
Metabolic Disturbances Involved in Cardiovascular Diseases: The Role of Mitochondrial Dysfunction, Altered Bioenergetics and Oxidative Stress

 , , , and
, , , and
Abstract
1. Introduction
2. Cellular Metabolism in Cardiovascular Health and Disease
3. Essential Hypertension
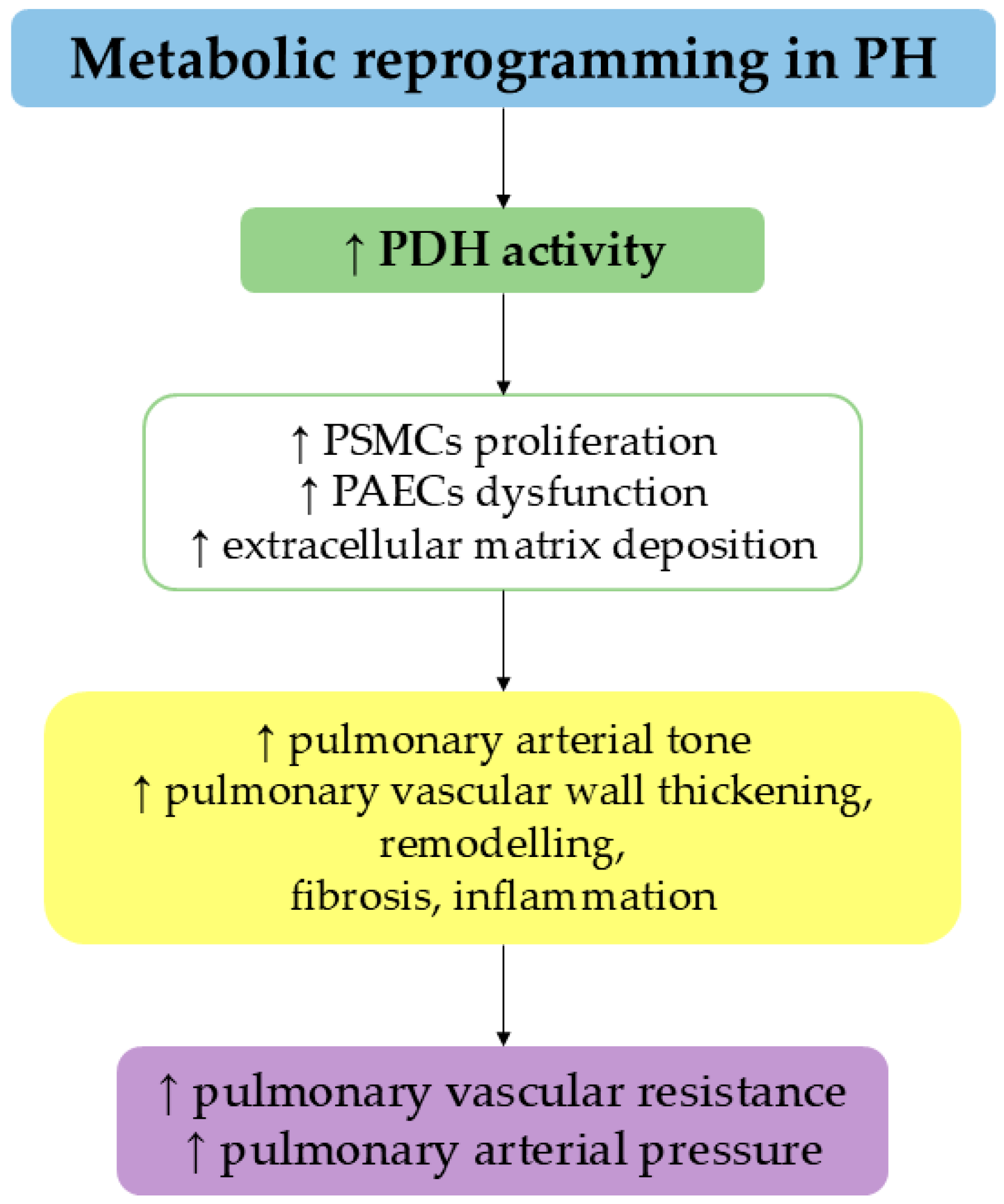
4. Pulmonary Hypertension
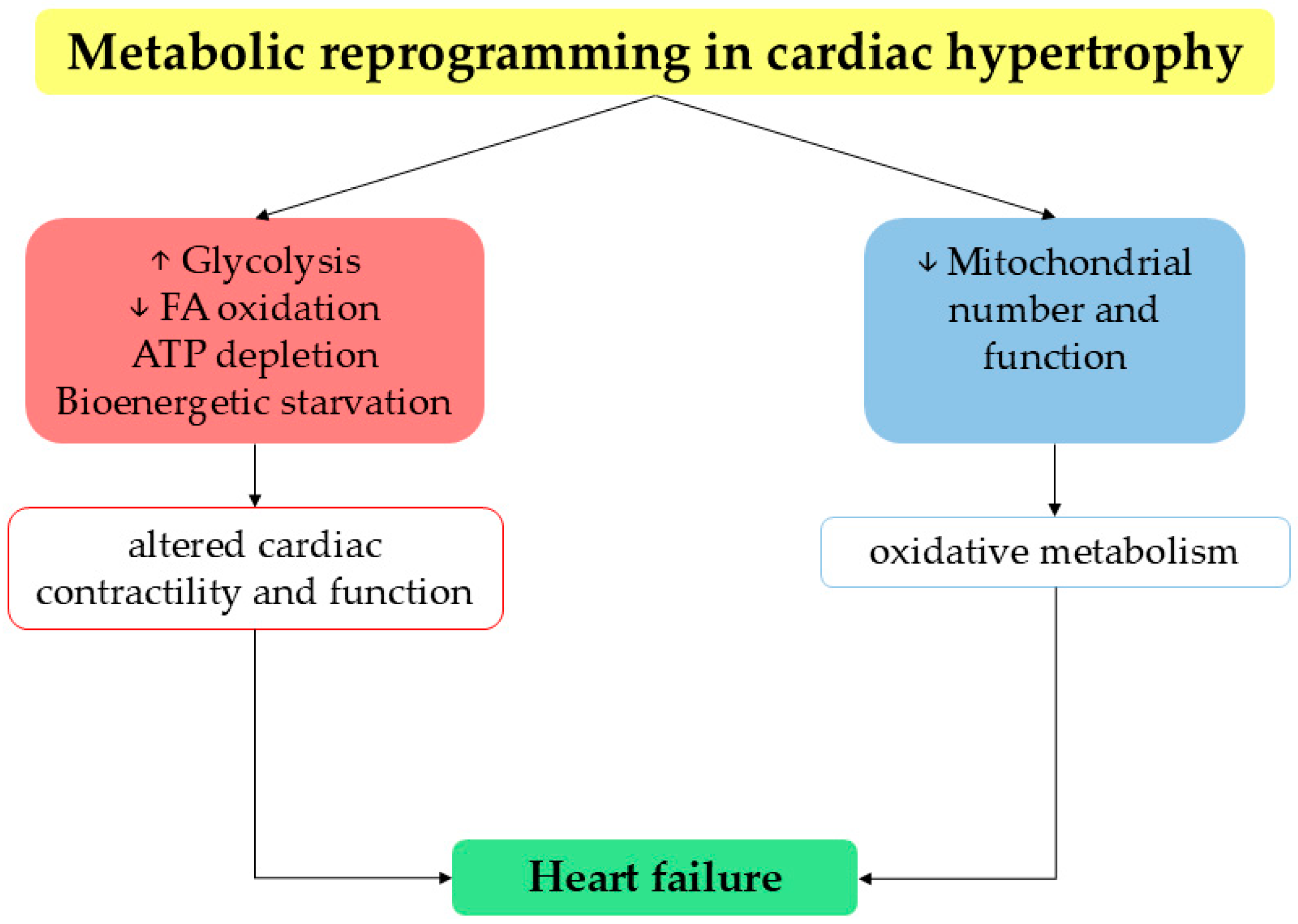
5. Heart Failure
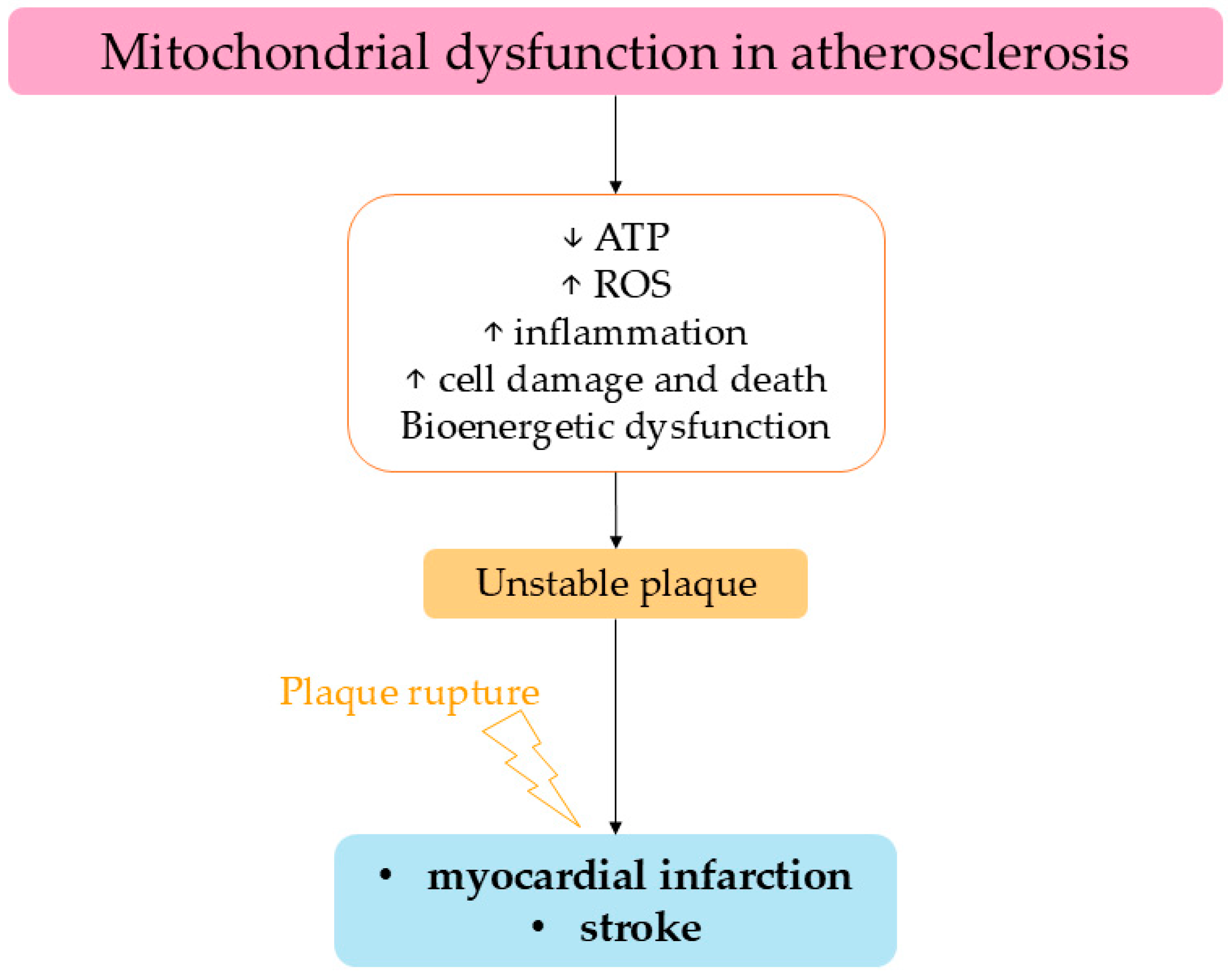
6. Atherosclerosis
7. Myocardial Infarction
8. Stroke
9. Therapeutic Perspectives Based on Recent Acquisitions on Metabolic Disturbances Involved in CVDs
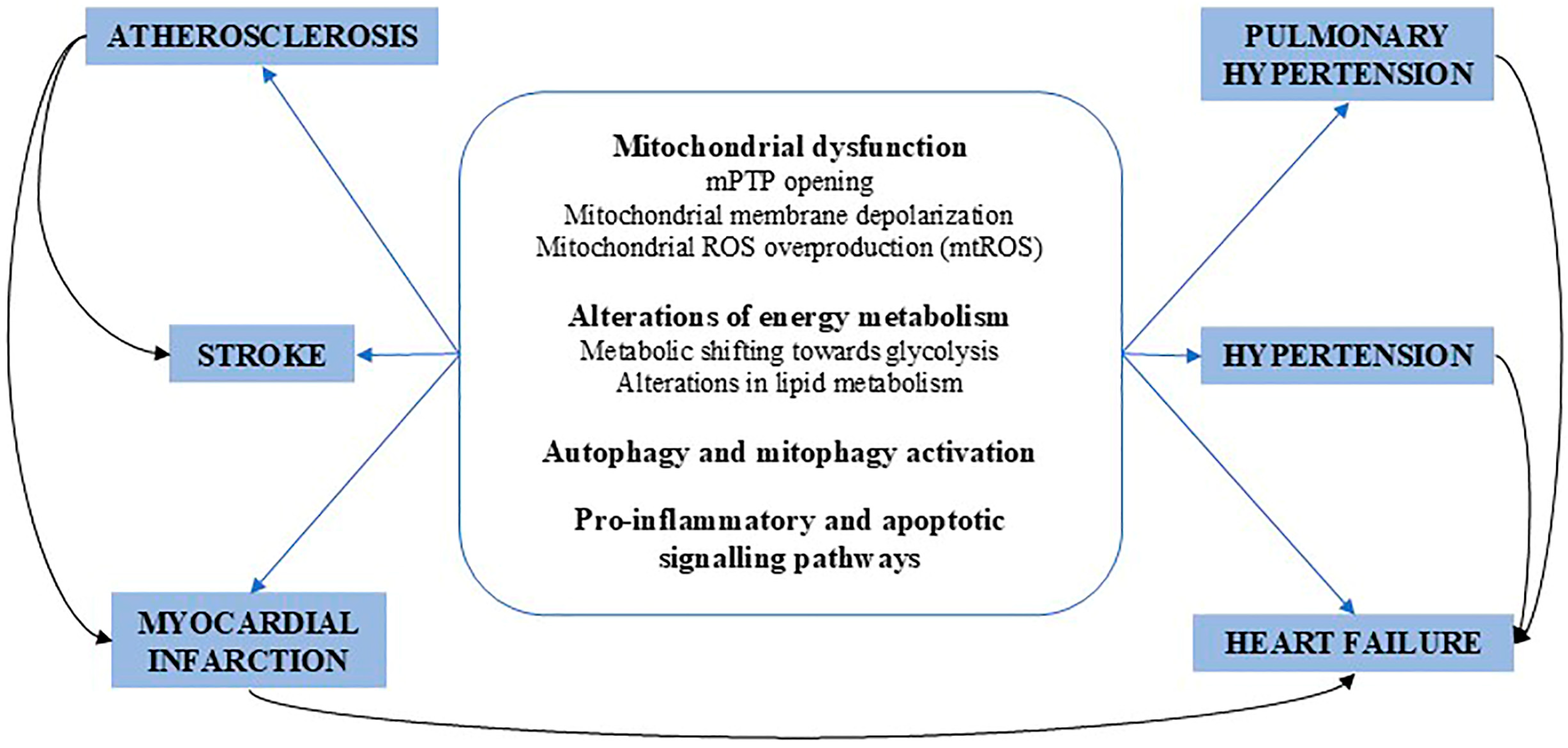
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATF3 | activating transcription factor 3 |
| AIF | apoptosis-inducing factor |
| ALOX12 | arachidonate 12-lipoxygenase |
| AMI | acute myocardial infarction |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| Ang II | angiotensin II |
| ANP | atrial natriuretic peptide |
| AOAA | aminooxyacetic acid |
| ApoE | apolipoprotein E |
| AR-C17 | 5-heptadecylresorcinol |
| ATP | adenosine triphosphate |
| BBB | blood–brain barrier |
| BDH1 | β-hydroxybutyrate dehydrogenase 1 |
| BP | blood pressure |
| CALCA | calcitonin gene-related peptide |
| CAD | coronary artery disease |
| CCR2+ monocytes | C-C chemokine receptor type 2-positive monocytes |
| cGMP | cyclic guanosine monophosphate |
| CKD | chronic kidney disease |
| CoA | coenzyme A |
| CPT-1 | carnitine palmitoyltranferase-1 |
| CVDs | cardiovascular diseases |
| DCM | dilated cardiomyopathy |
| DM | diabetes mellitus |
| DMF | dimethyl fumarate |
| DRAM1 | DNA damage-regulated autophagy modulator 2 |
| DRP1 | dynamin-related protein 1 |
| ECAR | extracellular acidification rate |
| ECM | extracellular matrix |
| ENO1 | alpha-enolase 1 |
| eNOS | endothelial nitric oxide synthase |
| EC | endothelial cell |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| FA | fatty acid |
| FAD | flavin adenine dinucleotide |
| FADH2 | flavin adenine dinucleotide hydrogenated |
| FAS | fatty acid synthase |
| FPER-1 | perhexiline |
| FtMt | mitochondrial ferritin |
| GLUT1 | glucose transporter 1 |
| Glc-1,6-BP | glucose-1,6 biphosphate |
| H3K27me3 | histone H3 lysine 27 trimethylation |
| HBP | hexosamine biosynthetic pathway |
| HDAC4 | histone deacetylase 4 |
| HDL | high-density lipoprotein |
| 12(S)-HETE | 12(S)-hydroxyicosa-5,8,10,14-tetraenoic acid |
| HF | heart failure |
| HFpEF | HF with preserved ejection fraction |
| HIF1α | hypoxia-inducible factor-1α |
| HIF2α | hypoxia-inducible factor 2α |
| IDH | isocitrate dehydrogenase |
| IL-6 | interleukin 6 |
| IL-10 | interleukin 10 |
| I/R | ischemia/reperfusion |
| IS | ischemic stroke |
| KLF7 | Krüppel-like factor 7 |
| LCACs | long-chain acylcarnitines |
| LDL | low-density lipoprotein |
| LDLR | low-density lipoprotein receptor |
| LPA | lysophosphatidic acid |
| LV | left ventricle |
| Mfn1 | mitofusin 1 |
| Mfn2 | mitofusin 2 |
| MI | myocardial infarction |
| miRNA | micro ribonucleic acid |
| MitoQ | mitoquinone mesylate |
| mPTP | mitochondrial permeability transition pore |
| mTOR | mechanistic target of rapamycin |
| mtROS | mitochondrial reactive oxygen species |
| NAD | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NASH | non-alcoholic steatohepatitis |
| NCOR1 | nuclear receptor corepressor 1 |
| NLRP3 | NLR family pyrin domain-containing 3 |
| NO | nitric oxide |
| NOX | NADPH oxidase |
| NPM1 | nucleophosmin 1 |
| Nppa | natriuretic peptide A (gene) |
| NPRA | type A natriuretic peptide receptor |
| OCR | oxygen consumption rate |
| OGD/R | oxygen–glucose deprivation and reperfusion |
| OPA1 | optic atrophy protein 1 |
| oxLDL | oxidized LDL |
| OXPHOS | oxidative phosphorylation |
| PASMCs | pulmonary artery smooth muscle cells |
| PAECs | pulmonary artery endothelial cells |
| PDH | pyruvate dehydrogenase |
| PECs | pulmonary endothelial cells |
| PH | pulmonary hypertension |
| PI3K/AKT | phosphatidylinositol 3-kinase/protein kinase B |
| PINK1 | PTEN-induced kinase 1 |
| PKG | cyclic GMP–dependent protein kinase |
| PKM2 | pyruvate kinase M2 |
| PKN1 | protein kinase N1 |
| PPP | pentose phosphate pathway |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | reactive oxygen species |
| RRAEC | rat renal artery endothelial cells |
| SCFA | short-chain fatty acid |
| sFLT-1 | soluble fms-like tyrosine kinase-1 |
| SGK | serum/glucocorticoid-regulated kinase |
| SGK1 | SGK isoform 1 |
| SGLT2i | sodium–glucose cotransporter 2-inhibitors |
| sMito | super mitochondria |
| SDH | succinate dehydrogenase |
| SOD2 | superoxide dismutase 2 |
| SOX17 | SRY-related HMG-box 17 |
| SS-HTN | salt-sensitive hypertension |
| T2D | type 2 diabetes mellitus |
| TCA | tricarboxylic acid cycle |
| TGRL | triglyceride-rich lipoprotein |
| TNF | tumor necrosis factor |
| TRPV1 | transient receptor potential cation channel subfamily V member 1 |
