
Hepatocytes as Model for Investigating Natural Senotherapeutic Compounds and Their Effects on Cell Cycle Dynamics and Genome Stability

 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Nuclear and Mitochondrial Genome Damage: Implications for Aging and Disease
2.1. Progeroid Syndromes and DNA Repair Defects
2.2. Telomere Shortening
2.3. Epigenetic Changes
2.4. Protein Misfolding
2.5. Mitochondria Function Defects
2.6. Calorie Restriction
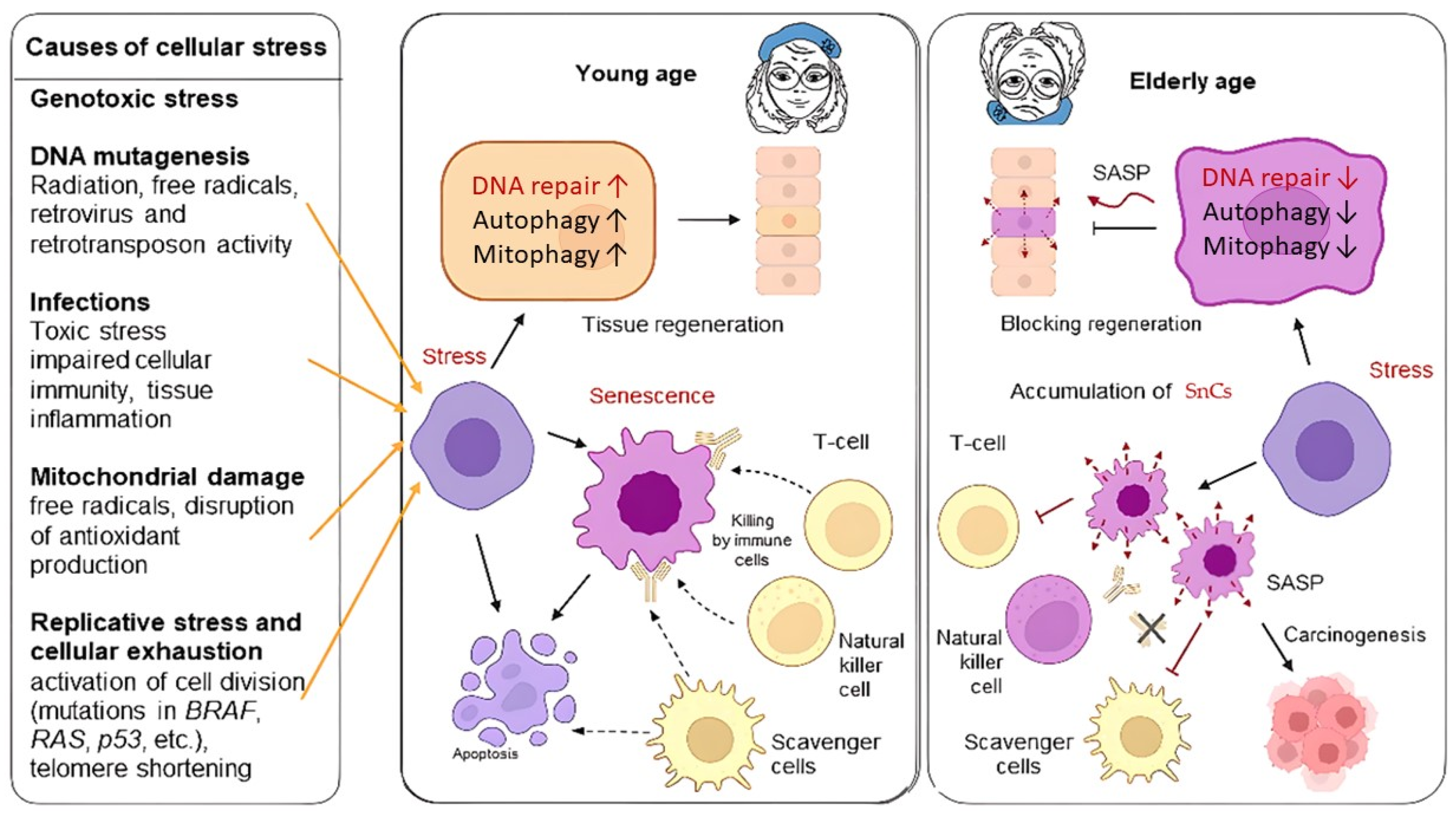
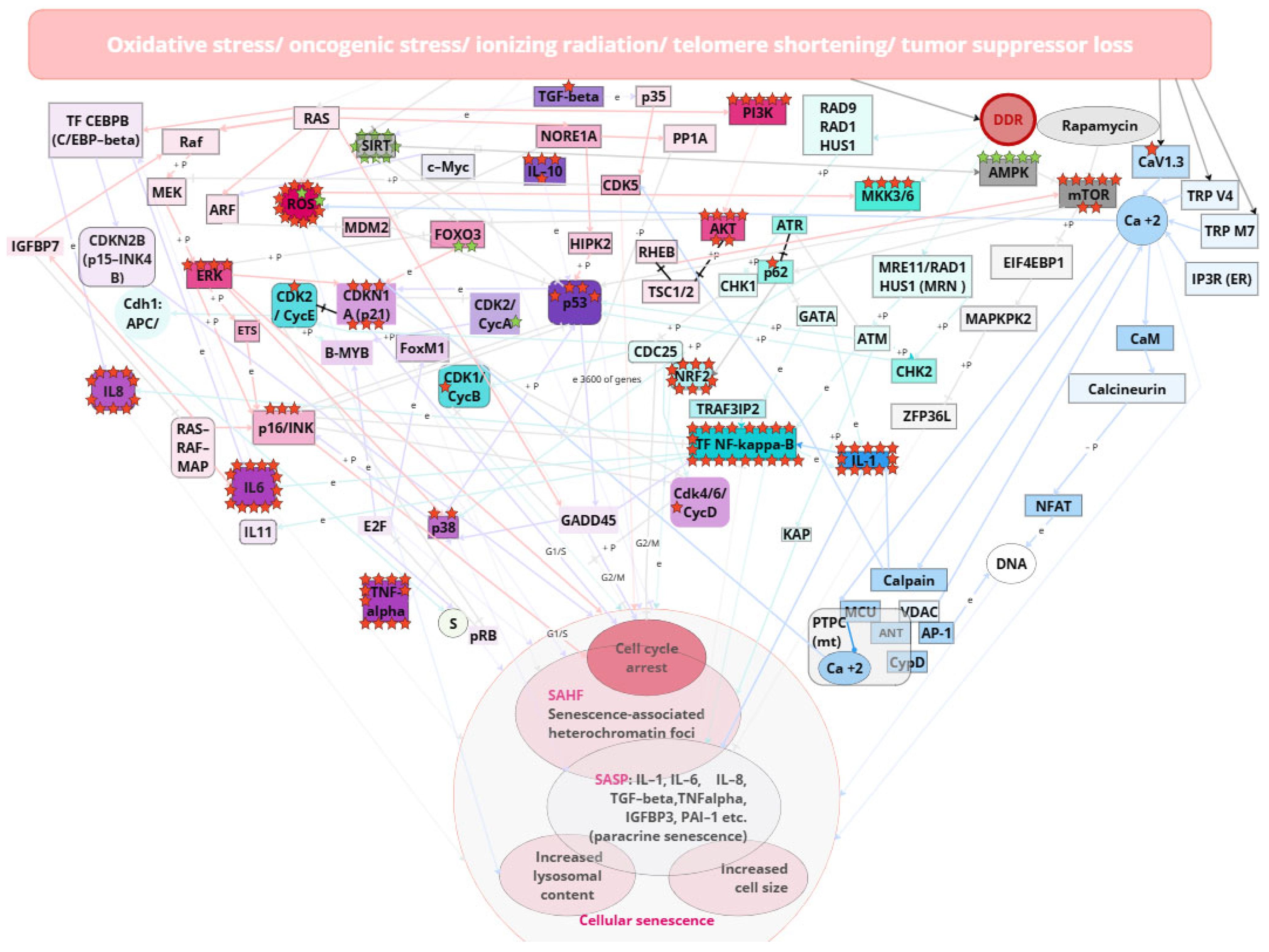
3. Senescent Phenotype of Cells
3.1. Calcium Signaling Pathway
3.2. DDR, ATR, ATM, p53, and NF-kB-Mediated Pathway
3.3. SASP
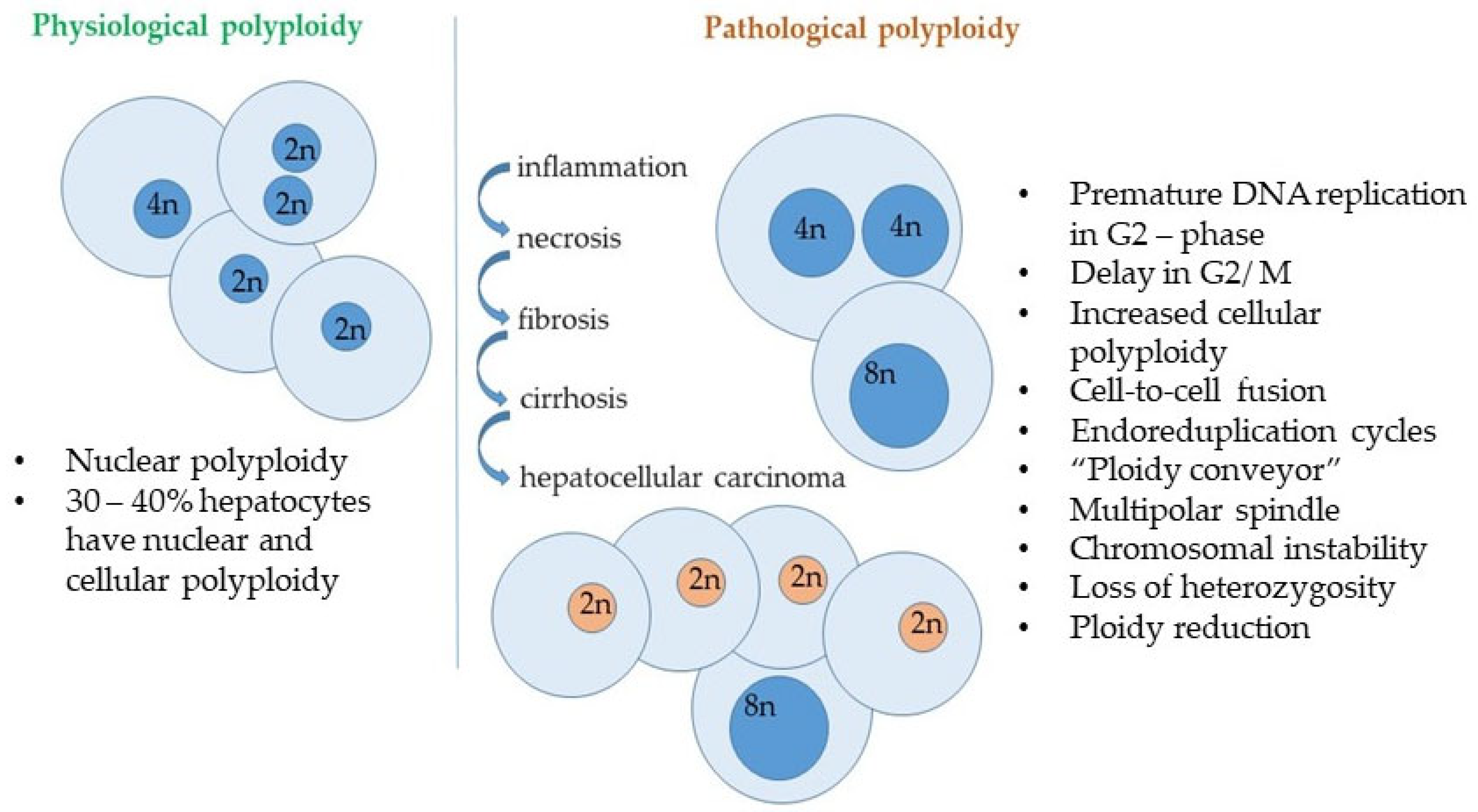
4. Senescence and DNA Stability in Liver Pathology
5. Senotherapeutic Compounds
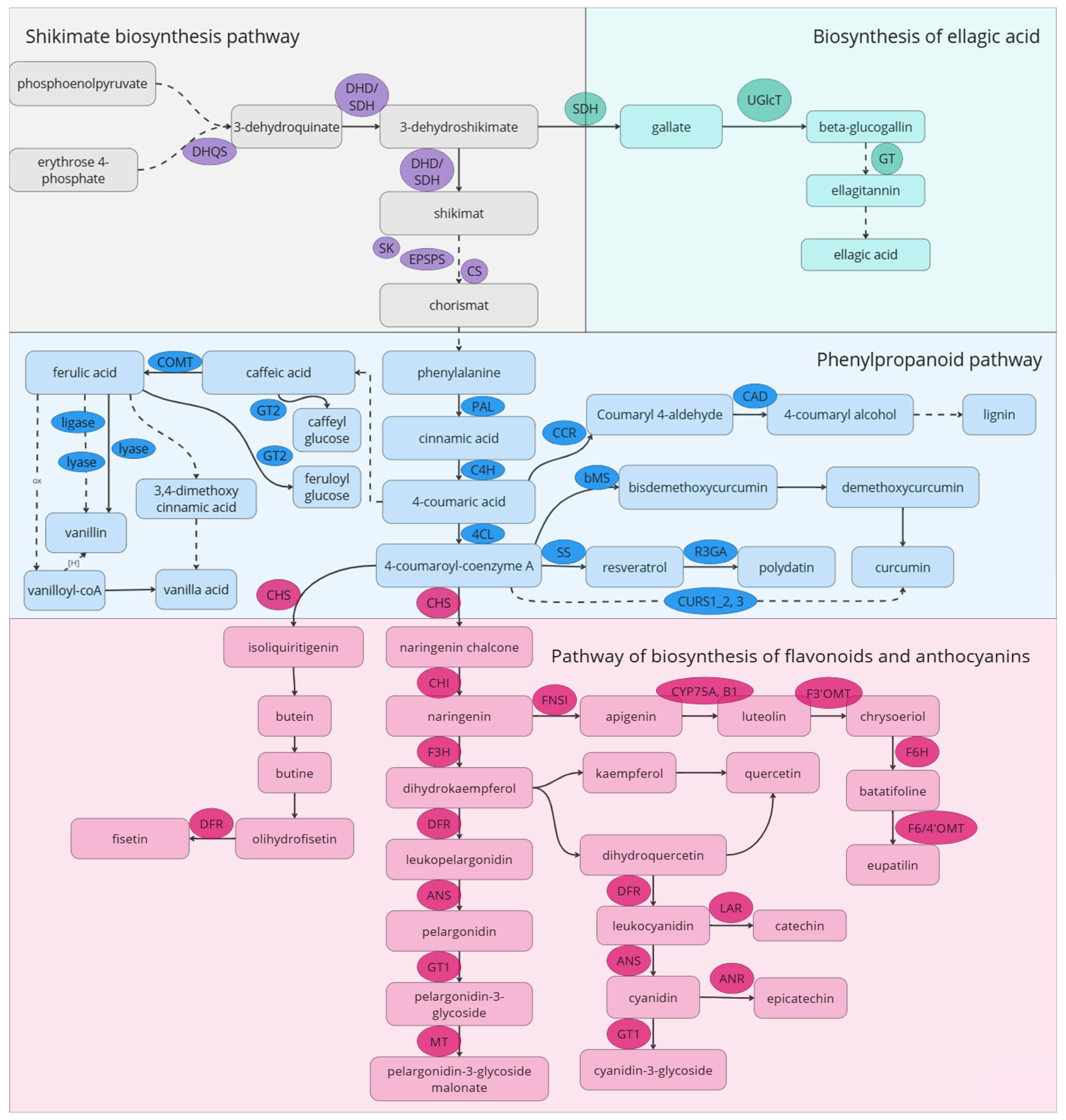
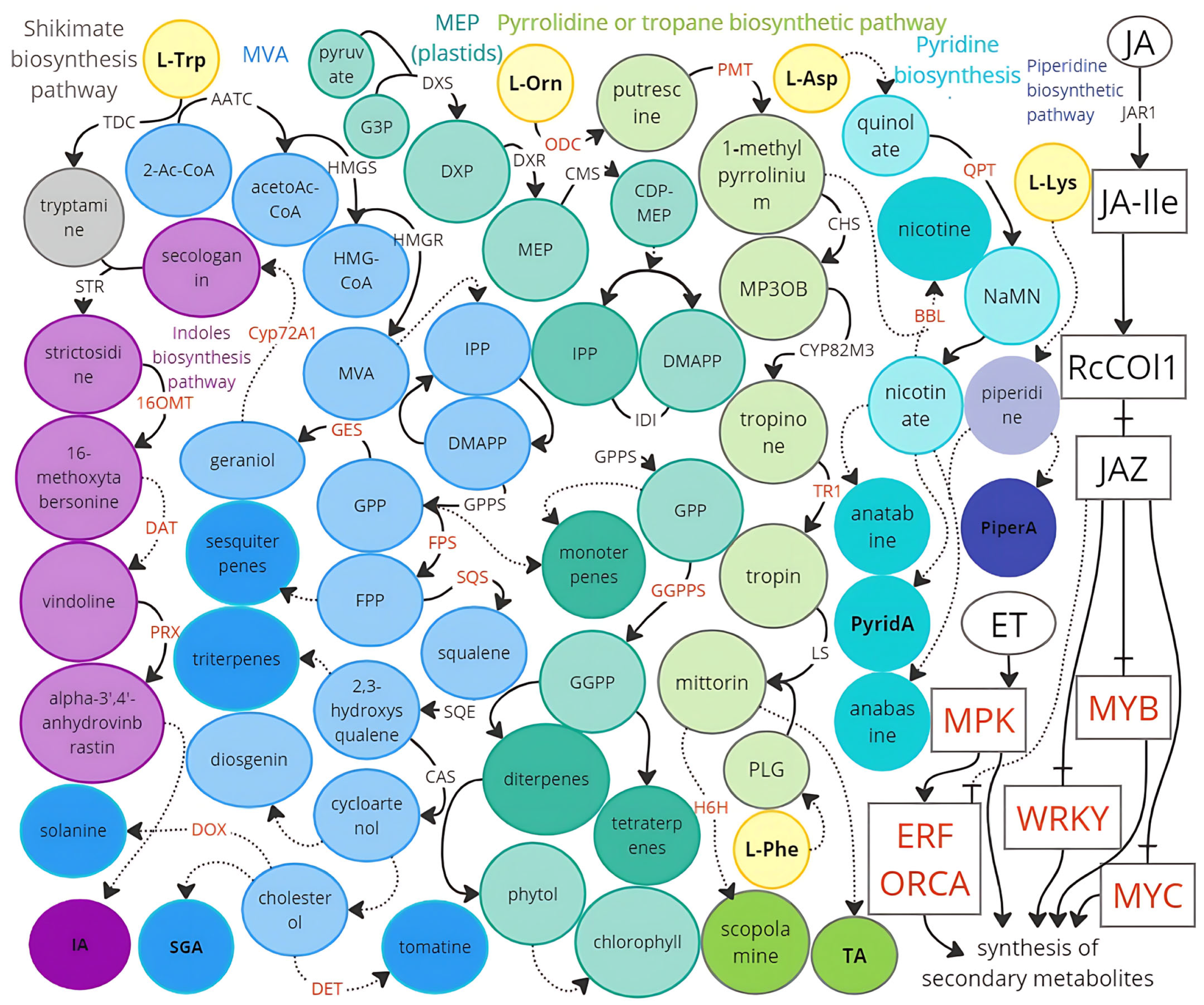
6. Biochemical Pathways Involved in the Accumulation of Senolytic Compounds: Potential Targets for Metabolic Engineering
6.1. Flavonoids Biosynthesis Pathway
6.2. Phenylpropanoid and Shikimate Pathways
6.3. Alkaloid Biosynthesis Pathways
6.4. Pseudoalkaloid Biosynthesis Pathways
7. Senotherapeutic Compounds and DNA Stability Maintenance
7.1. Resveratrol
7.2. Curcumin
7.3. Quercetin
7.4. Other Senotherapeutic Compounds
8. DNA Stability in Liver
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| SnCs | Senescent cells |
| ScMs | Secondary metabolites |
| ST | Senotherapeutic activity (senolytic or senomorphic) |
| SL | Senolytic activity or compound with senolytic activity |
| SM | Senomorphic activity or compound with senomorphic activity |
References
- Schumacher, B.; Pothof, J.; Vijg, J. The Central Role of DNA Damage in the Ageing Process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, K.; Vasilieva, M.; Minskaia, E.; Rybtsov, S.; Shevyrev, D. T-Cell Immunity against Senescence: Potential Role and Perspectives. Front. Immunol. 2024, 15, 1360109. [Google Scholar] [CrossRef] [PubMed]
- Karal-Ogly, D.D.; Shumeev, A.N.; Keburiya, V.V.; Mintel, M.V.; Rybtsov, S.A. Age-Related Changes in the Clustering of Blood Populations in Cynomolgus Monkeys Depend on Sex and Immune Status. Life 2023, 13, 316. [Google Scholar] [CrossRef] [PubMed]
- Boychenko, S.; Egorova, V.S.; Brovin, A.; Egorov, A.D. White-to-Beige and Back: Adipocyte Conversion and Transcriptional Reprogramming. Pharmaceuticals 2024, 17, 790. [Google Scholar] [CrossRef]
- Rybtsov, S.; Berezina, T.; Rybtsova, N. The Pre-Retirement Stress, Immune-Senescence, Inflammation and Biological Age. FEBS Open Bio 2021, 11, 103–507. [Google Scholar]
- Rybtsov, S.; Berezina, T. The Retirement Stress Increases Biological Age: Searching Stress-Induced Inflammatory and Immunosenescence Factors of Biological Aging Acceleration. Exp. Hematol. 2020, 88, S78. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V.; Berezina, T.N.; Rybtsov, S. Hematopoietic Stem Cells and the Immune System in Development and Aging. Int. J. Mol. Sci. 2023, 24, 5862. [Google Scholar] [CrossRef]
- Rybtsova, N.; Berezina, T.N.; Rybtsov, S. Molecular Markers of Blood Cell Populations Can Help Estimate Aging of the Immune System. Int. J. Mol. Sci. 2023, 24, 5708. [Google Scholar] [CrossRef]
- Pellarin, I.; Dall’Acqua, A.; Favero, A.; Segatto, I.; Rossi, V.; Crestan, N.; Karimbayli, J.; Belletti, B.; Baldassarre, G. Cyclin-Dependent Protein Kinases and Cell Cycle Regulation in Biology and Disease. Signal Transduct. Target. Ther. 2025, 10, 11. [Google Scholar]
- Simmons, C.; Grant, S.L.; Llorente-Ruiz, L.; Kerstetter, D.W.; Pimpley, M.; Latimer, J.J. Greater Expression of DNA Repair Pathways in Sharks vs. Rays/Skates Based on Transcriptomic Analyses. bioRxiv 2025. [Google Scholar] [CrossRef]
- Keane, M.; Semeiks, J.; Webb, A.E.; Li, Y.I.; Quesada, V.; Craig, T.; Madsen, L.B.; van Dam, S.; Brawand, D.; Marques, P.I.; et al. Insights into the Evolution of Longevity from the Bowhead Whale Genome. Cell Rep. 2015, 10, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Seluanov, A.; Gorbunova, V. Molecular Mechanisms Determining Lifespan in Short- and Long-Lived Species. Trends Endocrinol. Metab. 2017, 28, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, A.; Schumacher, B.; Shiloh, Y. Genome Instability: Linking Ageing and Brain Degeneration. Mech. Ageing Dev. 2017, 161, 4–18. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.-G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A Premature Aging Disease Caused by LMNA Gene Mutations. Ageing Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Abdallah, P.; Luciano, P.; Runge, K.W.; Lisby, M.; Géli, V.; Gilson, E.; Teixeira, M.T. A Two-Step Model for Senescence Triggered by a Single Critically Short Telomere. Nat. Cell Biol. 2009, 11, 988–993. [Google Scholar] [CrossRef]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of Telomere Length across Human Tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere Position Effect: Regulation of Gene Expression with Progressive Telomere Shortening over Long Distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cristina Cardoso, M.; Leonhardt, H. Recruitment of DNA Methyltransferase I to DNA Repair Sites. Proc. Natl. Acad. Sci. USA. 2005, 102, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meyer, D.H.; Schumacher, B. H3K4me2 Regulates the Recovery of Protein Biosynthesis and Homeostasis Following DNA Damage. Nat. Struct. Mol. Biol. 2020, 27, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Lopes, R.; Prasad, M.K. Beyond the Promise: Evaluating and Mitigating off-Target Effects in CRISPR Gene Editing for Safer Therapeutics. Front. Bioeng. Biotechnol. 2023, 11, 1339189. [Google Scholar] [CrossRef]
- Wei, Y.-N.; Hu, H.-Y.; Xie, G.-C.; Fu, N.; Ning, Z.-B.; Zeng, R.; Khaitovich, P. Transcript and Protein Expression Decoupling Reveals RNA Binding Proteins and MiRNAs as Potential Modulators of Human Aging. Genome Biol. 2015, 16, 41. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Dollé, M.E.T.; Reiling, E.; Jaarsma, D.; Payan-Gomez, C.; Bombardieri, C.R.; Wu, H.; Roks, A.J.M.; Botter, S.M.; Van Der Eerden, B.C.; et al. Restricted Diet Delays Accelerated Ageing and Genomic Stress in DNA-Repair-Deficient Mice. Nature 2016, 537, 427–431. [Google Scholar] [CrossRef]
- Kelmer Sacramento, E.; Kirkpatrick, J.M.; Mazzetto, M.; Baumgart, M.; Bartolome, A.; Di Sanzo, S.; Caterino, C.; Sanguanini, M.; Papaevgeniou, N.; Lefaki, M.; et al. Reduced Proteasome Activity in the Aging Brain Results in Ribosome Stoichiometry Loss and Aggregation. Mol. Syst. Biol. 2020, 16, e9596. [Google Scholar] [CrossRef]
- Williams, A.B.; Heider, F.; Messling, J.-E.; Rieckher, M.; Bloch, W.; Schumacher, B. Restoration of Proteostasis in the Endoplasmic Reticulum Reverses an Inflammation-Like Response to Cytoplasmic DNA in Caenorhabditis elegans. Genetics 2019, 212, 1259–1278. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature Ageing in Mice Expressing Defective Mitochondrial DNA Polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tétreault, M.; Komulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 Mutation Is Associated with PARP1 Hyperactivation and Cerebellar Ataxia. Nature 2017, 541, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Fizikova, A.Y.u.; Padkina, M.V.; Sambuk, E.V. The Absence of Cyclin-Dependent Protein Kinase Pho85 Affects Stability of Mitochondrial DNA in Yeast Saccharomyces cerevisiae. Russ. J. Genet. 2009, 45, 651–658. [Google Scholar] [CrossRef]
- Sambuk, E.V.; Popova, Y.G.; Fizikova, A.Y.; Padkina, M.V. Genetic Analysis of Pleiotropic Effects of Pho85 Mutations in Yeast Saccharomyces cerevisiae. Russ. J. Genet. 2003, 39, 871–877. [Google Scholar] [CrossRef]
- Sambuk, E.V.; Fizikova, A.Y.; Savinov, V.A.; Padkina, M.V. Acid Phosphatases of Budding Yeast as a Model of Choice for Transcription Regulation Research. Enzyme Res. 2011, 2011, 356093. [Google Scholar] [CrossRef]
- Spurrier, J.; Shukla, A.K.; McLinden, K.; Johnson, K.; Giniger, E. Altered Expression of the Cdk5 Activator-like Protein, Cdk5?, Causes Neurodegeneration, in Part by Accelerating the Rate of Aging. DMM Dis. Models Mech. 2018, 11, dmm031161. [Google Scholar] [CrossRef]
