
IgG:FcγRIIb Signaling on Mast Cells Blocks Allergic Airway Inflammation

 , and
, and
Abstract
1. Introduction
2. Results
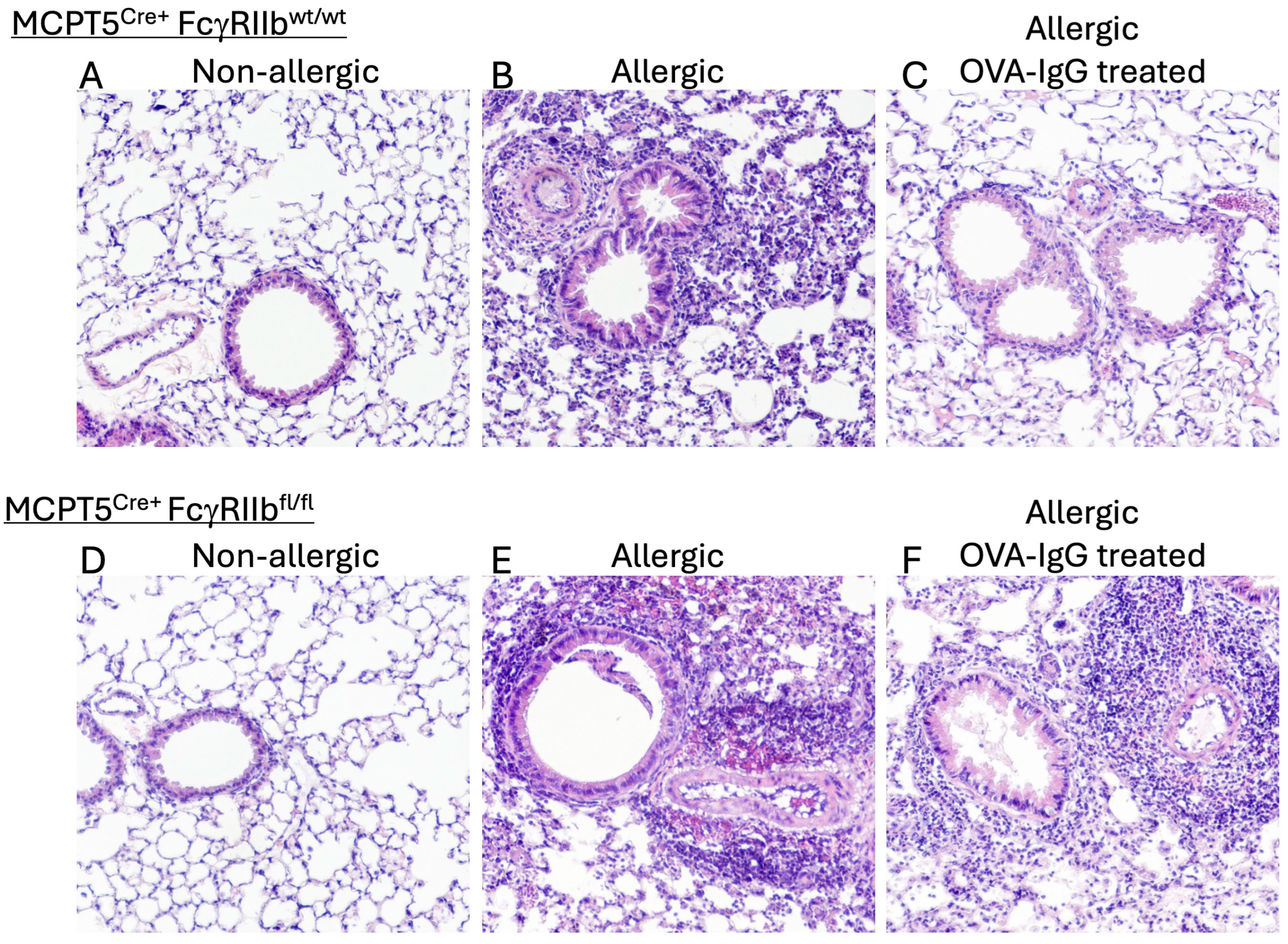
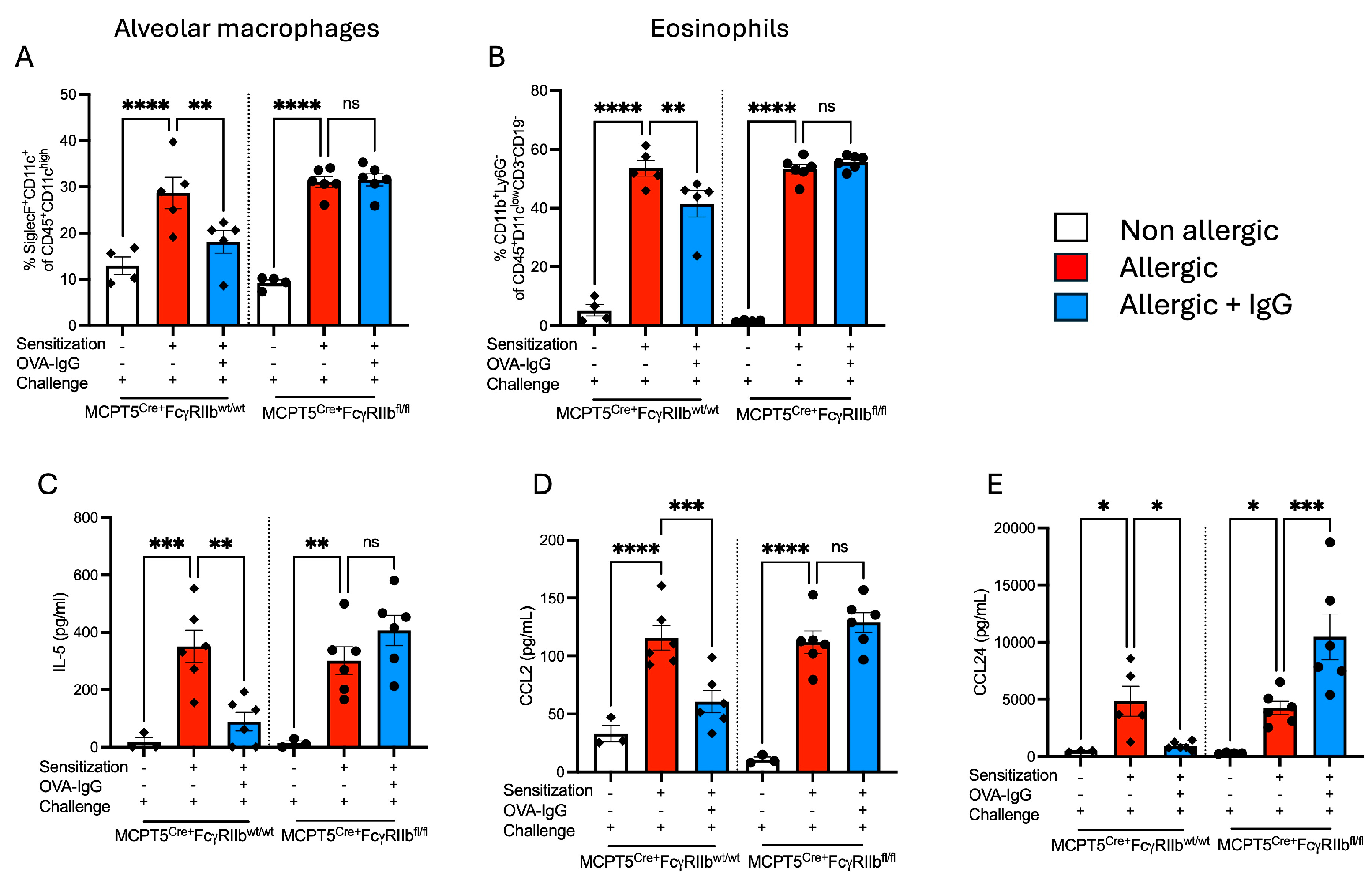
