inf. Model. 2023, 63, 3171–3185. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Yaacoub, J.C.; Gleave, J.; Fernandez, M.; Ton, A.-T.; Ban, F.; Stern, A.; Cherkasov, A. Artificial Intelligence–Enabled Virtual Screening of Ultra-Large Chemical Libraries with Deep Docking. Nat. Protoc. 2022, 17, 672–697. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dong, G.; Sheng, C. Structural Simplification: An Efficient Strategy in Lead Optimization. Acta Pharm. Sin. B 2019, 9, 880–901. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Shi, C.; Chen, J. GeneralizedDTA: Combining Pre-Training and Multi-Task Learning to Predict Drug-Target Binding Affinity for Unknown Drug Discovery. BMC Bioinform. 2022, 23, 367. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, D.; Alenicheva, A.; Grisoni, F. Exposing the Limitations of Molecular Machine Learning with Activity Cliffs. J. Chem. Inf. Model. 2022, 62, 5938–5951. [Google Scholar] [CrossRef] [PubMed]
- MacLean, F. Knowledge Graphs and Their Applications in Drug Discovery. Expert. Opin. Drug Discov. 2021, 16, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Slosky, L.M.; Caron, M.G.; Barak, L.S. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol. Sci. 2021, 42, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A Soft Tool for 3D Drug Design of Protein Targets by Artificial Intelligence and Classical Algorithm. Brief. Bioinform. 2021, 22, bbaa161. [Google Scholar] [CrossRef] [PubMed]
- Heid, E.; Greenman, K.P.; Chung, Y.; Li, S.-C.; Graff, D.E.; Vermeire, F.H.; Wu, H.; Green, W.H.; McGill, C.J. Chemprop: A Machine Learning Package for Chemical Property Prediction. J. Chem. Inf. Model. 2024, 64, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Staker, J.; Marshall, K.; Leswing, K.; Robertson, T.; Halls, M.D.; Goldberg, A.; Morisato, T.; Maeshima, H.; Ando, T.; Arai, H.; et al. De Novo Design of Molecules with Low Hole Reorganization Energy Based on a Quarter-Million Molecule DFT Screen: Part 2. J. Phys. Chem. A 2022, 126, 5837–5852. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef] [PubMed]
- B Fortela, D.L.; Mikolajczyk, A.P.; Carnes, M.R.; Sharp, W.; Revellame, E.; Hernandez, R.; Holmes, W.E.; Zappi, M.E. Predicting Molecular Docking of Per- and Polyfluoroalkyl Substances to Blood Protein Using Generative Artificial Intelligence Algorithm DiffDock. Biotechniques 2023, 76, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Krivák, R.; Hoksza, D. P2Rank: Machine Learning Based Tool for Rapid and Accurate Prediction of Ligand Binding Sites from Protein Structure. J. Cheminform. 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Sherieff, A.; Mohibbe Azam, M.; Sesha Maheswaramma, K. 3D Structure Prediction and Visualization of Protein of the Novel Strain of Rhodopseudomonas Faecalis. Int. J. Adv. Res. 2023, 11, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Fiscato, M.; Segler, M.H.S.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kreis, K.; Veccham, S.P.; Liu, M.; Reidenbach, D.; Paliwal, S.; Vahdat, A.; Nie, W. Molecule Generation with Fragment Retrieval Augmentation. arXiv 2024, arXiv:2411.12078. [Google Scholar]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Sumner, J.; Ling, L.H.; Quek, R.H.C.; Tan, A.T.H.; Teng, G.G.; Seetharaman, S.K.; Gollamudi, S.P.K.; Ho, D.; Motani, M. Personalised Dosing Using the CURATE.AI Algorithm: Protocol for a Feasibility Study in Patients with Hypertension and Type II Diabetes Mellitus. Int. J. Environ. Res. Public Health 2022, 19, 8979. [Google Scholar] [CrossRef] [PubMed]
- Londhe, V.Y.; Bhasin, B. Artificial Intelligence and Its Potential in Oncology. Drug Discov. Today 2019, 24, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A Public Database for Medicinal Chemistry, Computational Chemistry and Systems Pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Krasowski, A.; Muthas, D.; Sarkar, A.; Schmitt, S.; Brenk, R. DrugPred: A Structure-Based Approach To Predict Protein Druggability Developed Using an Extensive Nonredundant Data Set. J. Chem. Inf. Model. 2011, 51, 2829–2842. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Gouy, A.; Daub, J.T.; Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Nucleic Acids Res. 2017, 13, 2498–2504. [Google Scholar] [CrossRef]