- Zhuk, A.S.; Stepchenkova, E.I.; Inge-Vechtomov, S.G. ME Lobashev’s Physiological Theory of the Mutation Process and the Formation of Contemporary Views on Mutational Changes in Genetic Material. Ecol. Genet. 2023, 21, 329–342. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Dominick, G.; Bowman, J.; Li, X.; Miller, R.A.; Garcia, G.G. MTOR Regulates the Expression of DNA Damage Response Enzymes in Long-Lived Snell Dwarf, GHRKO, and PAPPA-KO Mice. Aging Cell 2017, 16, 52–60. [Google Scholar] [CrossRef]
- Ma, Y.; Vassetzky, Y.; Dokudovskaya, S. MTORC1 Pathway in DNA Damage Response. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1293–1311. [Google Scholar] [CrossRef] [PubMed]
- Adamowicz, M.; Vermezovic, J.; d’Adda di Fagagna, F. NOTCH1 Inhibits Activation of ATM by Impairing the Formation of an ATM-FOXO3a-KAT5/Tip60 Complex. Cell Rep. 2016, 16, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Kuehnemann, C.; Wiley, C.D. Senescent Cells at the Crossroads of Aging, Disease, and Tissue Homeostasis. Aging Cell 2024, 23, e13988. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Rybtsova, N.; Berezina, T.; Kagansky, A.; Rybtsov, S. Can Blood-Circulating Factors Unveil and Delay Your Biological Aging? Biomedicines 2020, 8, 615. [Google Scholar] [CrossRef]
- Kurgan, N.; Panova, E.; Silakova, L.; Kaganskii, A.; Rybtsov, S. Prospects for Assessing the Biological and Immunological Age of a Person by Blood Factors. Sci. Innov. Med. 2021, 6, 19–39. [Google Scholar] [CrossRef]
- Oda, T.; Gotoh, N.; Kasamatsu, T.; Handa, H.; Saitoh, T.; Sasaki, N. DNA Damage-Induced Cellular Senescence Is Regulated by 53BP1 Accumulation in the Nuclear Foci and Phase Separation. Cell Prolif. 2023, 56, e13398. [Google Scholar] [CrossRef]
- de Godoy, M.C.X.; Macedo, J.A.; Gambero, A. Researching New Drug Combinations with Senolytic Activity Using Senescent Human Lung Fibroblasts MRC-5 Cell Line. Pharmaceuticals 2024, 17, 70. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-ΚB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Perkins, N.D.; Gilmore, T.D. Good Cop, Bad Cop: The Different Faces of NF-ΚB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef]
- Buj, R.; Aird, K.M. P16: Cycling off the Beaten Path. Mol. Cell Oncol. 2019, 6, e1677140. [Google Scholar] [CrossRef]
- Gulaia, V.; Kumeiko, V.; Shved, N.; Cicinskas, E.; Rybtsov, S.; Ruzov, A.; Kagansky, A. Molecular Mechanisms Governing the Stem Cell’s Fate in Brain Cancer: Factors of Stemness and Quiescence. Front. Cell Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.Y.; Zhang, C.; Li, H.; Baker, D.J. Senescence Targeting Methods Impact Alzheimer’s Disease Features in 3xTg Mice. J. Alzheimer’s Dis. 2024, 97, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Hardy, P.B.; Chubinskaya, S.; Pearce, K.H.; Loeser, R.F. A Novel Small Molecule Screening Assay Using Normal Human Chondrocytes toward Osteoarthritis Drug Discovery. PLoS ONE 2024, 19, e0308647. [Google Scholar] [CrossRef]
- Cheng, W.; Fu, Y.; Lin, Z.; Huang, M.; Chen, Y.; Hu, Y.; Lin, Q.; Yu, B.; Liu, G. Lipoteichoic Acid Restrains Macrophage Senescence via β-Catenin/FOXO1/REDD1 Pathway in Age-Related Osteoporosis. Aging Cell 2024, 23, e14072. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Nakano, Y.; Johmura, Y. Functional Diversity of Senescent Cells in Driving Ageing Phenotypes and Facilitating Tissue Regeneration. J. Biochem. 2025, 177, 189–195. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Berezina, T.N.; Rybtsov, S. Acceleration of Biological Aging and Underestimation of Subjective Age Are Risk Factors for Severe COVID-19. Biomedicines 2021, 9, 913. [Google Scholar] [CrossRef]
- Takaya, K.; Asou, T.; Kishi, K. New Senolysis Approach via Antibody-Drug Conjugate Targeting of the Senescent Cell Marker Apolipoprotein D for Skin Rejuvenation. Int. J. Mol. Sci. 2023, 24, 5857. [Google Scholar] [CrossRef]
- Widjaja, A.A.; Lim, W.-W.; Viswanathan, S.; Chothani, S.; Corden, B.; Dasan, C.M.; Goh, J.W.T.; Lim, R.; Singh, B.K.; Tan, J.; et al. Inhibition of IL-11 Signalling Extends Mammalian Healthspan and Lifespan. Nature 2024, 632, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Martyshkina, Y.S.; Tereshchenko, V.P.; Bogdanova, D.A.; Rybtsov, S.A. Reliable Hallmarks and Biomarkers of Senescent Lymphocytes. Int. J. Mol. Sci. 2023, 24, 15653. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ding, Q.; Lin, Z.; Chen, X.; Chen, S.; Zhu, Y. New Insights of Epigenetics in Vascular and Cellular Senescence. J. Transl. Int. Med. 2021, 9, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally Occurring P16(Ink4a)-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Hoch, N.C. Tissue Specificity of DNA Damage and Repair. Physiology 2023, 38, 231–241. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef]
- Hayashi, K.; Horisaka, K.; Harada, Y.; Ogawa, Y.; Yamashita, T.; Kitano, T.; Wakita, M.; Fukusumi, T.; Inohara, H.; Hara, E.; et al. Polyploidy Mitigates the Impact of DNA Damage While Simultaneously Bearing Its Burden. Cell Death Discov. 2024, 10, 436. [Google Scholar] [CrossRef]
- Chinen, L.T.D.; Torres, J.A.; Calsavara, V.F.; Brito, A.B.C.; e Silva, V.S.; Novello, R.G.S.; Fernandes, T.C.; Decina, A.; Dachez, R.; Paterlini-Brechot, P. Circulating Polyploid Giant Cancer Cells, a Potential Prognostic Marker in Patients with Carcinoma. Int. J. Mol. Sci. 2024, 25, 9841. [Google Scholar] [CrossRef]
- Hosea, R.; Hillary, S.; Naqvi, S.; Wu, S.; Kasim, V. The Two Sides of Chromosomal Instability: Drivers and Brakes in Cancer. Signal Transduct. Target. Ther. 2024, 9, 75. [Google Scholar] [CrossRef]
- Mohamed Yusoff, A.A.; Mohd Khair, S.Z.N.; Abd Radzak, S.M. Mitochondrial DNA Copy Number Alterations: Key Players in the Complexity of Glioblastoma (Review). Mol. Med. Rep. 2025, 31, 78. [Google Scholar] [CrossRef]
- Usman, N.Y.; Rebrikov, D.V. Recombinant Adeno-Associated Viruses as a Gene Delivery Vehicle for the Use in Molecular Medicine. Bull. Russ. State Med. Univ. 2021, 5–10. [Google Scholar] [CrossRef]
- Franchini, M.; Mannucci, P.M. Past, Present and Future of Hemophilia: A Narrative Review. Orphanet J. Rare Dis. 2012, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, R.; Baboota, R.K.; Gogg, S.; Beguinot, F.; Blüher, M.; Nerstedt, A.; Smith, U. Increased Cell Senescence in Human Metabolic Disorders. J. Clin. Investig. 2023, 133, e169922. [Google Scholar] [CrossRef] [PubMed]
- Sekayan, T.; Dana, H.S.; von Drygalski, A. Etranacogene Dezaparvovec-Drlb Gene Therapy for Patients with Hemophilia B (Congenital Factor IX Deficiency). Expert Opin. Biol. Ther. 2023, 23, 1173–1184. [Google Scholar] [CrossRef]
- Minskaia, E.; Galieva, A.; Egorov, A.D.; Ivanov, R.; Karabelsky, A. Viral Vectors in Gene Replacement Therapy. Biochemistry 2023, 88, 2157–2178. [Google Scholar] [CrossRef]
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH Guidelines for the Management of Hemophilia, 3rd Edition. Haemophilia 2020, 26, 1–158. [Google Scholar] [CrossRef]
- Xu, Z.; Spencer, H.J.; Harris, V.A.; Perkins, S.J. An Updated Interactive Database for 1692 Genetic Variants in Coagulation Factor IX Provides Detailed Insights into Hemophilia B. J. Thromb. Haemost. 2023, 21, 1164–1176. [Google Scholar] [CrossRef]
- Carpintero-Fernández, P.; Borghesan, M.; Eleftheriadou, O.; Pan-Castillo, B.; Fafián-Labora, J.A.; Mitchell, T.P.; Yuste, A.; Ogrunc, M.; Nightingale, T.D.; Mayan, M.; et al. Genome Wide CRISPR/Cas9 Screen Identifies the Coagulation Factor IX (F9) as a Regulator of Senescence. Cell Death Dis. 2022, 13, 163. [Google Scholar] [CrossRef]
- Trappl, M.; Vostatek, R.; Salzmann, M.; Kraemmer, D.; Gebhart, J.; Hohensinner, P.; Pabinger, I.; Ay, C. Hemophilia Is Associated with Accelerated Biological Aging. Haematologica 2025. online ahead of print. [Google Scholar] [CrossRef]
- Addissouky, T.A. Polyploidy-Mediated Resilience in Hepatic Aging: Molecular Mechanisms and Functional Implication. Egypt. Liver J. 2024, 14, 83. [Google Scholar] [CrossRef]
- Arzumanian, V.A.; Pyatnitsky, M.A.; Vakhrushev, I.V.; Ptitsyn, K.G.; Radko, S.P.; Zgoda, V.G.; Kiseleva, O.I.; Poveryennaya, E.V. Molecular Profile of the HepG2 Tumor Cell Line. Biomed. Chem. Res. Methods 2024, 7, e00239. [Google Scholar] [CrossRef]
