
In Situ Electrochemical Atomic Force Microscopy Study of Interfacial Reactions on a Graphite Negative Electrode for Magnesium-Ion Batteries




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
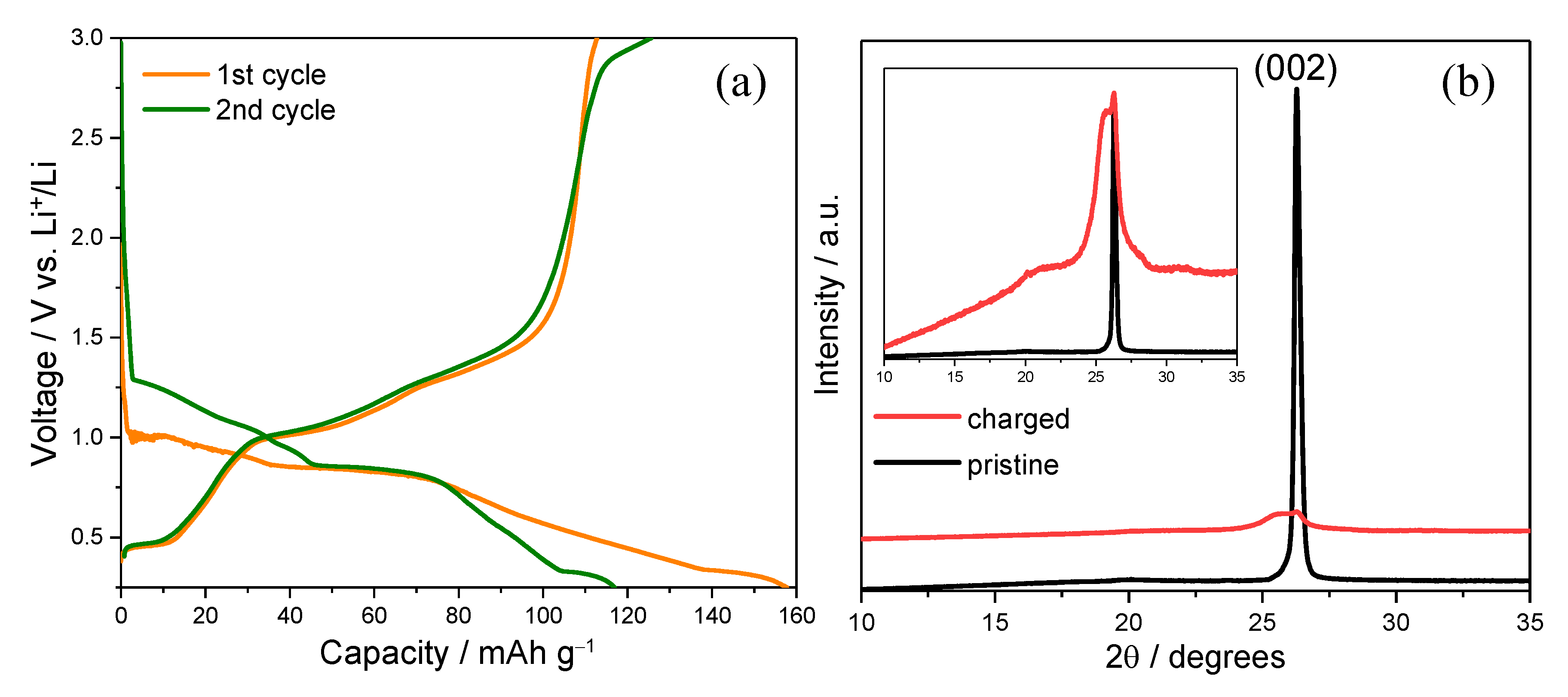
2.1. Electrochemical and Structural Evidence of Solvated Mg2+ Intercalation
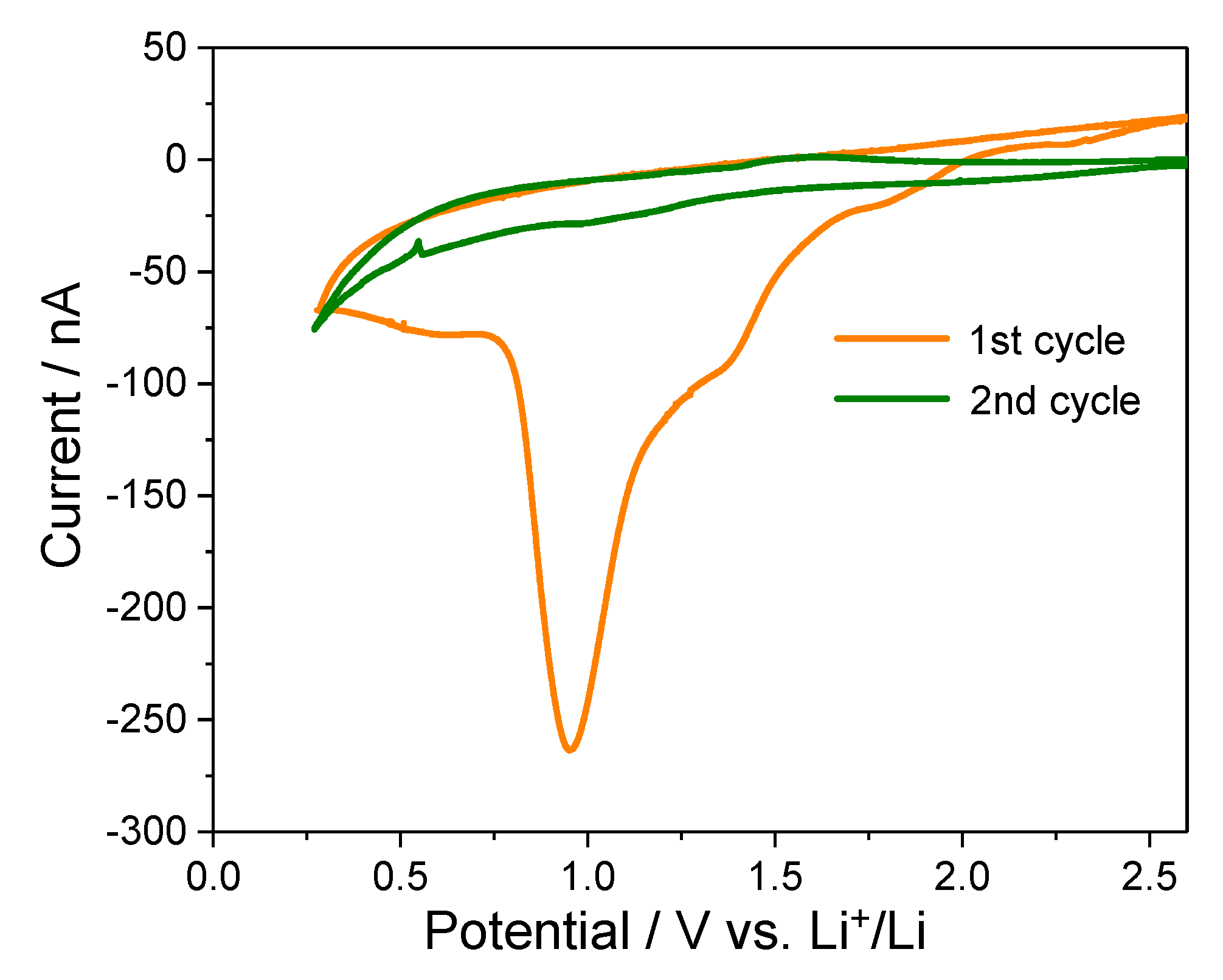
2.2. CV Analysis of HOPG Before In Situ Surface Imaging
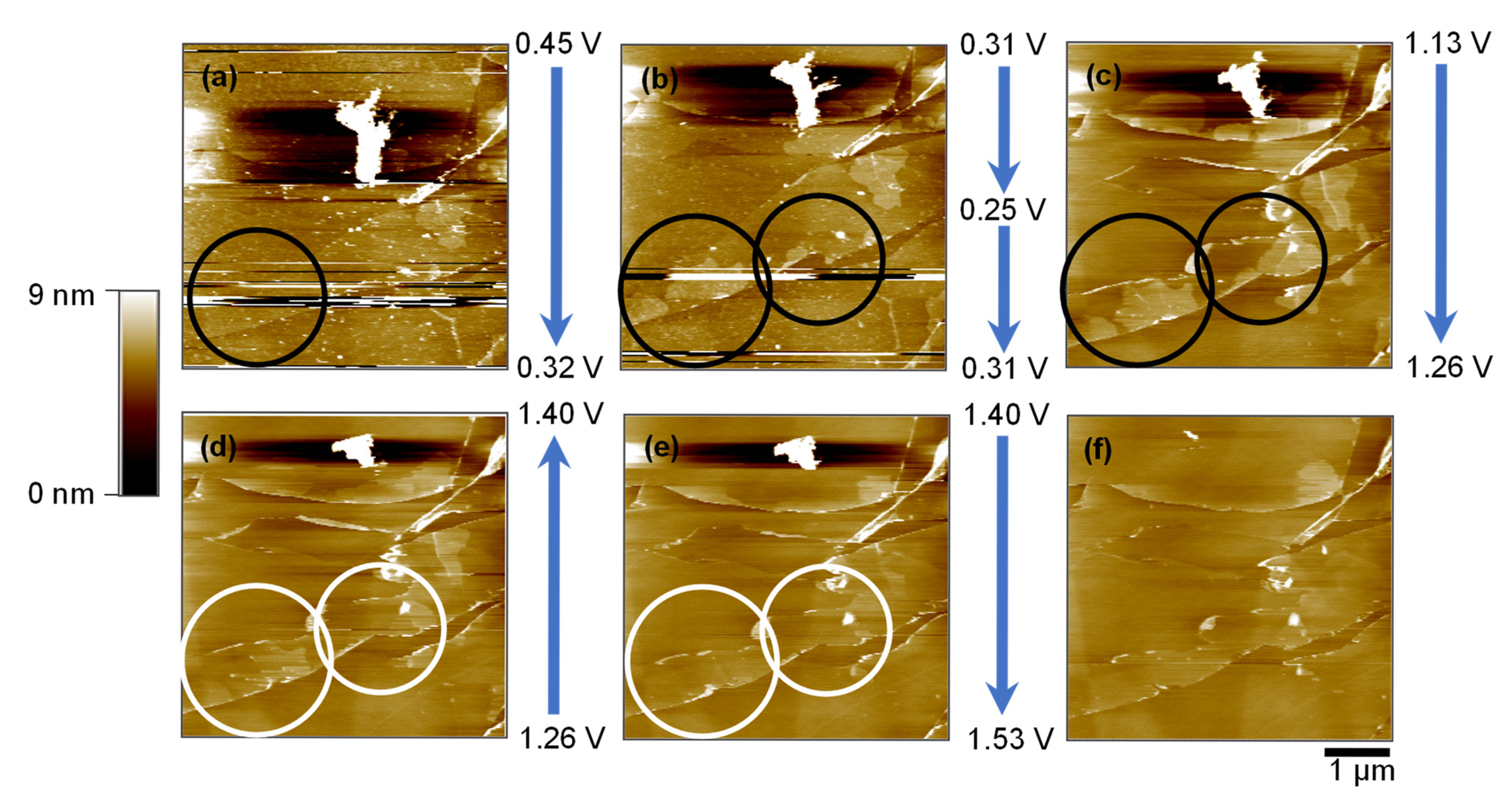
2.3. Surface Evolution on HOPG Basal Plane During CV Cycling
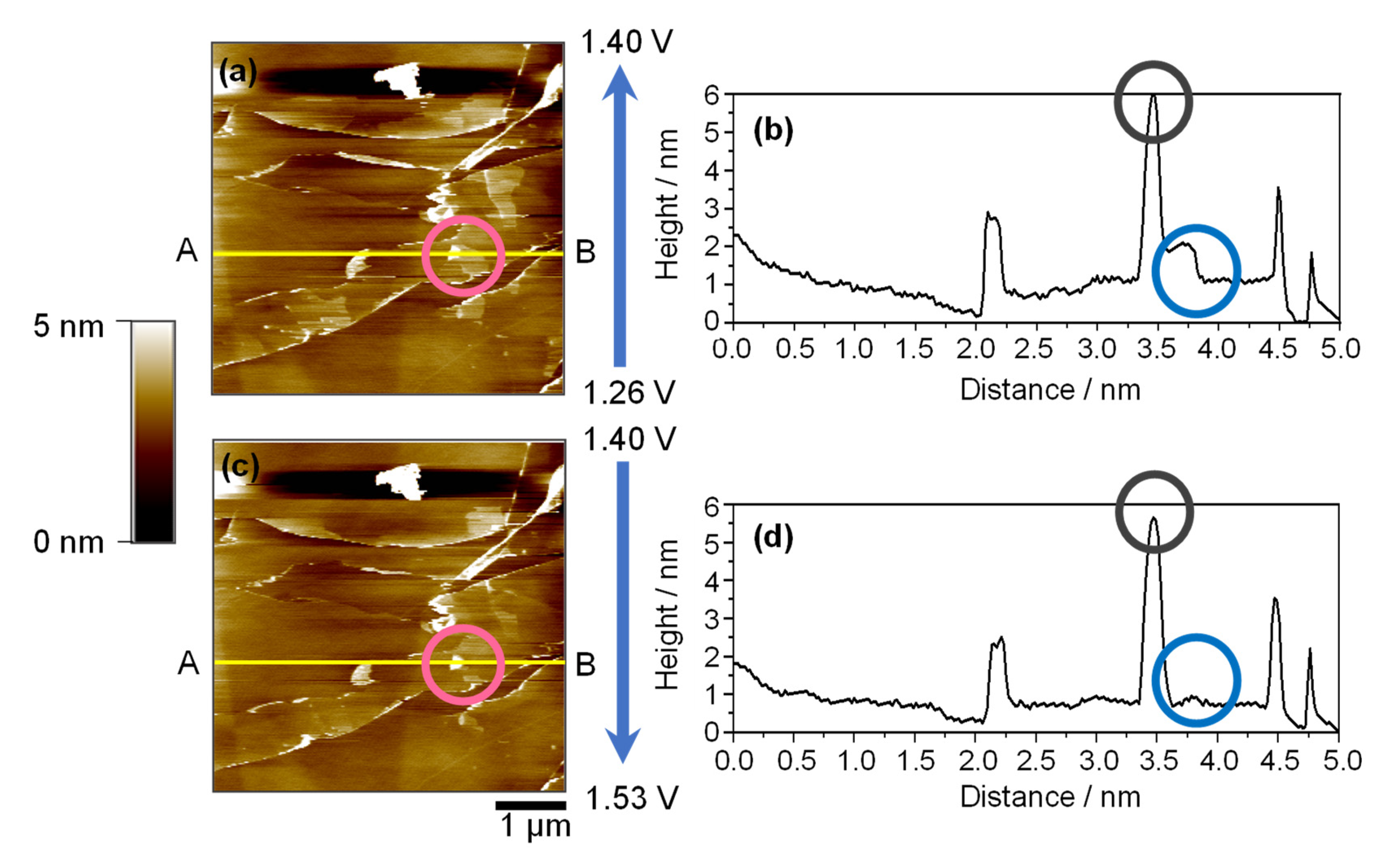
2.4. Morphological Evidence of Reversible Co-Intercalation: Hill and Blister Formation
2.5. Comparison of Solvated Ion Insertion Effects with LIB Systems
3. Materials and Methods
3.1. Electrode Fabrication and Electrochemical Testing
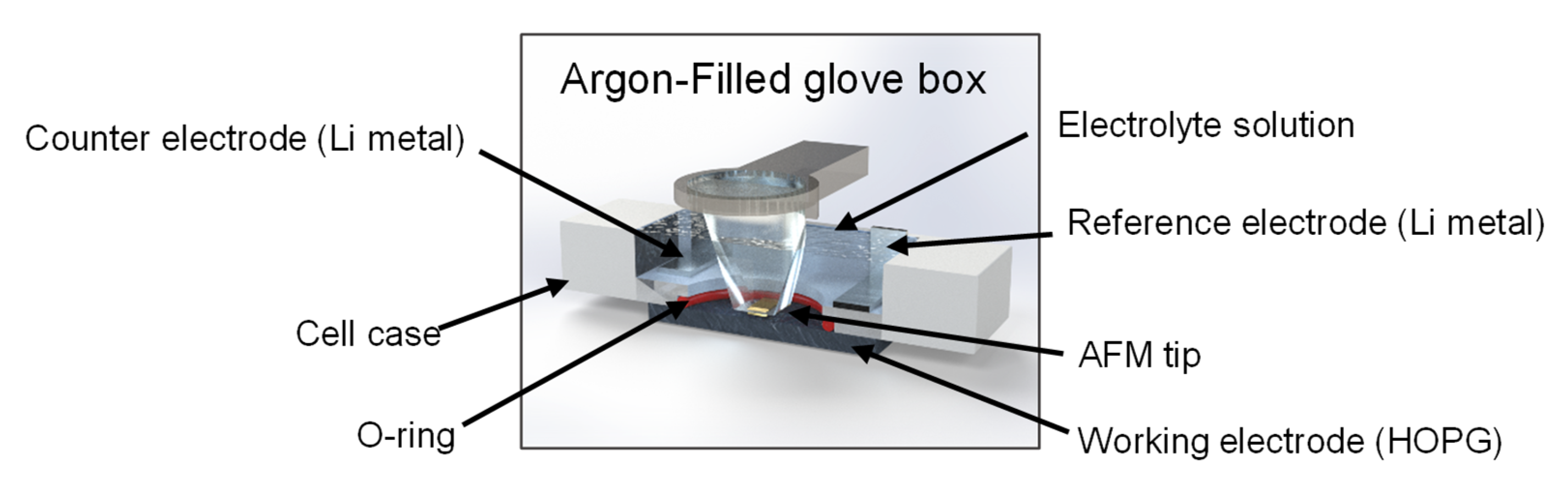
3.2. ECAFM Measurements
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zuo, X.; Zhu, J.; Müller-Buschbaum, P.; Cheng, Y. Silicon based lithium-ion battery anodes: A chronicle perspective review. Nano Energy 2017, 31, 113–143. [Google Scholar] [CrossRef]
- Choi, J.; Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Y.; Adelhelm, P.; Titirici, M.M.; Hu, Y.S. Intercalation chemistry of graphite: Recent advances and future prospects. Chem. Soc. Rev. 2019, 48, 4655–4687. [Google Scholar] [CrossRef] [PubMed]