2.1. Treatment with Allergen-Specific IgG Reduces Allergen-Induced Pulmonary Macrophage and Eosinophil Infiltration
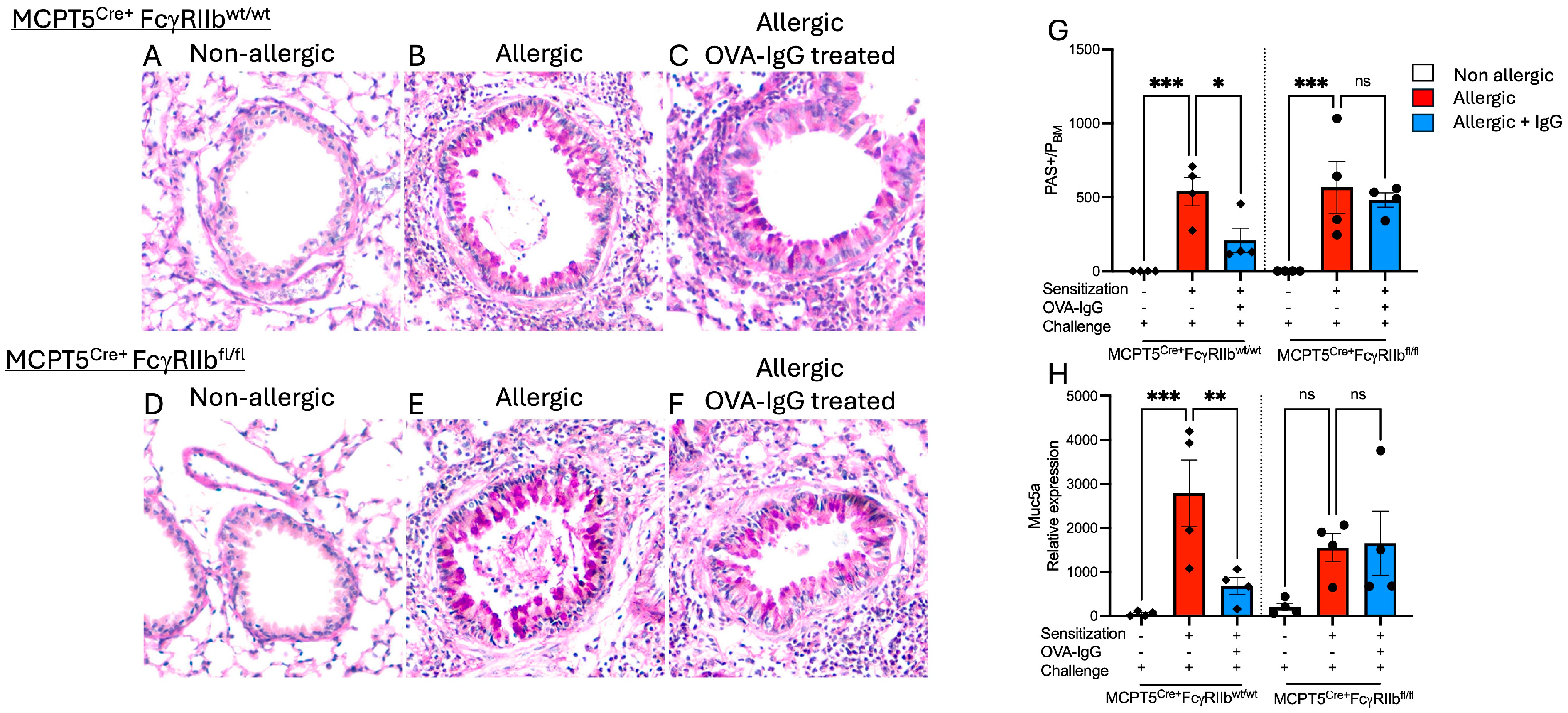
2.2. Goblet Cell Hyperplasia Induced Following Allergen Sensitization and Challenge Is Reduced by Allergen-Specific IgGF