- Carpintero-Fernández, P.; Borghesan, M.; Eleftheriadou, O.; Fafián-Labora, J.A.; Mitchell, T.P.; Nightingale, T.D.; Mayán, M.D.; O’Loghlen, A. The Coagulation Factor IX (F9) Loss of Function Prevents the Cell Cycle Arrest Induced by CDK4/6 Inhibitors Treatment. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fang, J.; de Bruin, A.; Villunger, A.; Schiffelers, R.; Lei, Z.; Sluijter, J.P.G. Cellular Polyploidy in Organ Homeostasis and Regeneration. Protein Cell 2023, 14, 560–578. [Google Scholar] [CrossRef]
- Yin, K.; Büttner, M.; Deligiannis, I.K.; Strzelecki, M.; Zhang, L.; Talavera-López, C.; Theis, F.; Odom, D.T.; Martinez-Jimenez, C.P. Polyploidisation Pleiotropically Buffers Ageing in Hepatocytes. J. Hepatol. 2024, 81, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.S.; Cunningham, R.P.; Miller, C.B.; Brown, L.A.; Cultraro, C.M.; Harned, A.; Narayan, K.; Hernandez, J.; Jenkins, L.M.; Lobanov, A.; et al. A Spatial Map of Hepatic Mitochondria Uncovers Functional Heterogeneity Shaped by Nutrient-Sensing Signaling. Nat. Commun. 2024, 15, 1799. [Google Scholar] [CrossRef]
- Gaikwad, S.; Senapati, S.; Haque, M.A.; Kayed, R. Senescence, Brain Inflammation, and Oligomeric Tau Drive Cognitive Decline in Alzheimer’s Disease: Evidence from Clinical and Preclinical Studies. Alzheimer’s Dement. 2024, 20, 709–727. [Google Scholar] [CrossRef]
- Mao, D.; Hinds, P.W. P35 Is Required for CDK5 Activation in Cellular Senescence. J. Biol. Chem. 2010, 285, 14671–14680. [Google Scholar] [CrossRef]
- Alexander, K.; Yang, H.-S.; Hinds, P.W. Cellular Senescence Requires CDK5 Repression of Rac1 Activity. Mol. Cell Biol. 2004, 24, 2808–2819. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Βgal) Activity, a Biomarker of Senescent Cells in Culture and in Vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Requejo-Aguilar, R. Cdk5 and Aberrant Cell Cycle Activation at the Core of Neurodegeneration. Neural Regen. Res. 2023, 18, 1186–1190. [Google Scholar] [CrossRef]
- Wong, D.P.; Fritz, C.E.; Feinberg, D.; Huang, A.Y.; Parameswaran, R. P35 Is a Crucial Player in NK-Cell Cytotoxicity and TGFβ-Mediated NK-Cell Dysfunction. Cancer Res. Commun. 2023, 3, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, B.; Li, S.; Wei, W.; Liu, X.; Cui, X.; Zhang, X.; Liu, N.; Yan, L.; Deng, Y.; et al. NKG2D-CAR T Cells Eliminate Senescent Cells in Aged Mice and Nonhuman Primates. Sci. Transl. Med. 2024, 15, eadd1951. [Google Scholar] [CrossRef] [PubMed]
- Moroz, V.D.; Gasanov, N.B.; Egorov, A.D.; Malogolovkin, A.S.; Nagornykh, M.O.; Subcheva, E.N.; Kolosova, E.S.; Fizikova, A.Y.; Ivanov, R.A.; Karabelsky, A.V. A method for the production of recombinant VSVs with confirmation of biological activity. Acta Naturae 2024, 16, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Lelarge, V.; Capelle, R.; Oger, F.; Mathieu, T.; Le Calvé, B. Senolytics: From Pharmacological Inhibitors to Immunotherapies, a Promising Future for Patients’ Treatment. NPJ Aging 2024, 10, 12. [Google Scholar] [CrossRef]
- Maeso-Díaz, R.; Du, K.; Pan, C.; Guy, C.D.; Oh, S.H.; Chen, T.; Wang, L.; Ko, D.C.; Tang, L.; Dutta, R.K.; et al. Targeting Senescent Hepatocytes Using the Thrombomodulin-PAR1 Inhibitor Vorapaxar Ameliorates NAFLD Progression. Hepatology 2023, 78, 1209–1222. [Google Scholar] [CrossRef]
- Yang, S.; Yang, R.; Wang, H.; Huang, Y.; Jia, Y. CDK5RAP3 Deficiency Restrains Liver Regeneration after Partial Hepatectomy Triggering Endoplasmic Reticulum Stress. Am. J. Pathol. 2020, 190, 2403–2416. [Google Scholar] [CrossRef]
- Wu, H.; Reizel, Y.; Wang, Y.J.; Lapiro, J.L.; Kren, B.T.; Schug, J.; Rao, S.; Morgan, A.; Herman, A.; Shekels, L.L.; et al. A Negative Reciprocal Regulatory Axis between Cyclin D1 and HNF4α Modulates Cell Cycle Progression and Metabolism in the Liver. Proc. Natl. Acad. Sci. USA 2020, 117, 17177–17186. [Google Scholar] [CrossRef]
- Li, M.; Tang, J.; Zhu, W.; Cheng, C.; Guo, L.; Liu, P.; Mo, Z. Medicine ® ATG9B-4 Accelerates the Proliferation and Migration of Liver Cancer Cells in an ARNTL-CDK5 Pathway-Dependent Manner A Case-Control Study. Medicine 2025, 104, e42227. [Google Scholar] [CrossRef]
- Ma, X.; Huang, T.; Chen, X.; Li, Q.; Liao, M.; Fu, L.; Huang, J.; Yuan, K.; Wang, Z.; Zeng, Y. Molecular Mechanisms in Liver Repair and Regeneration: From Physiology to Therapeutics. Signal Transduct. Target. Ther. 2025, 10, 63. [Google Scholar]
- Wang, Y.C.; Lee, W.C.; Liao, S.C.; Lee, L.C.; Su, Y.J.; Lee, C.T.; Chen, J.B. Mitochondrial DNA Copy Number Correlates with Oxidative Stress and Predicts Mortality in Nondiabetic Hemodialysis Patients. J. Nephrol. 2011, 24, 351–358. [Google Scholar] [CrossRef]
- Morris, J.P.; Baslan, T.; Soltis, D.E.; Soltis, P.S.; Fox, D.T. Integrating the Study of Polyploidy Across Organisms, Tissues, and Disease. Annu. Rev. Genet. 2024, 58, 297–318. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.; Riaz, M.; Arif, M. Environmental Stress and Secondary Metabolites in Plants: An Overview. In Plant Metabolites and Regulation Under Environmental Stress; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Kumar, R.; Sharma, A.; Kumari, A.; Gulati, A.; Padwad, Y.; Sharma, R. Epigallocatechin Gallate Suppresses Premature Senescence of Preadipocytes by Inhibition of PI3K/Akt/MTOR Pathway and Induces Senescent Cell Death by Regulation of Bax/Bcl-2 Pathway. Biogerontology 2019, 20, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Shi, Y.; Chen, Z.; Zhou, X.; Luo, P.; Hong, C.; Tian, N.; Wu, Y.; Zhou, Y.; Lin, Y.; et al. Apigenin Alleviates Intervertebral Disc Degeneration via Restoring Autophagy Flux in Nucleus Pulposus Cells. Front. Cell Dev. Biol. 2022, 9, 787278. [Google Scholar] [CrossRef]
- Yang, H.; Yang, X.; Rong, K.; Liang, J.; Wang, Z.; Zhao, J.; Zhang, P.; Li, Y.; Wang, L.; Ma, H.; et al. Eupatilin Attenuates the Senescence of Nucleus Pulposus Cells and Mitigates Intervertebral Disc Degeneration via Inhibition of the MAPK/NF-ΚB Signaling Pathway. Front. Pharmacol. 2022, 13, 940475. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, Y.; Liu, F.; Wang, J.; Liu, F.; Zhang, Q.; Li, J. Pharmacological Network Analysis of the Functions and Mechanism of Kaempferol from Du Zhong in Intervertebral Disc Degeneration (IDD). J. Orthop. Transl. 2023, 39, 135–146. [Google Scholar] [CrossRef]
- Xie, T.; Yuan, J.; Mei, L.; Li, P.; Pan, R. Luteolin Suppresses TNF-α-induced Inflammatory Injury and Senescence of Nucleus Pulposus Cells via the Sirt6/NF-κB Pathway. Exp. Ther. Med. 2022, 24, 469. [Google Scholar] [CrossRef]
- Barreca, D.; Smeriglio, A.; Bellocco, E.; Trombetta, D. Proanthocyanidins and Hydrolysable Tannins: Occurrence, Dietary Intake and Pharmacological Effects. Br. J. Pharmacol. 2017, 174, 1244. [Google Scholar]
- Hu, X.; Yang, Y.; Tang, S.; Chen, Q.; Zhang, M.; Ma, J.; Qin, J.; Yu, H. Anti-Aging Effects of Anthocyanin Extracts of Sambucus Canadensis Caused by Targeting Mitochondrial-Induced Oxidative Stress. Int. J. Mol. Sci. 2023, 24, 1528. [Google Scholar] [CrossRef]
- Deepika; Maurya, P.K. Health Benefits of Quercetin in Age-Related Diseases. Molecules 2022, 27, 2498. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, J.; Nisar, M.; Chen, T.; Xu, T.; Zheng, G.; Wang, C.; Jin, H.; Chen, J.; Gao, W.; et al. The Sirt1/P53 Axis in Diabetic Intervertebral Disc Degeneration Pathogenesis and Therapeutics. Oxidative Med. Cell Longev. 2019, 2019, 7959573. [Google Scholar] [CrossRef]
- Wu, X.; Cao, N.; Fenech, M.; Wang, X. Role of Sirtuins in Maintenance of Genomic Stability: Relevance to Cancer and Healthy Aging. DNA Cell Biol. 2016, 35, 542–575. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, L.C. Natural Coumarin Derivatives Activating Nrf2 Signaling Pathway as Lead Compounds for the Design and Synthesis of Intestinal Anti-Inflammatory Drugs. Pharmaceuticals 2023, 16, 511. [Google Scholar] [CrossRef] [PubMed]