| VSMC | vascular smooth muscle cell |
References
- Chandel, N.S. Glycolysis. Cold Spring Harb. Perspect. Biol. 2021, 13, 535. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef]
- Hardie, D.G. 100 years of the Warburg effect: A historical perspective. Endocr. Relat. Cancer 2022, 29, T1–T13. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Wisidagama, D.R.; Setoguchi, K.; Hong, J.S.; Van Horn, C.M.; Imam, S.S.; Vergnes, L.; Malone, C.S.; Koehler, C.M.; et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012, 7, 1068–1085. [Google Scholar] [CrossRef] [PubMed]
- Yoo, I.; Ahn, I.; Lee, J.; Lee, N. Extracellular flux assay (Seahorse assay): Diverse applications in metabolic research across biological disciplines. Mol. Cells 2024, 47, 100095. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Bohm, A.; Keuper, M.; Meile, T.; Zdichavsky, M.; Fritsche, A.; Haring, H.U.; de Angelis, M.H.; Staiger, H.; Franko, A. Increased mitochondrial respiration of adipocytes from metabolically unhealthy obese compared to healthy obese individuals. Sci. Rep. 2020, 10, 12407. [Google Scholar] [CrossRef]
- Baig, J.; Pradeepkiran, J.A.; Reddy, P.H. Methods to Study Mitochondria: Techniques Used to Study the Effects of Age-Related Diseases Including Alzheimer’s. Curr. Protoc. 2023, 3, e631. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Saddik, M.; Lopaschuk, G.D. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J. Biol. Chem. 1991, 266, 8162–8170. [Google Scholar] [CrossRef] [PubMed]
- Wisneski, J.A.; Stanley, W.C.; Neese, R.A.; Gertz, E.W. Effects of acute hyperglycemia on myocardial glycolytic activity in humans. J. Clin. Investig. 1990, 85, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., 3rd; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure with Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. J. Am. Heart Assoc. 2016, 5, 8. [Google Scholar] [CrossRef]
- Lanfear, D.E.; Gibbs, J.J.; Li, J.; She, R.; Petucci, C.; Culver, J.A.; Tang, W.H.W.; Pinto, Y.M.; Williams, L.K.; Sabbah, H.N.; et al. Targeted Metabolomic Profiling of Plasma and Survival in Heart Failure Patients. JACC Heart Fail. 2017, 5, 823–832. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Kruger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.R.F. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [Google Scholar] [CrossRef]
- Zhou, B.; Perel, P.; Mensah, G.A.; Ezzati, M. Global epidemiology, health burden and effective interventions for elevated blood pressure and hypertension. Nat. Rev. Cardiol. 2021, 18, 785–802. [Google Scholar] [CrossRef]
- Maaliki, D.; Itani, M.M.; Itani, H.A. Pathophysiology and genetics of salt-sensitive hypertension. Front. Physiol. 2022, 13, 1001434. [Google Scholar] [CrossRef]
- Domondon, M.; Polina, I.; Nikiforova, A.B.; Sultanova, R.F.; Kruger, C.; Vasileva, V.Y.; Fomin, M.V.; Beeson, G.C.; Nieminen, A.L.; Smythe, N.; et al. Renal Glomerular Mitochondria Function in Salt-Sensitive Hypertension. Front. Physiol. 2019, 10, 1588. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Ruperez, M.; Lorenzo, O.; Esteban, V.; Blanco, J.; Mezzano, S.; Egido, J. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int. Suppl. 2002, 82, S12–S22. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Pratico, D.; Ren, J. Angiotensin II: Role in oxidative stress, endothelial dysfunction, and diseases. Mol. Cell Endocrinol. 2024, 592, 112309. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, Y.; Li, W.; Han, C.; Qi, Z. MicroRNA and mRNA analysis of angiotensin II-induced renal artery endothelial cell dysfunction. Exp. Ther. Med. 2020, 19, 3723–3737. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Gallo, G.; Rubattu, S. Endocrine functions of the heart: From bench to bedside. Eur. Heart J. 2023, 44, 643–655. [Google Scholar] [CrossRef]
- Moro, C. Natriuretic peptides and fat metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 645–649. [Google Scholar] [CrossRef]
- Engeli, S.; Birkenfeld, A.L.; Badin, P.M.; Bourlier, V.; Louche, K.; Viguerie, N.; Thalamas, C.; Montastier, E.; Larrouy, D.; Harant, I.; et al. Natriuretic peptides enhance the oxidative capacity of human skeletal muscle. J. Clin. Investig. 2012, 122, 4675–4679. [Google Scholar] [CrossRef]
- Miyashita, K.; Itoh, H.; Tsujimoto, H.; Tamura, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Natriuretic peptides/cGMP/cGMP-dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes 2009, 58, 2880–2892. [Google Scholar] [CrossRef]
- Moriyama, T.; Kanmura, Y.; Lindahl, S.G. Atrial natriuretic peptide attenuation of renal ischemia-reperfusion injury after major surgery. J. Surg. Res. 2016, 201, 213–218. [Google Scholar] [CrossRef]
- Cherezova, A.; Sudarikova, A.; Vasileva, V.; Iurchenko, R.; Nikiforova, A.; Spires, D.R.; Zamaro, A.S.; Jones, A.C.; Schibalski, R.S.; Dong, Z.; et al. The effects of the atrial natriuretic peptide deficiency on renal cortical mitochondrial bioenergetics in the Dahl SS rat. FASEB J. 2024, 38, e23891. [Google Scholar] [CrossRef]