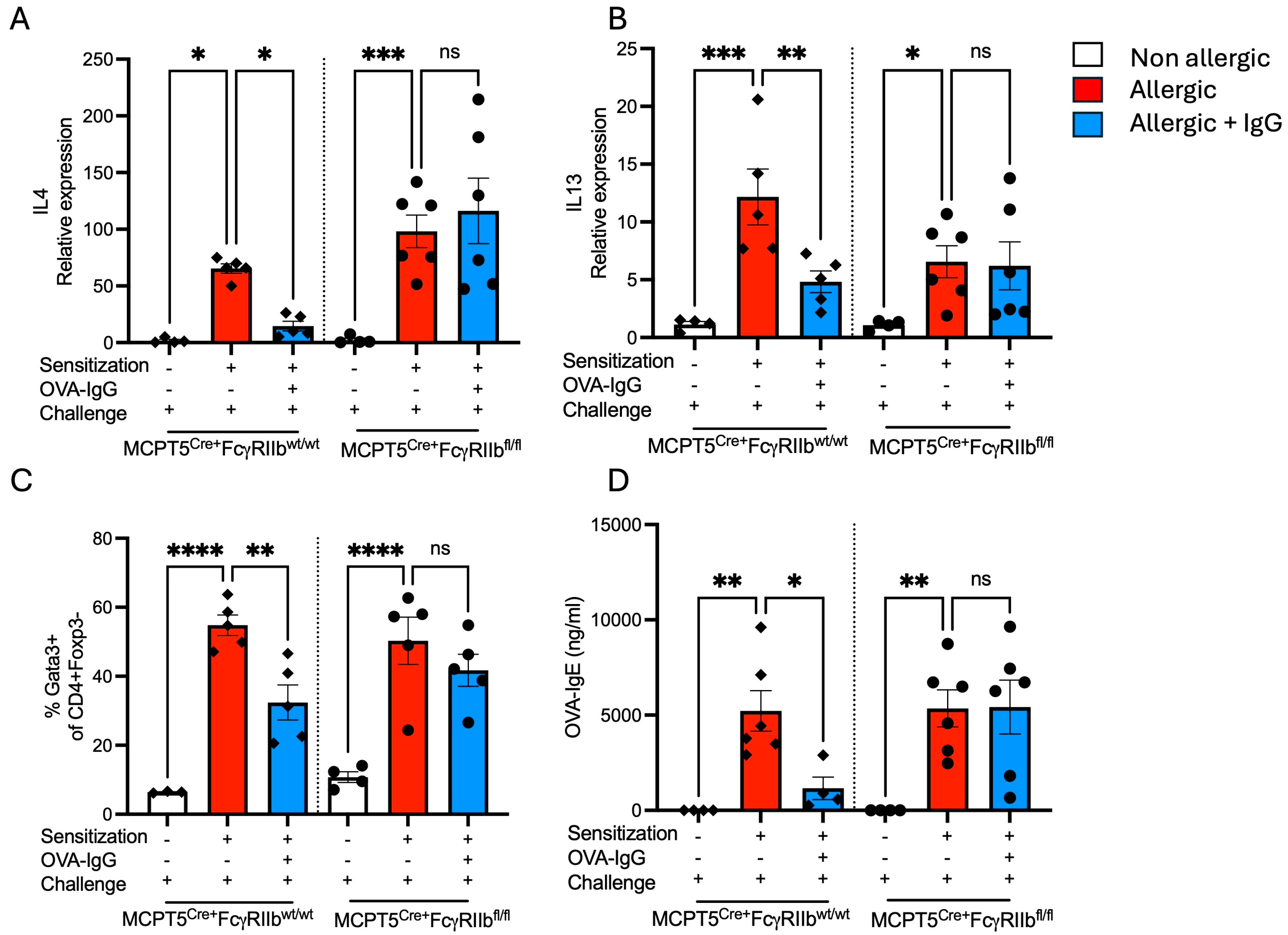
2.3. Allergen-Specific IgG Inhibits the Production of Type 2 Cytokines and IgE
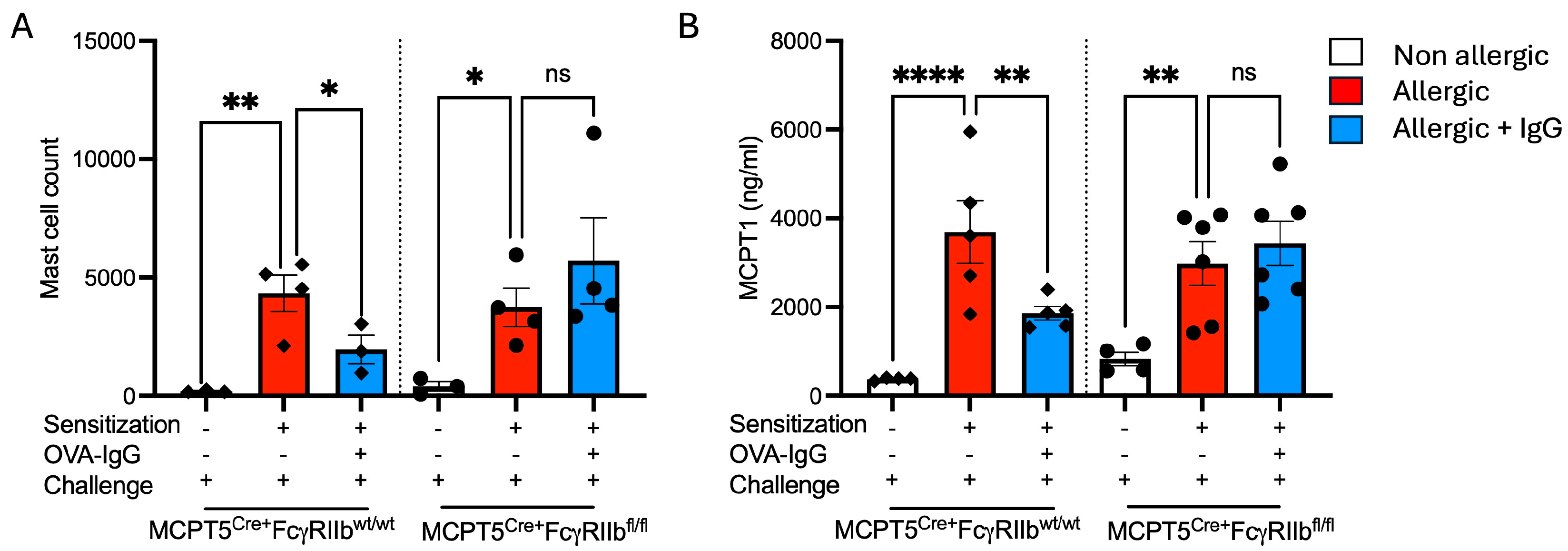
2.4. Expansion of Mucosal Mast Cells in the Allergic Lung Is Attenuated by Allergen-Specific IgG Treatment