- Meiners, F.; Hinz, B.; Boeckmann, L.; Secci, R.; Sueto, S.; Kuepfer, L.; Fuellen, G.; Barrantes, I. Computational Identification of Natural Senotherapeutic Compounds That Mimic Dasatinib Based on Gene Expression Data. Sci. Rep. 2024, 14, 6286. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The Curcumin Analog EF24 Is a Novel Senolytic Agent. Aging 2019, 11, 771–782. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin Is a Senotherapeutic That Extends Health and Lifespan. eBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New Agents That Target Senescent Cells: The Flavone, Fisetin, and the BCL-XL Inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Wang, J.; Nisar, M.; Huang, C.; Pan, X.; Lin, D.; Zheng, G.; Jin, H.; Chen, D.; Tian, N.; Huang, Q.; et al. Small Molecule Natural Compound Agonist of SIRT3 as a Therapeutic Target for the Treatment of Intervertebral Disc Degeneration. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, X.; Zhu, X.; Wang, C.; Xu, W. Myricetin Alleviated Hydrogen Peroxide-Induced Cellular Senescence of Nucleus Pulposus Cell through Regulating SERPINE1. J. Orthop. Surg. Res. 2023, 18, 143. [Google Scholar] [CrossRef]
- Xie, T.; Pan, R.; Huang, W.; Dong, S.; Wu, S.; Ye, Y. Myricetin Alleviates H2O2-Induced Senescence and Apoptosis in Rat Nucleus Pulposus-Derived Mesenchymal Stem Cells. Folia Histochem. Cytobiol. 2023, 61, 98–108. [Google Scholar] [CrossRef]
- Wang, J.; Huang, C.; Lin, Z.; Pan, X.; Chen, J.; Zheng, G.; Tian, N.; Yan, Y.; Zhang, Z.; Hu, J.; et al. Polydatin Suppresses Nucleus Pulposus Cell Senescence, Promotes Matrix Homeostasis and Attenuates Intervertebral Disc Degeneration in Rats. J. Cell Mol. Med. 2018, 22, 5720–5731. [Google Scholar] [CrossRef]
- Duta-Bratu, C.G.; Nitulescu, G.M.; Mihai, D.P.; Olaru, O.T. Resveratrol and Other Natural Oligomeric Stilbenoid Compounds and Their Therapeutic Applications. Plants 2023, 12, 2935. [Google Scholar] [CrossRef] [PubMed]
- Schwager, J.; Richard, N.; Widmer, F.; Raederstorff, D. Resveratrol Distinctively Modulates the Inflammatory Profiles of Immune and Endothelial Cells. BMC Complement. Altern. Med. 2017, 17, 309. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Banerjee, S.; Acosta, E.P.; Lillard, J.W.; Singh, R. Resveratrol Induces Cell Cycle Arrest and Apoptosis with Docetaxel in Prostate Cancer Cells via a P53/ P21 WAF1/CIP1 and P27 KIP1 Pathway. Oncotarget 2017, 8, 17216–17228. [Google Scholar] [CrossRef] [PubMed]
- Hedayati, N.; Safari, M.H.; Milasi, Y.E.; Kahkesh, S.; Farahani, N.; Khoshnazar, S.M.; Dorostgou, Z.; Alaei, E.; Alimohammadi, M.; Rahimzadeh, P.; et al. Modulation of the PI3K/Akt Signaling Pathway by Resveratrol in Cancer: Molecular Mechanisms and Therapeutic Opportunity. Discov. Oncol. 2025, 16. [Google Scholar] [CrossRef]
- Izzo, C.; Annunziata, M.; Melara, G.; Sciorio, R.; Dallio, M.; Masarone, M.; Federico, A.; Persico, M. The Role of Resveratrol in Liver Disease: A Comprehensive Review from in Vitro to Clinical Trials. Nutrients 2021, 13, 933. [Google Scholar] [CrossRef]
- Denissova, N.; Nasello, C.; Yeung, P.; Tischfield, J.; Brenneman, M. Resveratrol Protects Mouse Embryonic Stem Cells from Ionizing Radiation by Accelerating Recovery from DNA Strand Breakage. Carcinogenesis 2011, 33, 149–155. [Google Scholar] [CrossRef]
- Mannarino, M.; Cherif, H.; Li, L.; Sheng, K.; Rabau, O.; Jarzem, P.; Weber, M.H.; Ouellet, J.A.; Haglund, L. Toll-like Receptor 2 Induced Senescence in Intervertebral Disc Cells of Patients with Back Pain Can Be Attenuated by o-Vanillin. Arthritis Res. Ther. 2021, 23, 117. [Google Scholar] [CrossRef]
- Li, L.; Sheng, K.; Mannarino, M.; Jarzem, P.; Cherif, H.; Haglund, L. O-Vanillin Modulates Cell Phenotype and Extracellular Vesicles of Human Mesenchymal Stem Cells and Intervertebral Disc Cells. Cells 2022, 11, 3589. [Google Scholar] [CrossRef]
- Woo, J.; Shin, S.; Ji, H.; Ryu, D.; Cho, E.; Kim, Y.; Kim, J.; Park, D.; Jung, E.; Isatis Tinctoria, L. Leaf Extract Inhibits Replicative Senescence in Dermal Fibroblasts by Regulating MTOR-NF-ΚB-SASP Signaling. Nutrients 2022, 14, 1979. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, X.; Zhou, Y.; Liang, Z.; Chen, C.; Han, C.; Cao, X.; He, W.; Zhang, K.; Qin, A.; et al. Dehydrocostus Lactone Attenuates the Senescence of Nucleus Pulposus Cells and Ameliorates Intervertebral Disc Degeneration via Inhibition of STING-TBK1/NF-ΚB and MAPK Signaling. Front. Pharmacol. 2021, 12, 641098. [Google Scholar] [CrossRef]
- Xie, T.; Gu, X.; Pan, R.; Huang, W.; Dong, S. Evodiamine Ameliorates Intervertebral Disc Degeneration through the Nrf2 and MAPK Pathways. Cytotechnology 2024, 76, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Zhang, X.; Gao, Z.; Zhang, S.; Shi, P.; Zhang, X.; Song, L.; Hendrickson, H.; Zhou, D.; et al. Senolytic Activity of Piperlongumine Analogues: Synthesis and Biological Evaluation. Bioorg. Med. Chem. 2018, 26, 3925–3938. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, C.; Zhao, X.; Tan, H.; Li, C.; Deng, Y.; Chen, X.; Wu, Y.; Tian, N.; Zhang, X.; et al. 20-Deoxyingenol Alleviates Intervertebral Disc Degeneration by Activating TFEB in Nucleus Pulposus Cells. Biochem. Pharmacol. 2023, 218, 115865. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zuo, R.; Wang, Z.; Luo, L.; Wu, J.; Zhang, C.; Liu, M.; Shi, C.; Zhou, Y. Kinsenoside Ameliorates Intervertebral Disc Degeneration through the Activation of AKT-ERK1/2-Nrf2 Signaling Pathway. Aging 2019, 11, 7961–7977. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Smer-Barreto, V.; Quintanilla, A.; Elliott, R.J.R.; Dawson, J.C.; Sun, J.; Campa, V.M.; Lorente-Macías, Á.; Unciti-Broceta, A.; Carragher, N.O.; Acosta, J.C.; et al. Discovery of Senolytics Using Machine Learning. Nat. Commun. 2023, 14, 3445. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, D.; Yuan, Y.; Zheng, R.; Sun, M.; Jia, S.; Liu, J. Cycloastragenol: A Novel Senolytic Agent That Induces Senescent Cell Apoptosis and Restores Physical Function in TBI-Aged Mice. Int. J. Mol. Sci. 2023, 24, 6554. [Google Scholar] [CrossRef]
- Yasuda, S.; Horinaka, M.; Iizumi, Y.; Goi, W.; Sukeno, M.; Sakai, T. Oridonin Inhibits SASP by Blocking P38 and NF-ΚB Pathways in Senescent Cells. Biochem. Biophys. Res. Commun. 2022, 590, 55–62. [Google Scholar] [CrossRef]
- Li, J.; Bao, L.; Zha, D.; Zhang, L.; Gao, P.; Zhang, J.; Wu, X. Oridonin Protects against the Inflammatory Response in Diabetic Nephropathy by Inhibiting the TLR4/P38-MAPK and TLR4/NF-ΚB Signaling Pathways. Int. Immunopharmacol. 2018, 55, 9–19. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Chu, Z.; Chen, L.; Chen, J.; Yang, Y.; Tang, H.; Cheng, G.; Ma, A.; Zhang, Y.; et al. Oridonin Acts as a Novel Senolytic by Targeting Glutathione S-Transferases to Activate the ROS-P38 Signaling Axis in Senescent Cells. Chem. Commun. 2022, 58, 13250–13253. [Google Scholar] [CrossRef]
- Lämmermann, I.; Terlecki-Zaniewicz, L.; Weinmüllner, R.; Schosserer, M.; Dellago, H.; de Matos Branco, A.D.; Autheried, D.; Sevcnikar, B.; Kleissl, L.; Berlin, I.; et al. Blocking Negative Effects of Senescence in Human Skin Fibroblasts with a Plant Extract. NPJ Aging Mech. Dis. 2018, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowicz, P.; Roullet, N.; Bensadoun, P.; Bessou-Touya, S.; Lemaitre, J.-M.; Duplan, H. Reduction of Senescence-Associated Secretory Phenotype and Exosome-Shuttled MiRNAs by Haritaki Fruit Extract in Senescent Dermal Fibroblasts. Int. J. Cosmet. Sci. 2023, 45, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Shin, S.; Cho, E.; Ryu, D.; Garandeau, D.; Chajra, H.; Fréchet, M.; Park, D.; Jung, E. Senotherapeutic-like Effect of Silybum Marianum Flower Extract Revealed on Human Skin Cells. PLoS ONE 2021, 16, e0260545. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.I.; Jo, E.R.; Song, H. Urolithin A Attenuates Auditory Cell Senescence by Activating Mitophagy. Sci. Rep. 2022, 12, 7704. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A Induces Mitophagy and Prolongs Lifespan in C. elegans and Increases Muscle Function in Rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Liu, S.; D’Amico, D.; Shankland, E.; Bhayana, S.; Garcia, J.M.; Aebischer, P.; Rinsch, C.; Singh, A.; Marcinek, D.J. Effect of Urolithin A Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2144279. [Google Scholar] [CrossRef]