- Rubattu, S.; Evangelista, A.; Barbato, D.; Barba, G.; Stanzione, R.; Iacone, R.; Volpe, M.; Strazzullo, P. Atrial natriuretic peptide (ANP) gene promoter variant and increased susceptibility to early development of hypertension in humans. J. Hum. Hypertens. 2007, 21, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Kassi, E.; Pervanidou, P.; Kaltsas, G.; Chrousos, G. Metabolic syndrome: Definitions and controversies. BMC Med. 2011, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Boicean, A.; Ichim, C.; Sasu, S.M.; Todor, S.B. Key Insights into Gut Alterations in Metabolic Syndrome. J. Clin. Med. 2025, 14, 2678. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Wu, Q.; Yao, Q.; Jiang, K.; Yu, J.; Tang, Q. Short-chain fatty acid metabolism and multiple effects on cardiovascular diseases. Ageing Res. Rev. 2022, 81, 101706. [Google Scholar] [CrossRef]
- Mishra, A.; Betancourt, A.; Vidyadharan, V.A.; Blesson, C.S.; Belfort, M.; Yallampalli, C.; Chauhan, M. Calcitonin gene-related peptide protects from soluble fms-like tyrosine kinase-1-induced vascular dysfunction in a preeclampsia mouse model. Front. Physiol. 2023, 14, 1221684. [Google Scholar] [CrossRef]
- Runo, J.R.; Loyd, J.E. Primary pulmonary hypertension. Lancet 2003, 361, 1533–1544. [Google Scholar] [CrossRef]
- Hilton, L.R.; Ratsep, M.T.; VandenBroek, M.M.; Jafri, S.; Laverty, K.J.; Mitchell, M.; Theilmann, A.L.; Smart, J.A.; Hawke, L.G.; Moore, S.D.; et al. Impaired Interleukin-15 Signaling via BMPR2 Loss Drives Natural Killer Cell Deficiency and Pulmonary Hypertension. Hypertension 2022, 79, 2493–2504. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Wang, H.F.; Lian, T.Y.; Zhou, Y.P.; Xu, X.Q.; Guo, F.; Wei, Y.P.; Li, J.Y.; Sun, K.; Liu, C.; et al. Human Plasma IgG N-Glycome Profiles Reveal a Proinflammatory Phenotype in Chronic Thromboembolic Pulmonary Hypertension. Hypertension 2023, 80, 1929–1939. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef]
- Christou, H.; Khalil, R.A. Mechanisms of pulmonary vascular dysfunction in pulmonary hypertension and implications for novel therapies. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H702–H724. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Bonnet, S.; Haromy, A.; McMurtry, M.S.; Bleackley, R.C.; Michelakis, E.D. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFalpha contributes to the pathogenesis of pulmonary arterial hypertension. J. Mol. Med. 2011, 89, 771–783. [Google Scholar] [CrossRef]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Unger, S.A.; Horowitz, J.D. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem. Pharmacol. 1996, 52, 273–280. [Google Scholar] [CrossRef]
- Griffiths, K.; Grand, R.J.; Horan, I.; Certo, M.; Keeler, R.C.; Mauro, C.; Tseng, C.C.; Greig, I.; Morrell, N.W.; Zanda, M.; et al. Fluorinated perhexiline derivative attenuates vascular proliferation in pulmonary arterial hypertension smooth muscle cells. Vasc. Pharmacol. 2024, 156, 107399. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, X.; He, S.; Zhang, L.; Bai, J.; Qu, L.; Qi, J.; Zheng, X.; Zhu, X.; Mei, J.; et al. Ubiquitinated AIF is a major mediator of hypoxia-induced mitochondrial dysfunction and pulmonary artery smooth muscle cell proliferation. Cell Biosci. 2022, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Chen, J.; Zhao, Y.; Niu, Y.; Lin, S.; Chen, S.; Zong, Y.; Sun, X.; Xie, L.; Xiao, T. The Emerging Role of Fatty Acid Synthase in Hypoxia-Induced Pulmonary Hypertensive Mouse Energy Metabolism. Oxid. Med. Cell Longev. 2021, 2021, 9990794. [Google Scholar] [CrossRef] [PubMed]
- Yegambaram, M.; Sun, X.; Lu, Q.; Jin, Y.; Ornatowski, W.; Soto, J.; Aggarwal, S.; Wang, T.; Tieu, K.; Gu, H.; et al. Mitochondrial hyperfusion induces metabolic remodeling in lung endothelial cells by modifying the activities of electron transport chain complexes I and III. Free Radic. Biol. Med. 2024, 210, 183–194. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, J.; Zhang, R.; Zhang, M.; Cui, H.; Wang, L.; Cui, Y.; Wang, W.; Sun, Y.; Wang, C. Targeting Endothelial ENO1 (Alpha-Enolase) -PI3K-Akt-mTOR Axis Alleviates Hypoxic Pulmonary Hypertension. Hypertension 2023, 80, 1035–1047. [Google Scholar] [CrossRef]
- Sangam, S.; Sun, X.; Schwantes-An, T.H.; Yegambaram, M.; Lu, Q.; Shi, Y.; Cook, T.; Fisher, A.; Frump, A.L.; Coleman, A.; et al. SOX17 Deficiency Mediates Pulmonary Hypertension: At the Crossroads of Sex, Metabolism, and Genetics. Am. J. Respir. Crit. Care Med. 2023, 207, 1055–1069. [Google Scholar] [CrossRef]
- Pascual, F.; Coleman, R.A. Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim. Biophys. Acta 2016, 1861, 1425–1433. [Google Scholar] [CrossRef]