- Amendola, G.; Cosconati, S. PyRMD: A New Fully Automated AI-Powered Ligand-Based Virtual Screening Tool. J. Chem. Inf. Model. 2021, 61, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.; Trapotsi, M.-A.; Spjuth, O.; Singh, S.; Carreras-Puigvert, J.; Greene, N.; Bender, A.; Carpenter, A.E. Cell Painting: A Decade of Discovery and Innovation in Cellular Imaging. Nat. Methods 2025, 22, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Yeats, C.A.; Goater, R.J.; Abudahab, K.; Taylor, B.; Underwood, A.; Sánchez-Busó, L.; Wong, V.K.; Dyson, Z.A.; Nair, S.; et al. A Global Resource for Genomic Predictions of Antimicrobial Resistance and Surveillance of Salmonella Typhi at Pathogenwatch. Nat. Commun. 2021, 12, 2879. [Google Scholar] [CrossRef] [PubMed]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A Database for Classification of Antimicrobial Drug, Biocide and Metal Resistance Determinants in Metagenomic Sequence Data. Nucleic Acids Res. 2020, 48, D561–D569. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A Deep Learning Approach for Predicting Antibiotic Resistance Genes from Metagenomic Data. Microbiome 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T. Benford’s Law and Distributions for Better Drug Design. Expert. Opin. Drug Discov. 2024, 19, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Tardiel-Cyril, D.R.; Chai, D.; Generali, D.; Li, J.-R.; Vazquez-Perez, J.; Lim, J.M.; Morris, R.; Bullock, Z.N.; Davtyan, A.; et al. Artificial Intelligence-Powered Discovery of Small Molecules Inhibiting CTLA-4 in Cancer. BJC Rep. 2024, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackerman, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef] [PubMed]
- Xie, E.; Hasegawa, K.; Kementzidis, G.; Papadopoulos, E.; Aktas, B.H.; Deng, Y. An AI-Driven Framework for Discovery of BACE1 Inhibitors for Alzheimer’s Disease. bioRxiv 2024. [Google Scholar] [CrossRef]
- Burki, T. A New Paradigm for Drug Development. Lancet Digit. Health 2020, 2, e226–e227. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.J.; Robinson, B.W.S.; Smith, D.P.; Stebbing, J. The AI-Assisted Identification and Clinical Efficacy of Baricitinib in the Treatment of COVID-19. Vaccines 2022, 10, 951. [Google Scholar] [CrossRef] [PubMed]
- Vladimer, G.; Alt, I.; Sehlke, R.; Lobley, A.; Baumgärtler, C.; Stulic, M.; Hackner, K.; Dzurillova, L.; Petru, E.; Hadjari, L.; et al. 23P Enriching for Response: Patient Selection Criteria for A2AR Inhibition by EXS-21546 through Ex Vivo Modelling in Primary Patient Material. Immuno-Oncol. Technol. 2022, 16, 100128. [Google Scholar] [CrossRef]
- O’Connell, K.A.; Yosufzai, Z.B.; Campbell, R.A.; Lobb, C.J.; Engelken, H.T.; Gorrell, L.M.; Carlson, T.B.; Catana, J.J.; Mikdadi, D.; Bonazzi, V.R.; et al. Accelerating Genomic Workflows Using NVIDIA Parabricks. BMC Bioinform. 2023, 24, 221. [Google Scholar] [CrossRef] [PubMed]
- Ripabelli, G.; Salzo, A.; Tamburro, M. Safety of MRNA BNT162b2 COVID-19 (Pfizer-BioNtech) Vaccine in Children Aged 5–11 Years: Author’s Reply to Correspondence. Hum. Vaccin. Immunother. 2023, 19, 2168947. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Representation Type | Classical ML | DL Architectures | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| SMILES (1D strings) | SVM, RF, PLS, k-NN | RNN (LSTM, GRU), Transformers | Simple and compact, easy to store and parse, widely supported format | Non-unique representations, sensitive to syntax errors, lacks 3D stereochemical details | [13,14] |
| SELFIES (1D robust strings) | SVM, RF, PLS, k-NN | Transformers | 100% syntactically valid, maintains expressiveness | Less human readable than smiles | [15,16] |
| Molecular Graphs (2D atom-bond networks) | Graph kernels, SVM, RF | MPNN, GCN, GAT | Naturally encoding of atomic connectivity, capture local and global graph topology | Computationally expensive, high memory requirements | [17,18] |