- Singh, A.; D’Amico, D.; Andreux, P.A.; Fouassier, A.M.; Blanco-Bose, W.; Evans, M.; Aebischer, P.; Auwerx, J.; Rinsch, C. Urolithin A Improves Muscle Strength, Exercise Performance, and Biomarkers of Mitochondrial Health in a Randomized Trial in Middle-Aged Adults. Cell Rep. Med. 2022, 3, 100633. [Google Scholar] [CrossRef]
- Li, W.; Qin, L.; Feng, R.; Hu, G.; Sun, H.; He, Y.; Zhang, R. Emerging Senolytic Agents Derived from Natural Products. Mech. Ageing Dev. 2019, 181, 1–6. [Google Scholar] [CrossRef]
- Kusumoto, D.; Seki, T.; Sawada, H.; Kunitomi, A.; Katsuki, T.; Kimura, M.; Ito, S.; Komuro, J.; Hashimoto, H.; Fukuda, K.; et al. Anti-Senescent Drug Screening by Deep Learning-Based Morphology Senescence Scoring. Nat. Commun. 2021, 12, 257. [Google Scholar] [CrossRef]
- Fizikova, A.; Tikhonova, N.; Ukhatova, Y.; Ivanov, R.; Khlestkina, E. Applications of CRISPR/Cas9 System in Vegetatively Propagated Fruit and Berry Crops. Agronomy 2021, 11, 1849. [Google Scholar] [CrossRef]
- Fizikova, A.; Tukhuzheva, Z.; Zhokhova, L.; Tvorogova, V.; Lutova, L. A New Approach for CRISPR/Cas9 Editing and Selection of Pathogen-Resistant Plant Cells of Wine Grape Cv. ‘Merlot’. Int. J. Mol. Sci. 2024, 25, 10011. [Google Scholar] [CrossRef]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Tariq, H.; Asif, S.; Andleeb, A.; Hano, C.; Abbasi, B.H. Flavonoid Production: Current Trends in Plant Metabolic Engineering and De Novo Microbial Production. Metabolites 2023, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Kocábek, T.; Nath, V.S.; Khan, A.; Matoušek, J.; Hazzouri, K.M.; Sudalaimuthuasari, N.; Krofta, K.; Ludwig-Müller, J.; Amiri, K.M.A. The Multifaceted Roles of R2R3 Transcription Factor HlMYB7 in the Regulation of Flavonoid and Bitter Acids Biosynthesis, Development and Biotic Stress Tolerance in Hop (Humulus lupulus L.). Plant Physiol. Biochem. 2023, 197, 107636. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhao, W.; Li, R.; Guo, D.; Li, H.; Wang, Y.; Mei, W.; Peng, S. Identification and Characterization of Chalcone Isomerase Genes Involved in Flavonoid Production in Dracaena cambodiana. Front. Plant Sci. 2021, 12, 616396. [Google Scholar] [CrossRef]
- Elarabi, N.I.; Abdelhadi, A.A.; Sief-Eldein, A.G.M.; Ismail, I.A.; Abdallah, N.A. Overexpression of Chalcone Isomerase A Gene in Astragalus Trigonus for Stimulating Apigenin. Sci. Rep. 2021, 11, 24176. [Google Scholar] [CrossRef]
- Yin, Y.C.; Hou, J.M.; Tian, S.K.; Yang, L.; Zhang, Z.X.; Li, W.D.; Liu, Y. Overexpressing Chalcone Synthase (CHS) Gene Enhanced Flavonoids Accumulation in Glycyrrhiza Uralensis Hairy Roots. Bot. Lett. 2020, 167, 219–231. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, T.; Xin, Y.; Wang, G.; Xu, L.-A. Overexpression of the GbF3′H1 Gene Enhanced the Epigallocatechin, Gallocatechin, and Catechin Contents in Transgenic Populus. J. Agric. Food Chem. 2020, 68, 998–1006. [Google Scholar] [CrossRef]
- Sun, W.; Meng, X.; Liang, L.; Li, Y.; Zhou, T.; Cai, X.; Wang, L.; Gao, X. Overexpression of a Freesia Hybrida Flavonoid 3-O-Glycosyltransferase Gene, Fh3GT1, Enhances Transcription of Key Anthocyanin Genes and Accumulation of Anthocyanin and Flavonol in Transgenic Petunia (Petunia hybrida). Vitr. Cell Dev. Biol. Plant 2017, 53, 478–488. [Google Scholar] [CrossRef]
- Nguyen, T.N.L.; Hoang, T.T.H.; Nguyen, H.Q.; Tu, Q.T.; Tran, T.H.; Lo, T.M.T.; Vu, T.T.T.; Chu, H.M. Agrobacterium Tumefaciens–Mediated Genetic Transformation and Overexpression of the Flavonoid 3′5′-Hydroxylase Gene Increases the Flavonoid Content of the Transgenic Aconitum carmichaelii Debx. Plant. Vitr. Cell Dev. Biol. Plant 2022, 58, 93–102. [Google Scholar] [CrossRef]
- Yan, D.; Tajima, H.; Cline, L.C.; Fong, R.Y.; Ottaviani, J.I.; Shapiro, H.Y.; Blumwald, E. Genetic Modification of Flavone Biosynthesis in Rice Enhances Biofilm Formation of Soil Diazotrophic Bacteria and Biological Nitrogen Fixation. Plant Biotechnol. J. 2022, 20, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, T.; Xin, Y.; Wang, G.; Xu, L.-A. Overexpression of GbF3′5′H1 Provides a Potential to Improve the Content of Epicatechin and Gallocatechin. Molecules 2020, 25, 4836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Mei, X.; Li, Y.; Wu, J.; Li, Y.; Wang, H.; Huang, H.; Yang, M.; He, X.; et al. Phenolic Acids Released in Maize Rhizosphere During Maize-Soybean Intercropping Inhibit Phytophthora Blight of Soybean. Front. Plant Sci. 2020, 11, 886. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, J.; Pei, T.; Bai, Z.; Han, R.; Liang, Z. Overexpression of SmANS Enhances Anthocyanin Accumulation and Alters Phenolic Acids Content in Salvia miltiorrhiza and Salvia miltiorrhiza Bge f. alba Plantlets. Int. J. Mol. Sci. 2019, 20, 2225. [Google Scholar] [CrossRef]
- Bogs, J.; Downey, M.O.; Harvey, J.S.; Ashton, A.R.; Tanner, G.J.; Robinson, S.P. Proanthocyanidin Synthesis and Expression of Genes Encoding Leucoanthocyanidin Reductase and Anthocyanidin Reductase in Developing Grape Berries and Grapevine Leaves. Plant Physiol. 2005, 139, 652–663. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, Y.; Tan, H.; Chen, Q.; Huang, J. An Ubiquitin-like Protein SDE2 Negatively Affects Sucrose-Induced Anthocyanin Biosynthesis in Arabidopsis. Sci. Bull. 2017, 62, 1585–1592. [Google Scholar] [CrossRef]
- Chiriapkin, A.S.; Zolotykh, D.S.; Pozdnyakov, D.I. Review of Biological Activity of Flavonoids: Quercetin and Kaempferol. Juvenis Sci. 2023, 9, 5–20. [Google Scholar] [CrossRef]
- Chapman, J.M.; Muday, G.K. Flavonols Modulate Lateral Root Emergence by Scavenging Reactive Oxygen Species in Arabidopsis thaliana. J. Biol. Chem. 2021, 296, 100222. [Google Scholar] [CrossRef]
- Lin, X.; Xiao, M.; Luo, Y.; Wang, J.; Wang, H. The Effect of RNAi-Induced Silencing of FaDFR on Anthocyanin Metabolism in Strawberry (Fragaria × ananassa) Fruit. Sci. Hortic. 2013, 160, 123–128. [Google Scholar] [CrossRef]
- Kiokias, S.; Proestos, C.; Oreopoulou, V. Phenolic Acids of Plant Origin—A Review on Their Antioxidant Activity in Vitro (O/W Emulsion Systems) along with Their in Vivo Health Biochemical Properties. Foods 2020, 9, 534. [Google Scholar] [CrossRef]
- Landete, J.M. Ellagitannins, Ellagic Acid and Their Derived Metabolites: A Review about Source, Metabolism, Functions and Health. Food Res. Int. 2011, 44, 1150–1160. [Google Scholar] [CrossRef]
- Bar-Ya’akov, I.; Tian, L.; Amir, R.; Holland, D. Primary Metabolites, Anthocyanins, and Hydrolyzable Tannins in the Pomegranate Fruit. Front. Plant Sci. 2019, 10, 620. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.G. From Lignins to Tannins: Forty Years of Enzyme Studies on the Biosynthesis of Phenolic Compounds. Phytochemistry 2008, 69, 3018–3031. [Google Scholar] [CrossRef] [PubMed]
- Ono, N.N.; Bandaranayake, P.C.G.; Tian, L. Establishment of Pomegranate (Punica granatum) Hairy Root Cultures for Genetic Interrogation of the Hydrolyzable Tannin Biosynthetic Pathway. Planta 2012, 236, 931–941. [Google Scholar] [CrossRef]
- Chang, L.; Wu, S.; Tian, L. Effective Genome Editing and Identification of a Regiospecific Gallic Acid 4-O-Glycosyltransferase in Pomegranate (Punica granatum L.). Hortic. Res. 2019, 6, 123. [Google Scholar] [CrossRef]
- Habashi, R.; Hacham, Y.; Dhakarey, R.; Matityahu, I.; Holland, D.; Tian, L.; Amir, R. Elucidating the Role of Shikimate Dehydrogenase in Controlling the Production of Anthocyanins and Hydrolysable Tannins in the Outer Peels of Pomegranate. BMC Plant Biol. 2019, 19, 476. [Google Scholar] [CrossRef]
- Akagi, T.; Ikegami, A.; Tsujimoto, T.; Kobayashi, S.; Sato, A.; Kono, A.; Yonemori, K. DkMyb4 Is a Myb Transcription Factor Involved in Proanthocyanidin Biosynthesis in Persimmon Fruit. Plant Physiol. 2009, 151, 2028–2045. [Google Scholar] [CrossRef]
- Akagi, T.; Ikegami, A.; Suzuki, Y.; Yoshida, J.; Yamada, M.; Sato, A.; Yonemori, K. Expression Balances of Structural Genes in Shikimate and Flavonoid Biosynthesis Cause a Difference in Proanthocyanidin Accumulation in Persimmon (Diospyros kaki Thunb.) Fruit. Planta 2009, 230, 899–915. [Google Scholar] [CrossRef]