- van Bilsen, M.; van Nieuwenhoven, F.A.; van der Vusse, G.J. Metabolic remodelling of the failing heart: Beneficial or detrimental? Cardiovasc. Res. 2009, 81, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Scheubel, R.J.; Tostlebe, M.; Simm, A.; Rohrbach, S.; Prondzinsky, R.; Gellerich, F.N.; Silber, R.E.; Holtz, J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol. 2002, 40, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Linke, A.; Chandler, M.P.; Young, M.E.; Penn, M.S.; Gupte, S.; d’Agostino, C.; Hintze, T.H.; Stanley, W.C.; Recchia, F.A. Carnitine palmitoyl transferase-I inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovasc. Res. 2005, 66, 454–461. [Google Scholar] [CrossRef]
- Li, X.; Wu, F.; Gunther, S.; Looso, M.; Kuenne, C.; Zhang, T.; Wiesnet, M.; Klatt, S.; Zukunft, S.; Fleming, I.; et al. Inhibition of fatty acid oxidation enables heart regeneration in adult mice. Nature 2023, 622, 619–626. [Google Scholar] [CrossRef]
- Lionetti, V.; Matteucci, M.; Ribezzo, M.; Di Silvestre, D.; Brambilla, F.; Agostini, S.; Mauri, P.; Padeletti, L.; Pingitore, A.; Delsedime, L.; et al. Regional mapping of myocardial hibernation phenotype in idiopathic end-stage dilated cardiomyopathy. J. Cell Mol. Med. 2014, 18, 396–414. [Google Scholar] [CrossRef]
- He, X.; Zeng, H.; Chen, J.X. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J. Cell Physiol. 2019, 234, 2252–2265. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, J.X. Sirtuin 3, Endothelial Metabolic Reprogramming, and Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Pharmacol. 2019, 74, 315–323. [Google Scholar] [CrossRef]
- Song, Y.; Leem, J.; Dhanani, M.; McKirnan, M.D.; Ichikawa, Y.; Braza, J.; Harrington, E.O.; Hammond, H.K.; Roth, D.M.; Patel, H.H. Impact of blood factors on endothelial cell metabolism and function in two diverse heart failure models. PLoS ONE 2023, 18, e0281550. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Cerasuolo, F.A.; Avvisato, R.; Buonaiuto, A.; Maisto, M.; Bianco, A.; D’Argenio, V.; Mone, P.; Perrino, C.; D’Apice, S.; et al. L-Arginine supplementation as mitochondrial therapy in diabetic cardiomyopathy. Cardiovasc. Diabetol. 2024, 23, 450. [Google Scholar] [CrossRef]
- Feng, C.; Jiang, H.; Yang, X.; Cong, H.; Li, L.; Feng, J. GLUT1 Mediates the Metabolic Reprogramming and Inflammation of CCR2+ Monocytes/Macrophages from Patients with DCM. Front. Biosci. 2023, 28, 223. [Google Scholar] [CrossRef]
- Major, J.L.; Dewan, A.; Salih, M.; Leddy, J.J.; Tuana, B.S. E2F6 Impairs Glycolysis and Activates BDH1 Expression Prior to Dilated Cardiomyopathy. PLoS ONE 2017, 12, e0170066. [Google Scholar] [CrossRef]
- Gambardella, J.; Jankauskas, S.S.; Kansakar, U.; Varzideh, F.; Avvisato, R.; Prevete, N.; Sidoli, S.; Mone, P.; Wang, X.; Lombardi, A.; et al. Ketone Bodies Rescue Mitochondrial Dysfunction Via Epigenetic Remodeling. JACC Basic Transl. Sci. 2023, 8, 1123–1137. [Google Scholar] [CrossRef] [PubMed]
- Kansakar, U.; Nieves Garcia, C.; Santulli, G.; Gambardella, J.; Mone, P.; Jankauskas, S.S.; Lombardi, A. Exogenous Ketones in Cardiovascular Disease and Diabetes: From Bench to Bedside. J. Clin. Med. 2024, 13, 7391. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Kansakar, U.; Varzideh, F.; Wilson, S.; Mone, P.; Lombardi, A.; Gambardella, J.; Santulli, G. Heart failure in diabetes. Metabolism 2021, 125, 154910. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Bhattarai, S.; Ara, H.; Sun, G.; St Clair, D.K.; Bhuiyan, M.S.; Kevil, C.; Watts, M.N.; Dominic, P.; Shimizu, T.; et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. 2020, 37, 101740. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, L.; Liu, H.; Wang, J.; Li, J.; Yang, Y.; Wang, Y.; Cai, H.; Li, R.; Zhao, Y. Salsolinol Attenuates Doxorubicin-Induced Chronic Heart Failure in Rats and Improves Mitochondrial Function in H9c2 Cardiomyocytes. Front. Pharmacol. 2019, 10, 1135. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, L.; Wang, J.; Wang, J.; Wang, L.; Wang, R.; Li, R.; Liu, H.; Wei, S.; Li, H.; et al. Therapeutic effects of higenamine combined with [6]-gingerol on chronic heart failure induced by doxorubicin via ameliorating mitochondrial function. J. Cell Mol. Med. 2020, 24, 4036–4050. [Google Scholar] [CrossRef]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef]
- Bhandari, S. Impact of intravenous iron on cardiac and skeletal oxidative stress and cardiac mitochondrial function in experimental uraemia chronic kidney disease. Front. Biosci. 2021, 26, 442–464. [Google Scholar] [CrossRef]