- Jache, B.; Binder, J.O.; Abe, T.; Adelhelm, P. Layered FeS2 as intercalation cathode for rechargeable sodium batteries. Phys. Chem. Chem. Phys. 2016, 18, 14299–14316. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yu, M.; Feng, X. Two-dimensional materials and their derived architectures for advanced sodium batteries. Chem. Soc. Rev. 2021, 50, 2388–2407. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-Y.; Jeong, S.-K. Electrochemical Solvent Cointercalation into Graphite in Propylene Carbonate-Based Electrolytes: A Chronopotentiometric Characterization. J. Anal. Methods Chem. 2018, 2018, 9231857. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lim, K.; Yoon, G.; Park, J.; Lim, H.D.; Sung, Y.E.; Kang, K. Time-resolved observation of lithium intercalation into graphite using in-situ EC-AFM. Adv. Energy Mater. 2017, 7, 1700418. [Google Scholar] [CrossRef]
- Prabakar, S.; Ikhe, A.; Park, W.; Chung, K.Y.; Park, H.; Ahn, D.; Kwak, J.; Sohn, K.S.; Pyo, M. Magnesium-based battery systems: Current state and challenges. Adv. Sci. 2019, 6, 1902129. [Google Scholar] [CrossRef]
- Ling, C.; Banerjee, D.; Matsui, M. Study of the electrochemical deposition behavior of Mg in non-aqueous electrolytes. Electrochim. Acta 2012, 76, 270–274. [Google Scholar] [CrossRef]
- Yoo, H.D.; Shterenberg, I.; Gofer, Y.; Gershinsky, G.; Pour, N.; Aurbach, D. Mg rechargeable batteries: An on-going challenge. Energy Environ. Sci. 2013, 6, 2265–2279. [Google Scholar] [CrossRef]