- Bontpart, T.; Marlin, T.; Vialet, S.; Guiraud, J.L.; Pinasseau, L.; Meudec, E.; Sommerer, N.; Cheynier, V.; Terrier, N. Two Shikimate Dehydrogenases, VvSDH3 and VvSDH4, Are Involved in Gallic Acid Biosynthesis in Grapevine. J. Exp. Bot. 2016, 67, 3537–3550. [Google Scholar] [CrossRef]
- Ramírez, L.; Arrizon, J.; Sandoval, G.; Cardador, A.; Bello-Mendoza, R.; Lappe, P.; Mateos-Díaz, J.C. A New Microplate Screening Method for the Simultaneous Activity Quantification of Feruloyl Esterases, Tannases, and Chlorogenate Esterases. Appl. Biochem. Biotechnol. 2008, 151, 711–723. [Google Scholar] [CrossRef]
- Dai, X.; Liu, Y.; Zhuang, J.; Yao, S.; Liu, L.; Jiang, X.; Zhou, K.; Wang, Y.; Xie, D.; Bennetzen, J.L.; et al. Discovery and Characterization of Tannase Genes in Plants: Roles in Hydrolysis of Tannins. N. Phytol. 2020, 226, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huan, Q.; Li, K.; Qian, W. Single-Cell Transcriptome Atlas of the Leaf and Root of Rice Seedlings. J. Genet. Genom. 2021, 48, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, N.; Zhao, J.; Zhao, Y.; Xu, R.; Fu, F.; Pan, T.; Yu, Y.; Guo, Z.; She, W. Accumulation of Polyphenolics and Differential Expression of Genes Related to Shikimate Pathway during Fruit Development and Maturation of Chinese Olive (Canarium album). Agronomy 2023, 13, 895. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, Q.; Xiao, L.; Wang, Y.; Feng, J.; Bu, Q.; Xiao, Y.; Hao, K.; Guo, M.; Chen, W.; et al. Multiplexed CRISPR/Cas9-Mediated Knockout of Laccase Genes in Salvia miltiorrhiza Revealed Their Roles in Growth, Development, and Metabolism. Front. Plant Sci. 2021, 12, 647768. [Google Scholar] [CrossRef]
- Mo, R.; Huang, Y.; Yang, S.; Zhang, Q.; Luo, Z. Development of Agrobacterium-Mediated Transient Transformation in Persimmon (Diospyros kaki Thunb.). Sci. Hortic. 2015, 192, 29–37. [Google Scholar] [CrossRef]
- Kumar, S.R.; Shilpashree, H.B.; Nagegowda, D.A. Terpene Moiety Enhancement by Overexpression of Geranyl(Geranyl) Diphosphate Synthase and Geraniol Synthase Elevates Monomeric and Dimeric Monoterpene Indole Alkaloids in Transgenic Catharanthus roseus. Front. Plant Sci. 2018, 9, 942. [Google Scholar] [CrossRef]
- Zhu, H.; Li, C.; Gao, C. Applications of CRISPR–Cas in Agriculture and Plant Biotechnology. Nat. Rev. Mol. Cell Biol. 2020, 21, 661–677. [Google Scholar] [CrossRef]
- Gutiérrez-Grijalva, E.P.; López-Martínez, L.X.; Contreras-Angulo, L.A.; Elizalde-Romero, C.A.; Heredia, J.B. Plant Alkaloids: Structures and Bioactive Properties. In Plant-Derived Bioactives: Chemistry and Mode of Action; Springer: Singapore, 2020; pp. 85–117. ISBN 9789811523618. [Google Scholar]
- Ma, W.; Kang, X.; Liu, P.; Zhang, Y.; Lin, X.; Li, B.; Chen, Z. The Analysis of Transcription Factor CsHB1 Effects on Caffeine Accumulation in Tea Callus through CRISPR/Cas9 Mediated Gene Editing. Process Biochem. 2021, 101, 304–311. [Google Scholar] [CrossRef]
- Schachtsiek, J.; Stehle, F. Nicotine-Free, Nontransgenic Tobacco (Nicotiana tabacum L.) Edited by CRISPR-Cas9. Plant Biotechnol. J. 2019, 17, 2228–2230. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Mu, D.; Lu, Y.; Chen, W.; Zhang, Y.; Zhang, R.; Qin, Y.; Yuan, J.; Pan, L.; et al. Selection of Reference Genes in Evodia Rutaecarpa Var. Officinalis and Expression Patterns of Genes Involved in Its Limonin Biosynthesis. Plants 2023, 12, 3197. [Google Scholar] [CrossRef]
- Das, S.; Kwon, M.; Kim, J.Y. Enhancement of Specialized Metabolites Using CRISPR/Cas Gene Editing Technology in Medicinal Plants. Front. Plant Sci. 2024, 15, 1279738. [Google Scholar] [CrossRef] [PubMed]
- Rather, G.A.; Sharma, A.; Misra, P.; Kumar, A.; Kaul, V.; Lattoo, S.K. Molecular Characterization and Overexpression Analyses of Secologanin Synthase to Understand the Regulation of Camptothecin Biosynthesis in Nothapodytes nimmoniana (Graham.) Mabb. Protoplasma 2020, 257, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Singh, S.K.; Patra, B.; Sui, X.; Pattanaik, S.; Yuan, L. A Differentially Regulated AP2/ERF Transcription Factor Gene Cluster Acts Downstream of a MAP Kinase Cascade to Modulate Terpenoid Indole Alkaloid Biosynthesis in Catharanthus roseus. N. Phytol. 2017, 213, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, M.; Kumar, S.; Sinha, A.K. Overexpression of an Apoplastic Peroxidase Gene CrPrx in Transgenic Hairy Root Lines of Catharanthus roseus. Appl. Microbiol. Biotechnol. 2011, 90, 1005–1016. [Google Scholar] [CrossRef]
- Liu, D.H.; Ren, W.W.; Cui, L.J.; Zhang, L.; Liao, Z.; Tang, K.X. Enhanced Accumulation of Catharanthine and Vindoline in Catharanthus roseus Hairy Roots by Overexpression of Transcriptional Factor ORCA2. Afr. J. Biotechnol. 2011, 10, 3260–3268. [Google Scholar] [CrossRef]
- Tang, K.X.; Liu, D.H.; Wang, Y.L.; Cui, L.J.; Ren, W.W.; Sun, X.F. Overexpression of Transcriptional Factor ORCA3 Increases the Accumulation of Catharanthine and Vindoline in Catharanthus roseus Hairy Roots. Russ. J. Plant Physiol. 2011, 58, 415–422. [Google Scholar] [CrossRef]
- Suttipanta, N.; Pattanaik, S.; Kulshrestha, M.; Patra, B.; Singh, S.K.; Yuan, L. The Transcription Factor CrWRKY1 Positively Regulates the Terpenoid Indole Alkaloid Biosynthesis in Catharanthus roseus. Plant Physiol. 2011, 157, 2081–2093. [Google Scholar] [CrossRef]
- Hao, X.; Xie, C.; Ruan, Q.; Zhang, X.; Wu, C.; Han, B.; Qian, J.; Zhou, W.; Nützmann, H.-W.; Kai, G. The Transcription Factor OpWRKY2 Positively Regulates the Biosynthesis of the Anticancer Drug Camptothecin in Ophiorrhiza pumila. Hortic. Res. 2021, 8, 7. [Google Scholar] [CrossRef]
- Liu, J.; Gao, F.; Ren, J.; Lu, X.; Ren, G.; Wang, R. A Novel AP2/ERF Transcription Factor CR1 Regulates the Accumulation of Vindoline and Serpentine in Catharanthus roseus. Front. Plant Sci. 2017, 8, 2082. [Google Scholar] [CrossRef]
- Bunsupa, S.; Hanada, K.; Maruyama, A.; Aoyagi, K.; Komatsu, K.; Ueno, H.; Yamashita, M.; Sasaki, R.; Oikawa, A.; Saito, K.; et al. Molecular Evolution and Functional Characterization of a Bifunctional Decarboxylase Involved in Lycopodium Alkaloid Biosynthesis. Plant Physiol. 2016, 171, 2432–2444. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, H.; Song, Z.; Gao, Y.; Li, W.; Li, M.; Zhao, L.; Li, Y.; Wang, B. Ethylene Response Factor NtERF91 Positively Regulates Alkaloid Accumulations in Tobacco (Nicotiana tabacum L.). Biochem. Biophys. Res. Commun. 2019, 517, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, S.; Wang, J.; Zhou, Q.; Yang, C.; Bai, F.; Lan, X.; Chen, M.; Liao, Z. Engineering Tropane Alkaloid Production Based on Metabolic Characterization of Ornithine Decarboxylase in Atropa belladonna. ACS Synth. Biol. 2020, 9, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Li, S.; Yang, C.; Zhao, T.; Zhang, T.; Lan, X.; Chen, M.; Liao, Z. Overexpression of the AbSAUR1 Gene Enhanced Biomass Production and Alkaloid Yield in Atropa belladonna. Ind. Crops Prod. 2019, 140, 111705. [Google Scholar] [CrossRef]
- Zakaria, M.M.; Schemmerling, B.; Ober, D. CRISPR/Cas9-Mediated Genome Editing in Comfrey (Symphytum officinale) Hairy Roots Results in the Complete Eradication of Pyrrolizidine Alkaloids. Molecules 2021, 26, 1498. [Google Scholar] [CrossRef]
- Nakayasu, M.; Akiyama, R.; Lee, H.J.; Osakabe, K.; Osakabe, Y.; Watanabe, B.; Sugimoto, Y.; Umemoto, N.; Saito, K.; Muranaka, T.; et al. Generation of α-Solanine-Free Hairy Roots of Potato by CRISPR/Cas9 Mediated Genome Editing of the St16DOX Gene. Plant Physiol. Biochem. 2018, 131, 70–77. [Google Scholar] [CrossRef]
- Akiyama, R.; Lee, H.J.; Nakayasu, M.; Osakabe, K.; Osakabe, Y.; Umemoto, N.; Saito, K.; Muranaka, T.; Sugimoto, Y.; Mizutani, M. Characterization of Steroid 5α-Reductase Involved in α-Tomatine Biosynthesis in Tomatoes. Plant Biotechnol. 2019, 36, 253–263. [Google Scholar] [CrossRef]
- Broun, P. Transcription Factors as Tools for Metabolic Engineering in Plants. Curr. Opin. Plant Biol. 2004, 7, 202–209. [Google Scholar] [CrossRef]
- Sazegari, S.; Niazi, A.; Shahriari-Ahmadi, F.; Moshtaghi, N.; Ghasemi, Y. CrMYC1 Transcription Factor Overexpression Promotes the Production of Low Abundance Terpenoid Indole Alkaloids in Catharanthus roseus. Plant Omics 2018, 11, 30–36. [Google Scholar] [CrossRef]