- Santulli, G.; Varzideh, F.; Forzano, I.; Wilson, S.; Salemme, L.; de Donato, A.; Lombardi, A.; Rainone, A.; Nunziata, L.; Jankauskas, S.S.; et al. Functional and Clinical Importance of SGLT2-inhibitors in Frailty: From the Kidney to the Heart. Hypertension 2023, 80, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Butt, J.H.; Dewan, P.; Merkely, B.; Belohlavek, J.; Drozdz, J.; Kitakaze, M.; Inzucchi, S.E.; Kosiborod, M.N.; Martinez, F.A.; Tereshchenko, S.; et al. Efficacy and Safety of Dapagliflozin According to Frailty in Heart Failure With Reduced Ejection Fraction: A Post Hoc Analysis of the DAPA-HF Trial. Ann. Intern. Med. 2022, 175, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Varzideh, F.; Kansakar, U.; Santulli, G. SGLT2 inhibitors in cardiovascular medicine. Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, e67–e68. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Ren. Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Requena-Ibanez, J.A.; Santos-Gallego, C.G.; Rodriguez-Cordero, A.; Vargas-Delgado, A.P.; Mancini, D.; Sartori, S.; Atallah-Lajam, F.; Giannarelli, C.; Macaluso, F.; Lala, A.; et al. Mechanistic Insights of Empagliflozin in Nondiabetic Patients with HFrEF: From the EMPA-TROPISM Study. JACC Heart Fail. 2021, 9, 578–589. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Khan, M.S.; Marx, N.; Lam, C.S.P.; Schnaidt, S.; Ofstad, A.P.; Brueckmann, M.; Jamal, W.; et al. Effect of Empagliflozin on Cardiovascular and Renal Outcomes in Patients with Heart Failure by Baseline Diabetes Status: Results from the EMPEROR-Reduced Trial. Circulation 2021, 143, 337–349. [Google Scholar] [CrossRef]
- Mone, P.; Varzideh, F.; Jankauskas, S.S.; Pansini, A.; Lombardi, A.; Frullone, S.; Santulli, G. SGLT2 Inhibition via Empagliflozin Improves Endothelial Function and Reduces Mitochondrial Oxidative Stress: Insights from Frail Hypertensive and Diabetic Patients. Hypertension 2022, 79, 1633–1643. [Google Scholar] [CrossRef]
- Kalyani, R.R. Glucose-Lowering Drugs to Reduce Cardiovascular Risk in Type 2 Diabetes. N. Engl. J. Med. 2021, 384, 1248–1260. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Oppi, S.; Nusser-Stein, S.; Blyszczuk, P.; Wang, X.; Jomard, A.; Marzolla, V.; Yang, K.; Velagapudi, S.; Ward, L.J.; Yuan, X.M.; et al. Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARgamma signature. Eur. Heart J. 2020, 41, 995–1005. [Google Scholar] [CrossRef]
- Robinson, N.; Ganesan, R.; Hegedus, C.; Kovacs, K.; Kufer, T.A.; Virag, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Greenhalgh, R.M.; Davies, A.H. The symptomatic carotid plaque. Stroke 2000, 31, 774–781. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martin, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, N.; Tang, T.; Nie, S.; Zha, L.; Zhang, M.; Lv, B.; Lu, Y.; Jiao, J.; Li, J.; et al. Cholesterol suppresses human iTreg differentiation and nTreg function through mitochondria-related mechanisms. J. Transl. Med. 2023, 21, 224. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Li, Y.; Kuo, Y.M.; Andrews, A.J.; Nanayakkara, G.; Johnson, C.; Fu, H.; Shan, H.; Du, F.; et al. Mitochondrial Reactive Oxygen Species Mediate Lysophosphatidylcholine-Induced Endothelial Cell Activation. Arter. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1090–1100. [Google Scholar] [CrossRef]
- Kratzer, A.; Giral, H.; Landmesser, U. High-density lipoproteins as modulators of endothelial cell functions: Alterations in patients with coronary artery disease. Cardiovasc. Res. 2014, 103, 350–361. [Google Scholar] [CrossRef]
- Skalen, K.; Gustafsson, M.; Rydberg, E.K.; Hulten, L.M.; Wiklund, O.; Innerarity, T.L.; Boren, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef]
- Goyal, T.; Mitra, S.; Khaidakov, M.; Wang, X.; Singla, S.; Ding, Z.; Liu, S.; Mehta, J.L. Current Concepts of the Role of Oxidized LDL Receptors in Atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 150–159. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Treps, L.; Bodrug, N.; Carmeliet, P. Endothelial cell metabolism: A novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis 2016, 253, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Braczko, A.; Kutryb-Zajac, B.; Jedrzejewska, A.; Krol, O.; Mierzejewska, P.; Zabielska-Kaczorowska, M.; Slominska, E.M.; Smolenski, R.T. Cardiac Mitochondria Dysfunction in Dyslipidemic Mice. Int. J. Mol. Sci. 2022, 23, 1488. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Chen, Y.F.; Chan, H.C.; Chung, C.H.; Peng, H.Y.; Ho, Y.C.; Chen, C.H.; Chang, K.C.; Tang, C.H.; Lee, A.S. Role of apolipoprotein E in electronegative low-density lipoprotein-induced mitochondrial dysfunction in cardiomyocytes. Metabolism 2020, 107, 154227. [Google Scholar] [CrossRef]
- Olkowicz, M.; Karas, A.; Berkowicz, P.; Kaczara, P.; Jasztal, A.; Kurylowicz, Z.; Fedak, F.; Rosales-Solano, H.; Roy, K.S.; Kij, A.; et al. Upregulation of ALOX12-12-HETE pathway impairs AMPK-dependent modulation of vascular metabolism in ApoE/LDLR−/− mice. Pharmacol. Res. 2024, 210, 107478. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, Z.; Li, Q.; Wang, Z.; Liu, J.; Wang, J. 5-Heptadecylresorcinol Protects against Atherosclerosis in Apolipoprotein E-Deficient Mice by Modulating SIRT3 Signaling: The Possible Beneficial Effects of Whole Grain Consumption. Mol. Nutr. Food Res. 2022, 66, e2101114. [Google Scholar] [CrossRef]
- Chen, F.; Li, J.; Zheng, T.; Chen, T.; Yuan, Z. KLF7 Alleviates Atherosclerotic Lesions and Inhibits Glucose Metabolic Reprogramming in Macrophages by Regulating HDAC4/miR-148b-3p/NCOR1. Gerontology 2022, 68, 1291–1310. [Google Scholar] [CrossRef]