- Gregory, T.; Hoffman, R.; Winterton, R. Nonaqueous electrochemistry of magnesium. J. Electrochem. Soc. 1990, 3, 137–139. [Google Scholar] [CrossRef]
- Aurbach, D.; Lu, Z.; Schechter, A.; Gofer, Y.; Gizbar, H.; Turgeman, R.; Cohen, Y.; Moshkovich, M.; Levi, E. Prototype systems for rechargeable magnesium batteries. Nature 2000, 407, 724–727. [Google Scholar] [CrossRef] [PubMed]
- God, C.; Bitschnau, B.; Kapper, K.; Lenardt, C.; Schmuck, M.; Mautner, F.A.; Koller, S. Glyme-based electrolytes enabling magnesium intercalation into graphite. RSC Adv. 2017, 7, 14168–14175. [Google Scholar] [CrossRef]
- Kim, D.; Jung, S.; Ha, S.; Kim, Y.; Park, Y.; Ryu, J.; Han, Y.; Lee, K. Solvated magnesium ion intercalation into graphite from diglyme-based electrolyte. Chem. Mater. 2018, 30, 3199–3203. [Google Scholar] [CrossRef]
- Winter, M.; Besenhard, J.O.; Spahr, M.E.; Novák, P. Insertion electrode materials for rechargeable lithium batteries. Adv. Mater. 1998, 10, 725–763. [Google Scholar] [CrossRef]
- Shimizu, M.; Nakahigashi, A.; Arai, S. Intercalation/deintercalation of solvated Mg2+ into/from graphite interlayers. Phys. Chem. Chem. Phys. 2021, 23, 20317–20324. [Google Scholar] [CrossRef] [PubMed]
- Ponrouch, A.; Verrelli, R.; Barda, D.; Frontera, C.; Palacín, M.R. Electrochemical intercalation of calcium and magnesium in TiS2: Fundamental studies related to multivalent battery applications. Chem. Mater. 2018, 30, 7575–7583. [Google Scholar] [CrossRef]