- Singh, S.K.; Patra, B.; Paul, P.; Liu, Y.; Pattanaik, S.; Yuan, L. Revisiting the ORCA Gene Cluster That Regulates Terpenoid Indole Alkaloid Biosynthesis in Catharanthus roseus. Plant Sci. 2020, 293, 110408. [Google Scholar] [CrossRef]
- Zhang, Y.; Malzahn, A.A.; Sretenovic, S.; Qi, Y. The Emerging and Uncultivated Potential of CRISPR Technology in Plant Science. Nat. Plants 2019, 5, 778–794. [Google Scholar] [CrossRef]
- Pezzuto, J.M. Resveratrol: Twenty Years of Growth, Development and Controversy. Biomol. Ther. 2019, 27, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Ma, Y.; Liu, K.; Gao, J.; Li, S.; Sun, X.; Li, G. Resveratrol Induces DNA Damage-Mediated Cancer Cell Senescence through the DLC1–DYRK1A–EGFR Axis. Food Funct. 2023, 14, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Bermúdez-Cruz, R.M. Natural Compounds That Target DNA Repair Pathways and Their Therapeutic Potential to Counteract Cancer Cells. Front. Oncol. 2020, 10, 598174. [Google Scholar] [CrossRef] [PubMed]
- Erdemir Sayan, S.; Sreekumar, R.; Bhome, R.; Mirnezami, A.; Yagci, T.; Sayan, A.E. ERCC1 Abundance Is an Indicator of DNA Repair-Apoptosis Decision upon DNA Damage. Cell Death Discov. 2024, 10, 47. [Google Scholar] [CrossRef]
- Demeyer, A.; Benhelli-Mokrani, H.; Chenais, B.; Weigel, P.; Fleury, F. Inhibiting Homologous Recombination by Targeting RAD51 Protein. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188597. [Google Scholar] [CrossRef]
- Roy, M.; Sinha, D.; Chakraborty Mukherjee, S.; Biswas, J. Curcumin Prevents DNA Damage and Enhances the Repair Potential in a Chronically Arsenic-Exposed Human Population in West Bengal, India. Eur. J. Cancer Prev. 2011, 20, 123–131. [Google Scholar] [CrossRef]
- Tsai, K.D.; Lin, J.C.; Yang, S.M.; Tseng, M.J.; Hsu, J.D.; Lee, Y.J.; Cherng, J.M. Curcumin Protects against UVB-Induced Skin Cancers in SKH-1 Hairless Mouse: Analysis of Early Molecular Markers in Carcinogenesis. Evid. Based Complement. Altern. Med. 2012, 2012, 593952. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of P53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Eity, T.A.; Bhuia, M.; Chowdhury, R.; Ahmmed, S.; Sheikh, S.; Akter, R.; Islam, M.T. Therapeutic Efficacy of Quercetin and Its Nanoformulation Both the Mono- or Combination Therapies in the Management of Cancer: An Update with Molecular Mechanisms. J. Trop. Med. 2024, 2024, 5594462. [Google Scholar] [CrossRef]
- Li, C.; Wu, J.; Dong, Q.; Ma, J.; Gao, H.; Liu, G.; Chen, Y.; Ning, J.; Lv, X.; Zhang, M.; et al. The Crosstalk between Oxidative Stress and DNA Damage Induces Neural Stem Cell Senescence by HO-1/PARP1 Non-Canonical Pathway. Free Radic. Biol. Med. 2024, 223, 443–457. [Google Scholar] [CrossRef]
- Zhou, B.; Yang, Y.; Pang, X.; Shi, J.; Jiang, T.; Zheng, X. Quercetin Inhibits DNA Damage Responses to Induce Apoptosis via SIRT5/PI3K/AKT Pathway in Non-Small Cell Lung Cancer. Biomed. Pharmacother. 2023, 165, 115071. [Google Scholar] [CrossRef] [PubMed]
- Zahmatkesh, M.H.; Banparvar, M.; Saedi, H.S. Quercetin as a Radiosensitizer for Enhanced Efficacy of Radiotherapy in MCF-7 Breast Cancer Cells. J. Curr. Oncol. Med. Sci. 2024, 4, 925–935. [Google Scholar]
- Biyabani, A.; Mazidimoradi, A.; Ghorbani, F.; Allahqoli, L.; Salehiniya, H. The Effect of Quercetin on the Prevention and Treatment of Gynecologic Cancer. Clin. Exp. Obstet. Gynecol. 2024, 51, 205. [Google Scholar] [CrossRef]
- Lee, C.-F.; Yang, J.; Tsai, F.-J.; Chiang, N.-N.; Lu, C.-C.; Huang, Y.-S.; Chen, C.; Chen, F.-A. Kaempferol Induces ATM/P53-Mediated Death Receptor and Mitochondrial Apoptosis in Human Umbilical Vein Endothelial Cells. Int. J. Oncol. 2016, 48, 2007–2014. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, T.; Bao, Y.; Li, P.; Zhao, T.; Liu, Y.; Wang, H.; Sun, C. Genistein Implications in Radiotherapy: Kill Two Birds with One Stone. Molecules 2025, 30, 188. [Google Scholar] [CrossRef]
- Trivedi, S.; Hussain, U.; Agade, R.; Belgamwar, V. A Comprehensive Review on Exploring Thymoquinone as Novel Therapeutic Molecule for Clinical Management of Glioblastoma Multiforme. Pharmacol. Res. Nat. Prod. 2024, 5, 100107. [Google Scholar] [CrossRef]
- Shin, D.; Monga, S.P.S. Cellular and Molecular Basis of Liver Development. Compr. Physiol. 2013, 3, 799–815. [Google Scholar] [CrossRef]
- Wertheim, B.; Beukeboom, L.W.; van de Zande, L. Polyploidy in Animals: Effects of Gene Expression on Sex Determination, Evolution and Ecology. Cytogenet. Genome Res. 2013, 140, 256–269. [Google Scholar] [CrossRef]
- Broughton, K.M.; Khieu, T.; Nguyen, N.; Rosa, M.; Mohsin, S.; Quijada, P.; Wang, B.J.; Echeagaray, O.H.; Kubli, D.A.; Kim, T.; et al. Cardiac Interstitial Tetraploid Cells Can Escape Replicative Senescence in Rodents but Not Large Mammals. Commun. Biol. 2019, 2, 205. [Google Scholar] [CrossRef]
- Duelli, D.; Lazebnik, Y. Cell-to-Cell Fusion as a Link between Viruses and Cancer. Nat. Rev. Cancer 2007, 7, 968–976. [Google Scholar] [CrossRef]
- Hu, L.; Plafker, K.; Vorozhko, V.; Zuna, R.E.; Hanigan, M.H.; Gorbsky, G.J.; Plafker, S.M.; Angeletti, P.C.; Ceresa, B.P. Human Papillomavirus 16 E5 Induces Bi-Nucleated Cell Formation by Cell–Cell Fusion. Virology 2009, 384, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomavirus Oncoproteins: Pathways to Transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Gao, P.; Zheng, J. High-Risk HPV E5-Induced Cell Fusion: A Critical Initiating Event in the Early Stage of HPV-Associated Cervical Cancer. Virol. J. 2010, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.T.; Duronio, R.J. Endoreplication and Polyploidy: Insights into Development and Disease. Development 2013, 140, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in Liver Development, Homeostasis and Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 391–405. [Google Scholar] [CrossRef]
- Adachi, S.; Minamisawa, K.; Okushima, Y.; Inagaki, S.; Yoshiyama, K.; Kondou, Y.; Kaminuma, E.; Kawashima, M.; Toyoda, T.; Matsui, M.; et al. Programmed Induction of Endoreduplication by DNA Double-Strand Breaks in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 10004–10009. [Google Scholar] [CrossRef]
- Rieder, C.; Maiato, H. Stuck in Division or Passing ThroughWhat Happens When Cells Cannot Satisfy the Spindle Assembly Checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef]
- Balachandran, R.; Kipreos, E. Addressing a Weakness of Anticancer Therapy with Mitosis Inhibitors: Mitotic Slippage. Mol. Cell Oncol. 2017, 4, e1277293. [Google Scholar] [CrossRef]
- Kirsch-Volders, M.; Mišík, M.; Fenech, M. Tetraploidy in Normal Tissues and Diseases: Mechanisms and Consequences. Chromosoma 2025, 134, 3. [Google Scholar] [CrossRef]
- Ning, Y.; Zheng, M.; Zhang, Y.; Jiao, Y.; Wang, J.; Zhang, S. RhoA-ROCK2 Signaling Possesses Complex Pathophysiological Functions in Cancer Progression and Shows Promising Therapeutic Potential. Cancer Cell Int. 2024, 24, 339. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. P53: Guardian of Ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-Related Tubular Cell Hypertrophy and Progenitor Proliferation Recover Renal Function after Acute Kidney Injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, J.; Jackman, C.P.; Cox, A.H.; Trembley, M.A.; Balowski, J.J.; Cox, B.D.; De Simone, A.; Dickson, A.L.; Di Talia, S.; et al. Tension Creates an Endoreplication Wavefront That Leads Regeneration of Epicardial Tissue. Dev. Cell 2017, 42, 600-615.e4. [Google Scholar] [CrossRef]
- Øvrebø, J.I.; Edgar, B.A. Polyploidy in Tissue Homeostasis and Regeneration. Development 2018, 145, dev156034. [Google Scholar] [CrossRef]
- Kwon, A.-H.; Inada, Y.; Uetsuji, S.; Yamamura, M.; Hioki, K.; Yamamoto, M. Response of Fibronectin to Liver Regeneration after Hepatectomy. Hepatology 1990, 11, 593–598. [Google Scholar] [CrossRef]
- Kato, S.; Otsu, K.; Ohtake, K.; Kimura, Y.; Yashiro, T.; Suzuki, T.; Akamatsu, N. Concurrent Changes in Sinusoidal Expression of Laminin and Affinity of Hepatocytes to Laminin during Rat Liver Regeneration. Exp. Cell Res. 1992, 198, 59–68. [Google Scholar] [CrossRef]