| 3D Conformers (3D grids or point clouds) | RF, SVM | CNN, SE (3), SchNet, DimeNet, PaiNN | Encodes stereochemistry and spatial interactions | Sensitive to conformer generation, data- and computationally intensive | [19] |
| Fingerprints (fixed-length vectors, ECFP, MACCS) | SVM, RF, PLS, k-NN | MLP | Fast similarity search, interpretable binary features, Compact, fixed-length vectors | Ignore 3D detail, lose stereochemical and spatial details | [20] |
| Gene Expression Profiles | SVM, RF, LR, k-NN, PCA, PLS, | DNN, GNN, Autoencoders | Captures cellular state and pathway-level insights, reflects co-expression and regulatory activity | High dimensionality vs. small sample size, prone to overfitting, interpretability limits | [21] |
| Metabolite Profiles (NMR/MS) | PLS-DA, RF, SVM, PCA | Autoencoders, GNN, CNN | Biochemical and phenotype context, reflect pathway-level function | Instrument and batch variability, uneven metabolite coverage | [22,23] |
| Database (URL) | Scope | Key Statistics | Data Types | Classical ML | Deep Learning | Advantages | Disadvantages |
|---|---|---|---|---|---|---|---|
| ChEMBL https://www.ebi.ac.uk/chembl/ (accessed on 7 April 2025) | Curated bioactivity data: molecules, assays, mechanism of action, PD/PK | 2.5 M compounds, 1.7 M assays, 16 K targets | 2D structures, SMILES, InChI, physicochemical and bioactivity values (IC50/Ki/EC50) | PLS, RF, SVM | RNN, GNN, MPNN, GCN, Transformers, | High-quality curation, integrated chemical–biological data | Drug-like chemical bias, sparse 3D coverage, assay heterogeneity |
| PubChem https://pubchem.ncbi.nlm.nih.gov/ (accessed on 7 April 2025) | Open chemical resource for chemical structures, properties, bioassays, and literature | 119 M compounds, 330 M substances, 297 M bioactivities | Physicochemical properties, SMILES, InChI, bioassay results, substances metadata | k-NN, RF, SVM | Transformers, GNN | Largest public chemical repository, rich with biological links | Variable data quality, inconsistent annotations, extensive HTS noise |
| DrugBank https://go.drugbank.com/ (accessed on 7 April 2025) | Drug-centric database integrating chemistry, pharmacology, mechanisms, interactions, and ADMET | 17 K drug entries, 2991 approved drugs, 1726 approved biologics | SMILES, InChI, ADMET, pharmacodynamics/kinetics, drug-target interactions | PLS, RF, SVM | GNN, transformers | Deep integration of chemical and clinical information, curated manually | Pharmaceutical bias, licensing restrictions for some applications |
| DrugMatrix https://cebs.niehs.nih.gov/cebs/paper/15670 (accessed on 7 April 2025) | Toxicogenomic data from rodent models, including gene expression and pathology endpoints | 600 chemicals, Thousands of transcriptomic and pathology measurements | Microarray/RNA-seq gene expression, histopathology, clinical chemistry | SVM, RF, LR | DNN, autoencoders | Multimodal toxicological endpoints, standardized in vivo studies | Limited chemical diversity, rodent-specific applicability |
| BindingDB https://www.bindingdb.org/ (accessed on 7 April 2025) | Protein–ligand binding affinities for SAR, docking, and thermodynamics | 3 M affinity data, 1.3 M compounds, 9.5 K targets | Binding constants (Kd, Ki, IC50), ligand structures | RF, SVM | GNN, Siamese networks | High-quality affinity data, valuable for docking and SAR benchmarking | Limited to known targets, assay variability and noise |
| ZINC15 https://zinc15.docking.org/ (accessed on 7 April 2025) | Ready-to-dock compound library for virtual screening | 200 M molecules, 750 M purchasable compounds 37 B catalog size | 2D/3D small-molecules, vendor catalogs | k-NN, RF | GNN, CNN | Ultra-large-scale screening, diverse and purchasable compounds | No bioactivity data, requires significant indexing and storage |
| RCSB PDB https://www.rcsb.org/ (accessed on 7 April 2025) | 3D structures of macromolecules (proteins, RNA, complexes) | 236 K experimental structures, 1.06 M CSM | Atomic 3D coordinates (PDB/mmCIF), electron density, ligands | RF, SVM | CNN, SE (3)-equivariant nets | High-resolution structural gold standard, interactive visualization tools | Structural bias (e.g., solubility), conformational variability |