- Tchitchekova, D.S.; Monti, D.; Johansson, P.; Barde, F.; Randon-Vitanova, A.; Palacín, M.R.; Ponrouch, A. On the Reliability of Half-Cell Tests for Monovalent (Li+, Na+) and Divalent (Mg2+, Ca2+) Cation Based Batteries. J. Electrochem. Soc. 2017, 164, A1384–A1392. [Google Scholar] [CrossRef]
- Tan, S.; Xiong, F.; Wang, J.; An, Q.; Mai, L. Crystal regulation towards rechargeable magnesium battery cathode materials. Mater. Horiz. 2020, 7, 2289–2313. [Google Scholar] [CrossRef]
- Mizuno, Y.; Okubo, M.; Hosono, E.; Kudo, T.; Honma, I. Electrochemical Mg2+ intercalation into a bimetallic CuFe Prussian blue analog in aqueous electrolytes. J. Mater. Chem. A 2013, 1, 8859–8864. [Google Scholar] [CrossRef]
- Lee, H.J.; Shin, J.; Choi, J.W. Intercalated water and organic molecules for electrode materials of rechargeable batteries. Adv. Mater. 2018, 30, 1705851. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.K.; Inaba, M.; Mogi, R.; Iriyama, Y.; Abe, T.; Ogumi, Z. Surface film formation on a graphite negative electrode in lithium-ion batteries: Atomic force microscopy study on the effects of film-forming additives in propylene carbonate solutions. Langmuir 2001, 17, 8281–8286. [Google Scholar] [CrossRef]
- Zhu, H.; Russell, J.A.; Fang, Z.; Barnes, P.; Li, L.; Efaw, C.M.; Daniel, C. In situ analysis of solid electrolyte interphase formation and evolution on highly oriented pyrolytic and disordered graphite negative electrodes in lithium-ion batteries. Small 2021, 17, 2105292. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, W.; Gonzalez-Munoz, S.; Forcieri, L.; Yang, W.; Reece, M.J.; Brett, D.J.L.; Shearing, P.R. Nanoarchitecture factors of solid electrolyte interphase formation via 3D nano-rheology microscopy and surface force-distance spectroscopy. Nat. Commun. 2023, 14, 1562. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.K.; Inaba, M.; Iriyama, Y.; Abe, T.; Ogumi, Z. AFM study of surface film formation on a composite graphite electrode in lithium-ion batteries. J. Power Sources 2003, 119–121, 710–715. [Google Scholar] [CrossRef]
- Mahmood, A.; Bai, Z.; Wang, S.; Lei, Y.; Wang, S. Enabling high-performance multivalent metal-ion batteries: Current advances and future prospects. Chem. Soc. Rev. 2025, 54, 1778–1821. [Google Scholar] [CrossRef] [PubMed]
- Hoane, A.G.; Zheng, Q.; Maldonado, N.D.; Lutz, D.M.; Shao-Horn, Y. Impact of multivalent cations on interfacial layering in water-in-salt electrolytes. ACS Appl. Energy Mater. 2024, 7, 2367–2378. [Google Scholar] [CrossRef]
- Domi, Y.; Ochida, M.; Tsubouchi, S.; Yamada, K.; Momma, T. Electrochemical AFM observation of the HOPG edge plane in ethylene carbonate-based electrolytes containing film-forming additives. J. Electrochem. Soc. 2012, 159, A2071–A2076. [Google Scholar] [CrossRef]