- Nyunt, T.; Britton, M.; Wanichthanarak, K.; Budamagunta, M.; Voss, J.C.; Wilson, D.W.; Rutledge, J.C.; Aung, H.H. Mitochondrial oxidative stress-induced transcript variants of ATF3 mediate lipotoxic brain microvascular injury. Free Radic. Biol. Med. 2019, 143, 25–46. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Chen, X.; Wang, Z.; Wang, X.; Zhou, Q.; Fang, W.; Zheng, C. Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol. Cell Endocrinol. 2022, 545, 111560. [Google Scholar] [CrossRef]
- Karakasis, P.; Theofilis, P.; Patoulias, D.; Vlachakis, P.K.; Antoniadis, A.P.; Fragakis, N. Diabetes-Driven Atherosclerosis: Updated Mechanistic Insights and Novel Therapeutic Strategies. Int. J. Mol. Sci. 2025, 26, 2196. [Google Scholar] [CrossRef]
- Jha, J.C.; Ho, F.; Dan, C.; Jandeleit-Dahm, K. A causal link between oxidative stress and inflammation in cardiovascular and renal complications of diabetes. Clin. Sci. 2018, 132, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Barlovic, D.P.; Soro-Paavonen, A.; Jandeleit-Dahm, K.A. RAGE biology, atherosclerosis and diabetes. Clin. Sci. 2011, 121, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Todor, S.B.; Ichim, C.; Boicean, A.; Mihaila, R.G. Cardiovascular Risk in Philadelphia-Negative Myeloproliferative Neoplasms: Mechanisms and Implications—A Narrative Review. Curr. Issues Mol. Biol. 2024, 46, 8407–8423. [Google Scholar] [CrossRef]
- Martin, S.S.; Aday, A.W.; Allen, N.B.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Bansal, N.; Beaton, A.Z.; et al. 2025 Heart Disease and Stroke Statistics: A Report of US and Global Data from the American Heart Association. Circulation 2025, 151, e41–e660. [Google Scholar] [CrossRef]
- Andreadou, I.; Daiber, A.; Baxter, G.F.; Brizzi, M.F.; Di Lisa, F.; Kaludercic, N.; Lazou, A.; Varga, Z.V.; Zuurbier, C.J.; Schulz, R.; et al. Influence of cardiometabolic comorbidities on myocardial function, infarction, and cardioprotection: Role of cardiac redox signaling. Free Radic. Biol. Med. 2021, 166, 33–52. [Google Scholar] [CrossRef]
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation 2017, 136, 6–19. [Google Scholar] [CrossRef]
- Koncsos, G.; Varga, Z.V.; Baranyai, T.; Boengler, K.; Rohrbach, S.; Li, L.; Schluter, K.D.; Schreckenberg, R.; Radovits, T.; Olah, A.; et al. Diastolic dysfunction in prediabetic male rats: Role of mitochondrial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H927–H943. [Google Scholar] [CrossRef]
- Pagliaro, P.; Moro, F.; Tullio, F.; Perrelli, M.G.; Penna, C. Cardioprotective pathways during reperfusion: Focus on redox signaling and other modalities of cell signaling. Antioxid. Redox Signal. 2011, 14, 833–850. [Google Scholar] [CrossRef]
- Gao, X.M.; Su, Y.; Moore, S.; Han, L.P.; Kiriazis, H.; Lu, Q.; Zhao, W.B.; Ruze, A.; Fang, B.B.; Duan, M.J.; et al. Relaxin mitigates microvascular damage and inflammation following cardiac ischemia-reperfusion. Basic Res. Cardiol. 2019, 114, 30. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toan, S.; Zhou, H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis 2020, 23, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, S.; Kikuchi, Y.; Nakajima, M.; Kimura, H.; Tsuyama, S.; Uemura, K.; Yoshida, K. Endothelial NO Synthase (eNOS) phosphorylation regulates coronary diameter during ischemia-reperfusion in association with oxidative stress. Free Radic. Res. 2005, 39, 481–489. [Google Scholar] [CrossRef]
- Mouton, A.J.; Ma, Y.; Rivera Gonzalez, O.J.; Daseke, M.J., 2nd; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef]
- Su, H.X.; Li, P.B.; Shi, K.N.; Gao, J.; Zhang, H.J.; Li, H.H. The immunoproteasome subunit beta2i ameliorates myocardial ischemia/reperfusion injury by regulating Parkin-Mfn1/2-mediated mitochondrial fusion. Cell Mol. Life Sci. 2023, 80, 231. [Google Scholar] [CrossRef]
- Zhou, H.; Ren, J.; Toan, S.; Mui, D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res. Rev. 2021, 66, 101250. [Google Scholar] [CrossRef]
- Mohsin, A.A.; Chen, Q.; Quan, N.; Rousselle, T.; Maceyka, M.W.; Samidurai, A.; Thompson, J.; Hu, Y.; Li, J.; Lesnefsky, E.J. Mitochondrial Complex I Inhibition by Metformin Limits Reperfusion Injury. J. Pharmacol. Exp. Ther. 2019, 369, 282–290. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Zoungrana, L.I.; James, A.; Slotabec, L.; Didik, S.; Fatmi, M.K.; Krause-Hauch, M.; Lesnefsky, E.J.; Li, J. Administration of metformin rescues age-related vulnerability to ischemic insults through mitochondrial energy metabolism. Biochem. Biophys. Res. Commun. 2023, 659, 46–53. [Google Scholar] [CrossRef]