| UniProt https://www.uniprot.org/ (accessed on 7 April 2025) | Protein sequences and functional annotations | 252 M sequence entries, 573 K Swiss-Prot reviewed | FASTA sequences, GO terms, domains, PTMs | SVM, RF | Protein Transformers | Extensive coverage, manual curation in Swiss-Prot | Lower annotation quality in TrEMBL, redundancy |
| GEO https://www.ncbi.nlm.nih.gov/geo/ (accessed on 10 May 2025) | Gene expression datsets (microarray and RNA-Seq) across conditions and organisms | 7.8 M samples, 4 K Datasets, 27 K platforms | Expression matrices, sample phenotypes, metadata (GSE/GSM/GPL) | SVM, RF, LR, PCA/PLS | DNN, autoencoders | Broad context diversity, MIAME-compliant standardization | High dimensionality (p ≫ n), batch effects, requires preprocessing |
| TCGA (GDC) https://portal.gdc.cancer.gov/ (accessed on 10 May 2025) | Pan-cancer multiomics data and clinical metadata | 45 K patient cases, 1.1 M files >33 tumor types | Genomics, RNA-Seq, methylation, CNVs, proteomics, clinical annotations | SVM, RF, PLS-DA for biomarkers | DNN, GNN autoencoders | Large, deeply annotated cohorts across multiple cancers | Limited access controls, cross-platform heterogeneity |
| HMDB https://www.hmdb.ca/ (accessed on 10 May 2025) | Human metabolome: structures, biofluid concentrations, spectra, pathways | 220 K metabolites, 5700 MS/MS spectra, 1 K NMR spectra | Structures, biofluid levels, pathway and spectral data | PLS-DA, RF, SVM | GNN, Autoencoders | High-quality curation, spectral data, physiological relevance | Human-centric bias, delayed updates, clinical complexity |
| ArrayExpress https://www.ebi.ac.uk/arrayexpress/ (accessed on 10 May 2025) | Functional genomics data: expression profiling, microarrays, sequencing | 79 K experiments, 1.5 M profiles | Raw/processed data, experimental metadata | k-means/hierarchical, PLS-DA | DNN, GNN | Rich experiment metadata, linked to ENA | Inconsistent data formats, evolving standards, API complexity |
| GWAS Catalog https://www.ebi.ac.uk/gwas/ (accessed on 10 May 2025) | Curated genome-wide association studies and SNP-trait associations | 7 K publications, 799 K SNP-trait links, 118 K summary stats | Summary statistics, SNP–trait p-values, study metadata | LR on summary stats | Polygenic risk-score DL models | Manually curated associations, trait-level annotations | Limited to summary-level data, study variability and design bias |
| LINCS L1000 https://lincsproject.org/LINCS (accessed on 10 May 2025) | Gene expression signatures from chemical and genetic perturbations | 1.678 M signatures | L1000 landmark gene expression profiles (978 genes) | SVM, RF | DNN, autoencoders | Extensive perturbation atlas, standardized expression assay | Restricted to 978 genes, imputation for rest introduces noise |
| DisGeNET https://www.disgenet.org/ (accessed on 10 May 2025) | Gene–disease associations (GDA) from curated and text-mined sources | 1983 M associations linking 29 K genes to 42 K diseases | Gene–disease relationships, ontology mappings | Network-based random walks | GNN | Combines expert curation and literature mining | Text-mining false positives, heterogeneous evidence quality |
| STRING https://string-db.org/ (accessed on 1 June 2025) | Protein–protein interaction (PPI) networks based on experiments, predictions, and literature | 12 K organisms, 59 M proteins, 20 B interactions | Evidence-scored PPI networks | Random walk, network propagation | GNN | Comprehensive multi-evidence associations, user-friendly portal | Indirect interactions included, experimental coverage bias |
| STITCH http://stitch.embl.de/ (accessed on 1 June 2025) | Protein–chemical interaction networks from multiple sources | 2 K organisms, 0.5 M chemicals, 9.6 M proteins, 1.6 B interactions | Protein–chemical bipartite interaction networks | Similarity-based ML on chemical/protein profiles | GNN | Combines experimental, curated, and text-mined evidence | Noisy links from text-mining, variable confidence scores |