- Fukutsuka, T.; Kokumai, R.; Song, H.Y.; Abe, T. In-situ AFM observation of surface morphology of highly oriented pyrolytic graphite in propylene carbonate-based electrolyte solutions containing lithium and bivalent cations. J. Electrochem. Soc. 2016, 163, A2263–A2267. [Google Scholar] [CrossRef]
- Inaba, M.; Kawatate, Y.; Funabiki, A.; Jeong, S.K.; Abe, T.; Ogumi, Z. STM study on graphite/electrolyte interface in lithium-ion batteries: Solid electrolyte interface formation in trifluoropropylene carbonate solution. Electrochim. Acta 1999, 45, 99–105. [Google Scholar] [CrossRef]
- Danis, L.; Gateman, S.M.; Kuss, C. Nanoscale measurements of lithium-ion battery materials using scanning probe techniques. ChemElectroChem 2017, 4, 6–21. [Google Scholar] [CrossRef]
- Zhang, Z.; Said, S.; Smith, K.; Jervis, R.; Shearing, P. Characterizing batteries by in situ electrochemical atomic force microscopy: A critical review. Adv. Energy Mater. 2021, 11, 2101518. [Google Scholar] [CrossRef]
- Inaba, M.; Siroma, Z.; Kawatate, Y.; Funabiki, A.; Ogumi, Z. Electrochemical STM study on lithium intercalation and surface morphology changes of graphite electrode in EC-based electrolytes. J. Power Sources 1997, 68, 221–225. [Google Scholar] [CrossRef]
- Dini, D.; Cognigni, F.; Passeri, D.; Rossi, M.; Rinaldi, D. Multiscale characterization of Li-ion batteries through the combined use of atomic force microscopy and X-ray microscopy and considerations for a correlative analysis approach. J. Electrochem. Soc. 2021, 168, 110507. [Google Scholar] [CrossRef]