- Kong, Y.; Zhang, Q.; Wang, S.; Li, R.; Fu, C.; Wei, Q. Mitochondrial metabolism regulated macrophage phenotype in myocardial infarction. Biomed. Pharmacother. 2024, 180, 117494. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Li, J.; Lou, Y.; Chen, Q.; Yang, Y.; Zhang, R.; Liu, Z.; He, L.; Cheng, Y. Reprogramming macrophage metabolism following myocardial infarction: A neglected piece of a therapeutic opportunity. Int. Immunopharmacol. 2024, 142, 113019. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Duan, X.; Wang, B.; Zhan, Z. Targeting NPM1 Epigenetically Promotes Postinfarction Cardiac Repair by Reprogramming Reparative Macrophage Metabolism. Circulation 2024, 149, 1982–2001. [Google Scholar] [CrossRef]
- Zhao, P.; Zhou, W.; Zhang, Y.; Li, J.; Zhao, Y.; Pan, L.; Shen, Z.; Chen, W.; Hui, J. Aminooxyacetic acid attenuates post-infarct cardiac dysfunction by balancing macrophage polarization through modulating macrophage metabolism in mice. J. Cell Mol. Med. 2020, 24, 2593–2609. [Google Scholar] [CrossRef]
- Du, Y.; Gu, X.; Meng, H.; Aa, N.; Liu, S.; Peng, C.; Ge, Y.; Yang, Z. Muscone improves cardiac function in mice after myocardial infarction by alleviating cardiac macrophage-mediated chronic inflammation through inhibition of NF-kappaB and NLRP3 inflammasome. Am. J. Transl. Res. 2018, 10, 4235–4246. [Google Scholar]
- Du, Y.; Ge, Y.; Xu, Z.; Aa, N.; Gu, X.; Meng, H.; Lin, Z.; Zhu, D.; Shi, J.; Zhuang, R.; et al. Hypoxia-Inducible Factor 1 alpha (HIF-1alpha)/Vascular Endothelial Growth Factor (VEGF) Pathway Participates in Angiogenesis of Myocardial Infarction in Muscone-Treated Mice: Preliminary Study. Med. Sci. Monit. 2018, 24, 8870–8877. [Google Scholar] [CrossRef]
- Wang, X.; Meng, H.; Chen, P.; Yang, N.; Lu, X.; Wang, Z.M.; Gao, W.; Zhou, N.; Zhang, M.; Xu, Z.; et al. Beneficial effects of muscone on cardiac remodeling in a mouse model of myocardial infarction. Int. J. Mol. Med. 2014, 34, 103–111. [Google Scholar] [CrossRef]
- Gu, X.; Bao, N.; Zhang, J.; Huang, G.; Zhang, X.; Zhang, Z.; Du, Y.; Meng, H.; Liu, J.; Wu, P.; et al. Muscone ameliorates myocardial ischemia-reperfusion injury by promoting myocardial glycolysis. Heliyon 2023, 9, e22154. [Google Scholar] [CrossRef]
- Mouton, A.J.; Flynn, E.R.; Moak, S.P.; Aitken, N.M.; Omoto, A.C.M.; Li, X.; da Silva, A.A.; Wang, Z.; do Carmo, J.M.; Hall, J.E. Dimethyl fumarate preserves left ventricular infarct integrity following myocardial infarction via modulation of cardiac macrophage and fibroblast oxidative metabolism. J. Mol. Cell Cardiol. 2021, 158, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Dasgupta, C.; Mulder, C.; Zhang, L. MicroRNA-210 Controls Mitochondrial Metabolism and Protects Heart Function in Myocardial Infarction. Circulation 2022, 145, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Borden, A.; Kurian, J.; Nickoloff, E.; Yang, Y.; Troupes, C.D.; Ibetti, J.; Lucchese, A.M.; Gao, E.; Mohsin, S.; Koch, W.J.; et al. Transient Introduction of miR-294 in the Heart Promotes Cardiomyocyte Cell Cycle Reentry After Injury. Circ. Res. 2019, 125, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Trevelyan, C.J.; MacCannell, A.D.V.; Stewart, L.; Tarousa, T.; Taylor, H.A.; Murray, M.; Bageghni, S.A.; Hemmings, K.E.; Drinkhill, M.J.; Roberts, L.D.; et al. MiR-214-3p regulates Piezo1, lysyl oxidases and mitochondrial function in human cardiac fibroblasts. Matrix Biol. 2024, 132, 34–46. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Murray, V.; Berge, E.; del Zoppo, G.J. Thrombolysis for acute ischaemic stroke. Cochrane Database Syst. Rev. 2014, 2014, CD000213. [Google Scholar] [CrossRef]
- Bai, J.; Lyden, P.D. Revisiting cerebral postischemic reperfusion injury: New insights in understanding reperfusion failure, hemorrhage, and edema. Int. J. Stroke 2015, 10, 143–152. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Zhang, Y.; Xiao, Z.B.; Zhang, Y.B.; Zhang, J.; Li, Z.Q.; Zhu, Y.B. CFTR prevents neuronal apoptosis following cerebral ischemia reperfusion via regulating mitochondrial oxidative stress. J. Mol. Med. 2018, 96, 611–620. [Google Scholar] [CrossRef]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med. 2020, 146, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 447. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef]
- Wang, P.; Cui, Y.; Liu, Y.; Li, Z.; Bai, H.; Zhao, Y.; Chang, Y.Z. Mitochondrial ferritin alleviates apoptosis by enhancing mitochondrial bioenergetics and stimulating glucose metabolism in cerebral ischemia reperfusion. Redox Biol. 2022, 57, 102475. [Google Scholar] [CrossRef]
- Xue, T.; Ji, J.; Sun, Y.; Huang, X.; Cai, Z.; Yang, J.; Guo, W.; Guo, R.; Cheng, H.; Sun, X. Sphingosine-1-phosphate, a novel TREM2 ligand, promotes microglial phagocytosis to protect against ischemic brain injury. Acta Pharm. Sin. B 2022, 12, 1885–1898. [Google Scholar] [CrossRef]
- Huang, X.X.; Li, L.; Jiang, R.H.; Yu, J.B.; Sun, Y.Q.; Shan, J.; Yang, J.; Ji, J.; Cheng, S.Q.; Dong, Y.F.; et al. Lipidomic analysis identifies long-chain acylcarnitine as a target for ischemic stroke. J. Adv. Res. 2024, 61, 133–149. [Google Scholar] [CrossRef]