- Otsu, K.; Kato, S.; Ohtake, K.; Akamatsu, N. Alteration of Rat Liver Proteoglycans during Regeneration. Arch. Biochem. Biophys. 1992, 294, 544–549. [Google Scholar] [CrossRef]
- Gallai, M.; Sebestyén, A.; Nagy, P.; Kovalszky, I.; Ónody, T.; Thorgeirsson, S.S. Proteoglycan Gene Expression in Rat Liver after Partial Hepatectomy. Biochem. Biophys. Res. Commun. 1996, 228, 690–694. [Google Scholar] [CrossRef]
- Liu, B.; Paranjpe, S.; Bowen, W.C.; Bell, A.W.; Luo, J.H.; Yu, Y.P.; Mars, W.M.; Michalopoulos, G.K. Investigation of the Role of Glypican 3 in Liver Regeneration and Hepatocyte Proliferation. Am. J. Pathol. 2009, 175, 717–724. [Google Scholar] [CrossRef]
- Schuppan, M.; Somasundaram, R.; Hahn, G.E. Matrix as a Modulator of Hepatic Fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A. Transforming Growth Factor β in Tissue Fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Choi, Y.; Ha, S.-K.; Song, B.-J. Cytochrome P450-2E1 Promotes Aging-Related Hepatic Steatosis, Apoptosis and Fibrosis through Increased Nitroxidative Stress. Free Radic. Biol. Med. 2016, 91, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, P.D.; Duncan, A.W. Differential Roles for Diploid and Polyploid Hepatocytes in Acute and Chronic Liver Injury. Semin. Liver Dis. 2021, 41, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kim, J.; Lee, C.; Oh, D.; Han, J.; Kim, T.-J.; Kim, S.-W.; Seo, Y.-S.; Oh, S.; Jung, Y. Tumor Necrosis Factor-Inducible Gene 6 Reprograms Hepatic Stellate Cells into Stem-like Cells, Which Ameliorates Liver Damage in Mouse. Biomaterials 2019, 219, 119375. [Google Scholar] [CrossRef]
- Tang, C.; Chen, H.; Jiang, L.; Liu, L. Liver Regeneration: Changes in Oxidative Stress, Immune System, Cytokines, and Epigenetic Modifications Associated with Aging. Oxidative Med. Cell Longev. 2022, 2022, 9018811. [Google Scholar] [CrossRef]
- Yang, Y.; Jn-Simon, N.; He, Y.; Sun, C.; Zhang, P.; Hu, W.; Tian, T.; Zeng, H.; Basha, S.; Huerta, A.S.; et al. A BCL-XL/BCL-2 PROTAC Effectively Clears Senescent Cells in the Liver and Reduces MASH-Driven Hepatocellular Carcinoma in Mice. Nat. Aging 2025, 5, 386–400. [Google Scholar] [CrossRef]
- Lukkunaprasit, T.; Tansawet, A.; Boonmanunt, S.; Sobhonslidsuk, A.; McKay, G.J.; Attia, J.; Thakkinstian, A. An Updated Meta-Analysis of Effects of Curcumin on Metabolic Dysfunction-Associated Fatty Liver Disease Based on Available Evidence from Iran and Thailand. Sci. Rep. 2023, 13, 5824. [Google Scholar] [CrossRef]
- Xiong, F.; Zhang, Y.; Li, T.; Tang, Y.; Song, S.Y.; Zhou, Q.; Wang, Y. A Detailed Overview of Quercetin: Implications for Cell Death and Liver Fibrosis Mechanisms. Front. Pharmacol. 2024, 15, 1389179. [Google Scholar] [CrossRef]
- Zhang, L.; De Cecco, M.; Lee, M.; Hao, X.; Maslov, A.Y.; Montagna, C.; Campisi, J.; Dong, X.; Sedivy, J.M.; Vijg, J. Analysis of Somatic Mutations in Senescent Cells Using Single-Cell Whole-Genome Sequencing. Aging Biol. 2023, 1, 20230005. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Plants | ST | Targets | References |
|---|---|---|---|---|
| Epigallocatechin gallate 1 | Green tea | SM | ↓ PI3k/Akt/mTOR, ROS, Cox2, NF-κB, IL-6, TNF-α and ↑ AMPK | [103] |
| Apigenin 1 | Lamiaceae | SM | AMPK-mTOR-TFEB, NF-κB subunit p65 and IκB | [104] |
| Eupatilin 1 | Wormwood | SM | MAPK-NF-κB, ↓ e p21, p53 | [105] |
| Kaempferol 1 | Apples, grapes, tomatoes, green tea, etc. | SM | MAPK, NF-κB subunit p65 and IκB | [106] |
| Luteolin 1 | Celery, parsley, broccoli, apple peels, chrysanthemum flowers | SM | ↑ SIRT6, ↓ NF-κB | [107] |
| Proanthocyanidins 1 | Fruits, bark, leaves, seeds of many plants | SM | ↓ PI3K-Akt | [108,109] |
| Anthocyanins 1 | Canadian elderberries | SL | ↓ PI3K/AKT/mTOR | [109] |
| Quercetin 1 | Rosaceae, Theaceae, Brassicaceae, Asparagaceae, Ericaceae, Moraceae | SL SM | ↓ NF-κB, ↑ SIRT-1, ↓ COX and lipoxygenase; mTOR, PI3K/Akt, p53/p21/serpins | [4,110] |
| Butein 2 | Dahlias, coreopsis | SM | Sirt1-p53 | [111,112] |
| p-Coumaric acid 2 | Peanuts, beans, tomatoes, sweet clover, carrots, basil, garlic, strawberries | SM | ↓ Nrf2-NF-κB | [113] |
| Gallic 2 and ellagic acids 2 | Blackberries, cloudberries, pomegranates, raspberries, strawberries, chestnuts, walnuts | SL | ↓ e BCL-2, ↓ NF-κB, ↓ TNFα, ↓ IL-1β, ↓ IL-6, ↓ ROS | [48] |
| Curcumin 2 | Turmeric | SM | AMPK-mTOR-ULK1↑ | [114] |
| SM SL | p70/S6K, Akt-LC3-II-SQSTM1/p62 JNK, ↑ Nrf2, ↓ NF-κB, and e pro-inflammatory cytokines, ↓ e IL-1β, TNF-α, IL-10 | [115] | ||
| Fisetin 2 | Lacquer tree (Anacardiaceae), strawberries, apples, persimmons, grapes | SL SM | ↑ SIRT1, ↓ IL-6, TNF-alpha, ↓ NF-κB and Nrf2 | [116,117] |
| Honokiol 2 (“hou po”) | Bark and leaves of magnolias | SM | ↑ AMPK-PGC-1α-SIRT3 | [118] |
| Myricetin 2 | Myricaceae, Polygonaceae, Primulaceae | SM | SERPINE1 SIRT1-PGC-1α, ↓ IL-1β and ↓ IL-6, ↓ e p21, p16 | [119,120] |
| Polydatin 2 | Vitaceae, Liliaceae, Fabaceae | SL | Nrf2-HO-1 | [121] |
| Resveratrol 2 | Grapes, raspberries, mulberries, pistachios, and peanuts | SM SL | SIRT1, ROS-NF-κB ROS-PI3K-Akt | [122,123,124,125,126,127] |
| Vanillin 2 | Vanilla orchid, Korean pine fruits, mango | SL SM | TLR-2, NF-κB, Nrf2, ↓ SASP | [128,129] |
| Gingerol A, 6-Shogaol | Ginger | SL | Caspase-3, ↓ Bcl-XL, ↓ IL-6 | [100] |
| Avenanthramide C 2 | Oats, Isatis tinctoria L. leaf extract | SL SM | ↓ p21 CDKN1A and p16INK4A, SASP, ↑ AMPK, ↓ mTOR, MAPK, and IκBα, ↓ NFκB | [130] |
| Dehydrocostus lactone 4 | Costus, sunflower | SM | STING-TBK1-NF-κB, MAPK | [131] |
| Evodiamine 3 | Dried immature fruits of Evodia | SM | Nrf2-HO-1, MAPK | [132] |
| Piperlongumine 3 | Long pepper (Piperaceae) | SL | Inactivation of OXR-1 and ROS-reducing enzyme | [114,133] |
| 20-Deoxyingenol 4 | Bark of Erythroxylum tree | SM | TFEB-mediated autophagy | [134] |
| Kinsenoside 4 | Anectochilus (Orchidaceae) | SM | Akt-ERK1/2-Nrf2 | [135] |
| Morroniside 4 | Cornelian cherry | SM | ROS-Hippo-Mst1/2 and Lats1/2-YAP/TAZ-p53 | [108] |
| Proscillaridin A 4, Digoxin 4 | Wooly and purple foxglove | SL | Na+/K+ATPase, apoptosis | [136] |
| Oleandrin 4 | Oleander | SL | ↑ e NOXA, ↓ p16 and p21, and pro-inflammatory cytokines IL1α, IL1β, and IL8 | [137] |
| Astragaloside 4 | Astragalus membranaceus | SL | STING/NF-κB | [138] |
| Oridonin 4 | Lamiaceae | SL SM | ↓ IL-6 and IL-8, ↓ p38, p65 (NF-κB), glutathione S-transferase | [139,140,141] |
| Active substance not identified | Common goldenrod | SL | ↓ SASP | [142] |
| Cocktail of substances | Fruits of Terminalia chebula (haritaki) | SL SM | ↓ CSF3, CXCL1, IL-1β, IL-6, and IL-8, ↓ miR 29a-3p, miR 30a-3p, miR 34a-5p, miR 24a-3p | [143] |
| Active substance not identified | Extract of flowers of Silybum marianum | SL | ↓ IL-6, MMP-1 | [144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fizikova, A.; Prokhorova, A.; Churikova, D.; Konstantinov, Z.; Ivanov, R.; Karabelsky, A.; Rybtsov, S. Hepatocytes as Model for Investigating Natural Senotherapeutic Compounds and Their Effects on Cell Cycle Dynamics and Genome Stability. Int. J. Mol. Sci. 2025, 26, 6794. https://doi.org/10.3390/ijms26146794
Fizikova A, Prokhorova A, Churikova D, Konstantinov Z, Ivanov R, Karabelsky A, Rybtsov S. Hepatocytes as Model for Investigating Natural Senotherapeutic Compounds and Their Effects on Cell Cycle Dynamics and Genome Stability. International Journal of Molecular Sciences. 2025; 26(14):6794. https://doi.org/10.3390/ijms26146794
Chicago/Turabian StyleFizikova, Anastasia, Anna Prokhorova, Daria Churikova, Zahar Konstantinov, Roman Ivanov, Alexander Karabelsky, and Stanislav Rybtsov. 2025. "Hepatocytes as Model for Investigating Natural Senotherapeutic Compounds and Their Effects on Cell Cycle Dynamics and Genome Stability" International Journal of Molecular Sciences 26, no. 14: 6794. https://doi.org/10.3390/ijms26146794
APA StyleFizikova, A., Prokhorova, A., Churikova, D., Konstantinov, Z., Ivanov, R., Karabelsky, A., & Rybtsov, S. (2025). Hepatocytes as Model for Investigating Natural Senotherapeutic Compounds and Their Effects on Cell Cycle Dynamics and Genome Stability. International Journal of Molecular Sciences, 26(14), 6794. https://doi.org/10.3390/ijms26146794