- Inaba, M.; Jeong, S.K.; Ogumi, Z. In situ scanning probe microscopy of interfacial phenomena in batteries. Interface 2011, 20, 55–59. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.; Nogales, P.M.; Lee, S.; Yang, S.; Jeong, S.-K. In Situ Electrochemical Atomic Force Microscopy Study of Interfacial Reactions on a Graphite Negative Electrode for Magnesium-Ion Batteries. Int. J. Mol. Sci. 2025, 26, 6793. https://doi.org/10.3390/ijms26146793
Yoon S, Nogales PM, Lee S, Yang S, Jeong S-K. In Situ Electrochemical Atomic Force Microscopy Study of Interfacial Reactions on a Graphite Negative Electrode for Magnesium-Ion Batteries. International Journal of Molecular Sciences. 2025; 26(14):6793. https://doi.org/10.3390/ijms26146793
Chicago/Turabian StyleYoon, Sungjae, Paul Maldonado Nogales, Sangyup Lee, Seunga Yang, and Soon-Ki Jeong. 2025. "In Situ Electrochemical Atomic Force Microscopy Study of Interfacial Reactions on a Graphite Negative Electrode for Magnesium-Ion Batteries" International Journal of Molecular Sciences 26, no. 14: 6793. https://doi.org/10.3390/ijms26146793
APA StyleYoon, S., Nogales, P. M., Lee, S., Yang, S., & Jeong, S.-K. (2025). In Situ Electrochemical Atomic Force Microscopy Study of Interfacial Reactions on a Graphite Negative Electrode for Magnesium-Ion Batteries. International Journal of Molecular Sciences, 26(14), 6793. https://doi.org/10.3390/ijms26146793