- Bhattarai, S.; Sharma, S.; Subedi, U.; Ara, H.; Shum, A.; Milena, M.; Bhuiyan, M.S.; Kidambi, S.; Sun, H.; Miriyala, S.; et al. The ATX-LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion. Int. J. Mol. Sci. 2022, 23, 4138. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Zhang, Y.; Xu, L.; Huang, Z.; Zou, P.; Clemons, G.A.; Li, C.; Citadin, C.T.; Zhang, Q.; Lee, R.H. The role of serum/glucocorticoid-regulated kinase 1 in brain function following cerebral ischemia. J. Cereb. Blood Flow Metab. 2024, 44, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Zur Nedden, S.; Safari, M.S.; Weber, D.; Kuenkel, L.; Garmsiri, C.; Lang, L.; Orset, C.; Freret, T.; Haelewyn, B.; Hotze, M.; et al. Protein kinase N1 deficiency results in upregulation of cerebral energy metabolism and is highly protective in in vivo and in vitro stroke models. Metabolism 2024, 161, 156039. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Ravussin, E. Caloric restriction in humans: Impact on physiological, psychological, and behavioral outcomes. Antioxid. Redox Signal. 2011, 14, 275–287. [Google Scholar] [CrossRef]
- Monsour, M.; Gorsky, A.; Nguyen, H.; Castelli, V.; Lee, J.Y.; Borlongan, C.V. Enhancing oxidative phosphorylation over glycolysis for energy production in cultured mesenchymal stem cells. Neuroreport 2022, 33, 635–640. [Google Scholar] [CrossRef]
- Gorsky, A.; Monsour, M.; Nguyen, H.; Castelli, V.; Lee, J.Y.; Borlongan, C.V. Metabolic Switching of Cultured Mesenchymal Stem Cells Creates Super Mitochondria in Rescuing Ischemic Neurons. Neuromol. Med. 2023, 25, 120–124. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Biological Activity | Suitable CVD to Treat |
|---|---|---|
| CALCA | It inhibits sFLT-1 | Preeclampsia |
| FPER-1 | It regulates PASMCs proliferation through inhibition of PI2K/Akt axis | |
| AIF | Mitochondrial oxidoreductase | Pulmonary Hypertension |
| FAS | Its inhibition improves mitochondrial function, mitochondrial respiratory capacity and increases ATP level reducing oxidative stress | Pulmonary Hypertension |
| ENO1 | Its inhibition improves endothelial and mitochondrial dysfunction in PAECs through PI3K-Akt-mTOR pathway, and it improves right ventricular function in vivo | |
| CPT-I | Its inhibition regulates mitochondrial FA oxidation and prevents LV dysfunction | Heart Failure |
| It increases mitochondrial respiration and biogenesis in vitro; it preserves cardiac diastolic function in vivo | ||
| Salsolinol | It improves mitochondrial respiratory function and energy metabolism by inhibiting the excessive activation of a specific mitochondrial calcium uniporter | |
| MitoTEMPO | It restores mitochondrial function and reduces mtROS | Atherosclerosis |
| KLF7 | It reduces atherosclerosis by limiting glucose metabolic reprogramming through HDAC4 activation | |
| NPM1 | Its inhibition improves myocardial damage and promotes cardiac remodeling post infarction | Myocardial Infarction |
| Muscone | It exerts anti-inflammatory activity, anti-apoptotic property and promotes glycolytic flux | |
| DMF | It exerts anti-inflammatory effects and improves oxidative phosphorylation | |
| FtMt | It improves mitochondrial function, protects from neuronal apoptosis and OGD/R energy stress | Stroke |
| Autotaxin-LPA | Its inhibition restores cell permeability and mitochondrial function | |
| SGK1 | Its inhibition improves mitochondrial function, basal respiration and neuronal survival | |
| PKN1 | Its inhibition exerts positive regulation of cerebral glycolytic flux and of mitochondrial respiration and it increases ATP levels. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrangelo, D.; Lopa, C.; Litterio, M.; Cotugno, M.; Rubattu, S.; Lombardi, A. Metabolic Disturbances Involved in Cardiovascular Diseases: The Role of Mitochondrial Dysfunction, Altered Bioenergetics and Oxidative Stress. Int. J. Mol. Sci. 2025, 26, 6791. https://doi.org/10.3390/ijms26146791
Pietrangelo D, Lopa C, Litterio M, Cotugno M, Rubattu S, Lombardi A. Metabolic Disturbances Involved in Cardiovascular Diseases: The Role of Mitochondrial Dysfunction, Altered Bioenergetics and Oxidative Stress. International Journal of Molecular Sciences. 2025; 26(14):6791. https://doi.org/10.3390/ijms26146791
Chicago/Turabian StylePietrangelo, Donatella, Caroline Lopa, Margherita Litterio, Maria Cotugno, Speranza Rubattu, and Angela Lombardi. 2025. "Metabolic Disturbances Involved in Cardiovascular Diseases: The Role of Mitochondrial Dysfunction, Altered Bioenergetics and Oxidative Stress" International Journal of Molecular Sciences 26, no. 14: 6791. https://doi.org/10.3390/ijms26146791
APA StylePietrangelo, D., Lopa, C., Litterio, M., Cotugno, M., Rubattu, S., & Lombardi, A. (2025). Metabolic Disturbances Involved in Cardiovascular Diseases: The Role of Mitochondrial Dysfunction, Altered Bioenergetics and Oxidative Stress. International Journal of Molecular Sciences, 26(14), 6791. https://doi.org/10.3390/ijms26146791