| KEGG https://www.kegg.jp/ (accessed on 1 June 2025) | Integrated genomic, chemical, and pathway database | 19 K compounds, 11 K glycans, 15 K reaction 8.2 K enzyme | Metabolic/reaction pathways, enzyme, drug, disease mappings | Network-based ML (e.g., random walks, PLS) | DNN, GNN | Pathway-based integrative multiomics, manually curated maps | FTP access requires subscription, slower update cycle |
| METLIN https://metlin.scripps.edu/ (accessed on 1 June 2025) | Experimental MS/MS spectra for metabolite identification | 960 K compounds | MS/MS spectra, neutral-loss data, precursor ions | spectral matching (cosine similarity) | CNN, deep Siamese nets | Largest public MS/MS repository, regular updates | No quantitative concentration data, preprocessing (e.g., peak picking) required |
| Expression Atlas https://www.ebi.ac.uk/gxa/home (accessed on 1 June 2025) | Gene and protein expression across conditions and species (baseline and differential) | 66 species, 4 K studies, 159 K assays | RNA-Seq, microarray, proteomics matrices | SVM, RF, LR, PCA/PLS | DNN, autoencoders, GNN | Standardized analysis pipelines, high cross-study comparability | Batch effects, occasional metadata incompleteness |
| Bgee https://bgee.org/ (accessed on 1 June 2025) | Healthy baseline gene expression across tissues and species | 52 species, 31 K RNA-Seq libraries 56 K unique conditions | Anatomical expression calls, ontology annotations | LR, RF on calls | DNN, autoencoders | Emphasizes healthy baselines, ontology-based integration | No disease data, limited to selected model organisms |
| MetaboLights https://www.ebi.ac.uk/metabolights/ (accessed on 1 June 2025) | Public metabolomics repository covering diverse platforms | 26 K compounds | Raw and processed NMR/MS spectra, concentrations, pathway roles | PLS-DA, RF, SVM, PCA | Autoencoders, GNN | Platform-agnostic, widely accepted for metabolomics publication | Metadata heterogeneity, identification pipelines still evolving |
| Type | Methods | Advantages | Disadvantages | Applications | Examples |
|---|---|---|---|---|---|
| Supervised | SVR (RBF kernel), Ridge, Lasso, Elastic Net, GPR | SVR captures nonlinear relationships, Ridge/Lasso/Elastic Net mitigate overfitting by regularization, GPR quantifies uncertainty | SVR requires kernel, Ridge/Lasso assume linearity, GPR scale poorly with data size | Predicting potency (IC50, logP), permeability (PAMPA/Caco-2), active learning in lead optimization | SVR on HIV protease inhibitors [34]; phenolics [35] |
| Unsupervised | PCA, k-Means, Hierarchical Clustering, SOM | Visualize chemical space, Reduces noise/dimensionality | Clustering may not reflect bioactivity, SOM requires tuning | Scaffold hopping, SAR exploration, Chemical diversity analysis | SOM for SAR maps [36]; PCA for dimensionality reduction [37] |
| Ensemble | RF, XGBoost, LightGBM | Robust to noise, RF require minimal tuning, High accuracy (GBM), Feature importance | Computationally intensive, Risk of overfitting without tuning | QSAR/QSPR modeling, Multi-objective scoring, Bioactivity and toxicity prediction | RF for anti-malarials [38] XGBoost for Fyn kinase inhibitors [39]; RF for QSAR [40] |
| Kernel-Based Methods | SVM, KRR | Effective for high dimensional data, Captures nonlinear patterns | High Computational cost, Complex hyperparameter tuning | HTS classification, toxicity profiling, low-data QSAR modeling | SVM for VEGFR2 inhibitors [41]; KRR for PLK1 inhibitors [42] |
| Probabilistic and Bayesian | Naïve Bayes (NB), Bayesian Networks (BN) | Fast training, handles large libraries efficiently, Bayesian networks allow causal modeling | NB assumes feature independence, BN requires expert knowledge | Early-stage virtual screening, toxicity triage, Mechanism-based interpretation | BN for QSAR interpretation [43] NB reduced PI3Kγ screening cost [44] |
| Instance-Based models | k-NN | Intuitive and Simple, no training phase, handles multi-class problems | Prediction slows with large dataset, Suffers from curse of dimensionality | Similarity-based screening, local SAR activity estimation | k-NN for QSAR [45] |
| Multitask and Transfer Learning | joint RF, BN, kernel models, GPR | Boosts low data performance, Reduces experimental needs, Ideal for rare/novel targets | Risk of negative transfer, Requires related bioassays | One-shot modeling, Cross-target predictions | One-shot GPR on GPCRs [46] |
| SBVS Tools | Mechanism | URL |
|---|---|---|
| MtiOpenScreen * | Web-based platform using AutoDock for structure-based virtual screening (SBVS) | https://bioserv.rpbs.univ-paris-diderot.fr/services/MTiOpenScreen/ (accessed on 23 May 2025) |
| FlexX-Scan ** | High-throughput docking tool employing incremental construction algorithms to accelerate docking | https://www.biosolveit.de/products/ (accessed on 23 May 2025) |
| DockM8 v1.0.3 * | Consensus scoring method combining multiple docking scoring functions to improve virtual screening accuracy | https://drugbud-suite.github.io/dockm8-web/ (accessed on 23 May 2025) |
| BindScope * (PlayMolecule) | Deep learning-based approach using CNNs to predict binding affinities on a large scale | https://open.playmolecule.org/landing BindScope (accessed on 23 May 2025) |
| GeauxDock * | Monte Carlo-based docking tool using hybrid scoring functions combining physics- and knowledge-based potentials | https://www.brylinski.org/geauxdock (accessed on 23 May 2025) |
| EasyVS * | Web-based tool for molecule library curation and docking-based virtual screening | https://bio.tools/easyvs (accessed on 23 May 2025) |
| DEKOIS 2.0 * | Provides decoy sets to benchmark and challenge VS pipelines, aiding performance assessment | http://www.dekois.com (accessed on 23 May 2025) |
| PL-PatchSurfer2 * | Uses 3D Zernike descriptors for local surface matching between ligands and receptor pockets | https://kiharalab.org/plps2/ (accessed on 23 May 2025) |
| SPOT-Ligand 2 * | Template-based screening approach enhanced by a large, diverse binding homology library | https://sparks-lab.org/server/spot-ligand2/ (accessed on 23 May 2025) |
| Gypsum-DL * | Open-source tool for generating 3D structures of small-molecules in various tautomeric and ionization states | https://durrantlab.pitt.edu/gypsum-dl/ (accessed on 23 May 2025) |
| ENRI * | Tool for selecting optimal protein conformations to enhance docking outcomes | https://github.com/fibonaccirabbits/enri (accessed on 23 May 2025) |
| LBVS Tools | Mechanism | URL |
|---|---|---|
| LBS-comparison * | Performance of eleven ligand binding site prediction methods were compared | https://github.com/bartongroup/LBS-comparison |
| VSFlow * | RDKit-based tool for substructure, fingerprint, and shape-based ligand screening | https://github.com/czodrowskilab/VSFlow |
| MolProphet ** | Implements 2D and 3D similarity algorithms for lead identification and profiling | https://molprophet.com/ |
| PharmScreen ** | LBVS using quantum mechanics-derived hydrophobic molecular field descriptors for 3D alignment | https://pharmacelera.com/pharmscreen/ |
| LiSiCA * | Software for 2D/3D ligand similarity using graph-based algorithms | http://insilab.org/lisica/ |
| Name | Description | Type | Access Link |
|---|---|---|---|
| CARD * | Curated database of AMR genes and mechanisms | Resistance genes | https://card.mcmaster.ca/ https://github.com/arpcard/rgi |
| ResFinder * | Tool for identifying acquired AMR genes and chromosomal mutations mediating antimicrobial resistance | Resistance gene detection | http://genepi.food.dtu.dk/resfinder |
| MEGARes * | Hierarchical classification of AMR genes for metagenomics | Metagenomics and AMR | https://www.meglab.org/ |
| PATRIC * | Comprehensive bacterial bioinformatics resource | Pathogen and AMR database | https://www.bv-brc.org |
| ARG-ANNOT * | Annotated reference gene database for AMR genes | Resistance gene curation | https://www.mediterranee-infection.com/acces-ressources/base-de-donnees/arg-annot-2/ |
| DeepARG * | DL-based tool to predict AMR genes from DNA/protein sequences | Resistance prediction | https://github.com/gaarangoa/deeparg |
| Pathogenwatch * | Surveillance platform for AMR and pathogen genomics | Genomic surveillance | https://pathogen.watch/ |
| MLAMP ** | ML tool novel antimicrobial peptides with notable antibacterial potency | Antimicrobial design | https://github.com/jkwang93/AMP-Designer |
| Platform | Description | URL |
|---|---|---|
| PandaOmics * | Cloud-based AI platform integrating multiomics and literature mining to prioritize novel disease targets; demonstrated by identifying 28 ALS candidates validated through Drosophila models. | https://pharma.ai/pandaomics |
| Open Targets ** | Consortium-based resource combining genetic, transcriptomic, proteomic, and NLP-derived evidence; employs XGBoost-based L2G scoring and knowledge graphs to prioritize GWAS loci for drug targeting. | https://platform.opentargets.org/ |
| BenevolentAI ** | Utilizes a proprietary ML engine and knowledge graph to normalize and analyze scientific literature, patents, and proprietary datasets for explainable target hypothesis generation. | https://www.benevolent.com/benevolent-platform/ |
| Recursion OS ** | Integrates high-content imaging, omics, and chemical data with GNN to create maps of Biology, revealing novel targets and expediting preclinical drug candidate identification. | https://www.recursion.com/technology |
| AtomNet ** (Atomwise) | Deep learning platform utilizing CNN trained on extensive compound libraries for structure-based virtual screening; achieved 74% hit rate across 318 targets and identified clinical candidate REC-3565. | https://www.atomwise.com/how-we-do-it/ |
| Chemistry42 ** | Generative AI suite (VAE, GAN, RNN) integrated with physics-based modeling for de novo molecular design and scaffold optimization, part of the Insilico Medicine Pharma.AI ecosystem. | https://insilico.com/chemistry42 |
| In Clinico ** | Transformer-based ensemble model that predicts Phase II–III clinical trial success (ROC AUC = 0.88) using multimodal clinical and molecular data; validated prospectively with 79% accuracy. | https://pharma.ai/inclinico |
| CTO 2.0 ** (ConcertAI) | SaaS platform for oncology and hematology trials leveraging real-world EHR and claims data to optimize trial eligibility, endpoint definition, and site selection. | https://www.concertai.com/clinical-trial-optimization |
| Deep 6 AI ** | NLP-powered platform that extracts insights from unstructured EHRs, pathology reports, and clinical notes to rapidly identify trial-eligible patients, streamlining recruitment processes. | https://deep6.ai/ |
| Saama Technologies ** | AI analytics suite for patient stratification, cross-platform data integration, and compliance monitoring, supporting enhanced recruitment and data quality throughout clinical trials. | https://www.saama.com/platform/products/data-hub/ |
| Medidata AI ** (Trials Analytics) | Real-time predictive analytics integrated into clinical infrastructure to forecast patient enrollment, site performance, and dropout risk based on industry benchmarks. | https://www.medidata.com/en/clinical-trial-products/medidata-ai/clinical-trial-analytics/ |
| Unlearn.ai ** | Employs Bayesian time-series modeling to create digital twins of trial participants, enabling in silico simulations, adaptive trial designs, and regulatory-aligned power calculations. | https://www.unlearn.ai/ |
| Trials.ai ** | NLP and ML-driven protocol optimization tool that analyzes previous trial data to refine eligibility criteria and endpoints, reducing protocol amendments and accelerating regulatory approval. | https://trials.ai/ |
| PhaseV ** | Platform offering AI-powered dashboards for trial design, risk forecasting, site selection, and scenario analysis at the portfolio level for sponsors and CROs. | https://www.phasevtrials.com/solutions |
| WCG AI Solutions ** | Embeds generative AI into clinical trial operations to support site feasibility, dropout risk prediction, and digital recruitment within a compliant, unified workflow. | https://www.wcgclinical.com/insights/generative-ai-the-path-to-unlocking-value/ |
| Worldwide Clinical Trials ** | CRO integrating AI-powered predictive analytics and patient segmentation tools into trial operations to optimize cohort selection and accelerate development timelines. | https://www.wct.com/ |
| Category | Aspect | Description |
|---|---|---|
| Benefits | Large and Diverse Datasets | FL enables the integration of data from multiple organizations, increasing dataset diversity and size, which enhances model robustness and generalizability |
| Privacy-Preserving Collaboration | FL allows for collaborative model training without sharing raw data, ensuring compliance with privacy regulations (e.g., HIPAA, GDPR) | |
| Improved Model Accuracy | Exposure to varied data distributions improves predictive performance and reduces overfitting | |
| Faster Drug Development | Leveraging distributed data accelerates target discovery, compound screening, and lead optimization | |
| Secure Knowledge Sharing | Institutions can share model insights without compromising proprietary data, promoting pre-competitive collaboration | |
| Addressing Data Scarcity | Combines small, fragmented datasets from multiple sources, enhancing modeling in rare diseases or under-researched conditions | |
| Applications | Drug Target Identification | FL integrates genomic, proteomic, and clinical data across sources to identify and validate new drug targets |
| Drug Efficacy Prediction | Models trained on federated clinical datasets can predict patient-specific drug responses, aiding precision medicine | |
| Drug Safety Prediction | Federated models can detect adverse events early by analyzing pharmacovigilance data from EHRs and other distributed sources | |
| Mechanism of Action (MoA) Analysis | Helps predict molecular interactions and mechanisms of new or repurposed drugs, supporting rational drug design | |
| Clinical Trial Optimization | Enhances trial design and execution by aggregating insights from multiple study sites and patient populations | |
| Real-Time Pharmacovigilance | Enables continuous monitoring of drug safety signals using real-world data while preserving data privacy | |
| Challenges | Data Heterogeneity | Variability in data types, formats, and distributions across institutions complicates model training and aggregation |
| Communication Overhead | Sharing frequent model updates across sites can result in high bandwidth and computational demands | |
| Security and Adversarial Threats | FL is vulnerable to model inversion, poisoning, and gradient leakage, requiring advanced security protocols (e.g., differential privacy, secure aggregation) | |
| Regulatory and Ethical Constraints | FL systems must comply with national and international data privacy laws, necessitating rigorous auditing and consent processes | |
| Model Convergence and Optimization | Heterogeneous data and hardware require specialized optimization strategies (e.g., adaptive FedAvg) to ensure reliable model convergence |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manan, A.; Baek, E.; Ilyas, S.; Lee, D. Digital Alchemy: The Rise of Machine and Deep Learning in Small-Molecule Drug Discovery. Int. J. Mol. Sci. 2025, 26, 6807. https://doi.org/10.3390/ijms26146807
Manan A, Baek E, Ilyas S, Lee D. Digital Alchemy: The Rise of Machine and Deep Learning in Small-Molecule Drug Discovery. International Journal of Molecular Sciences. 2025; 26(14):6807. https://doi.org/10.3390/ijms26146807
Chicago/Turabian StyleManan, Abdul, Eunhye Baek, Sidra Ilyas, and Donghun Lee. 2025. "Digital Alchemy: The Rise of Machine and Deep Learning in Small-Molecule Drug Discovery" International Journal of Molecular Sciences 26, no. 14: 6807. https://doi.org/10.3390/ijms26146807
APA StyleManan, A., Baek, E., Ilyas, S., & Lee, D. (2025). Digital Alchemy: The Rise of Machine and Deep Learning in Small-Molecule Drug Discovery. International Journal of Molecular Sciences, 26(14), 6807. https://doi.org/10.3390/ijms26146807