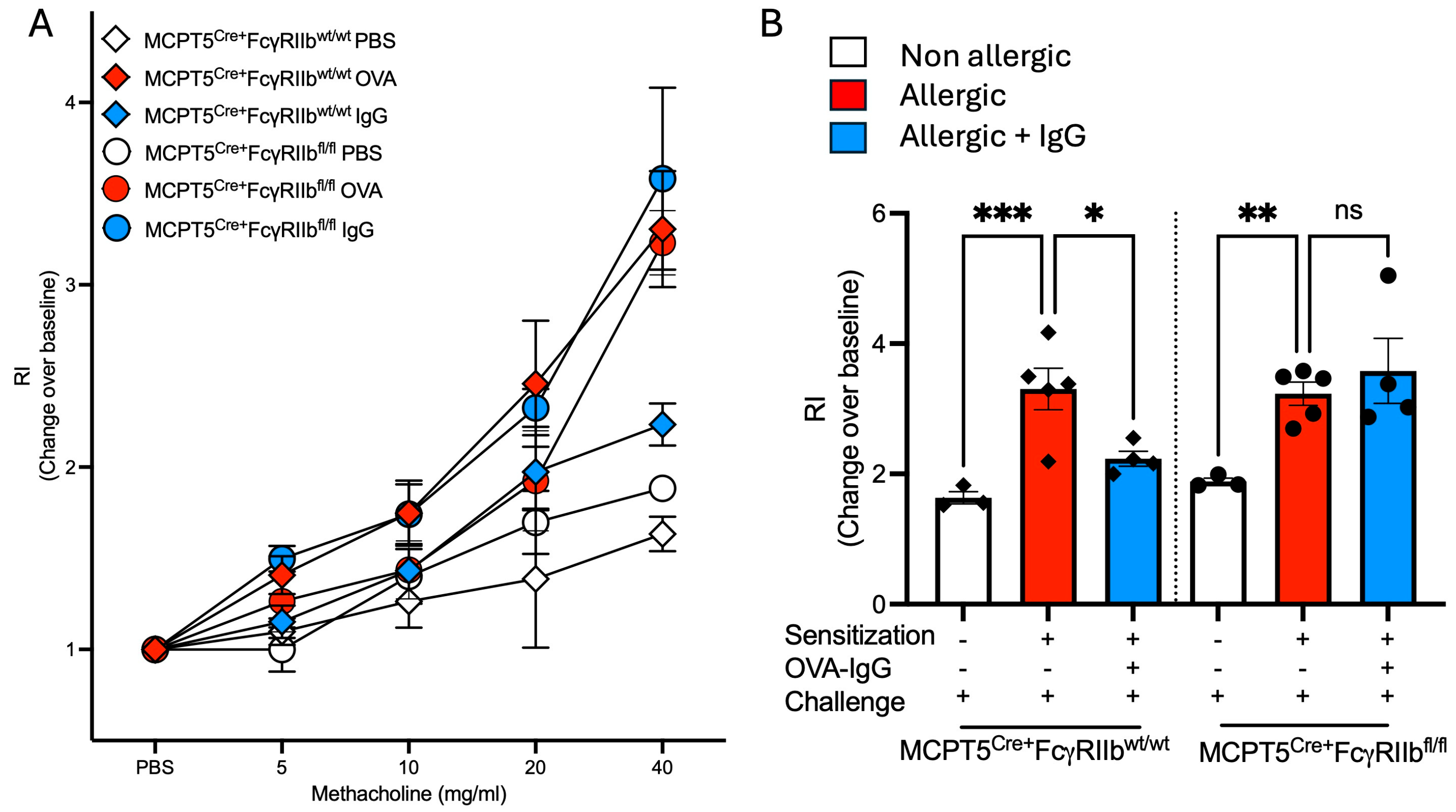
2.5. Increases in Airway Responsiveness Following Allergen Sensitization and Challenge Are Reduced with Allergen-Specific IgG Treatment
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Allergic Asthma Protocol
4.3. Bronchoalveolar Lavage Fluid (BALF) Collection
4.4. CCL2, CCL24 and IL-5 ELISA
4.5. Lung Immune Cell Phenotyping
4.6. MCPT1 ELISA
4.7. OVA-IgE ELISA
4.8. RNA Extraction, cDNA Synthesis, and RT-qPCR
4.9. Generation of Ovalbumin-Specific IgG Antibodies
4.10. Histology
4.11. Measurement of Goblet Cell Hyperplasia
4.12. Measurement of Airway Hyperresponsiveness
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AHR | airway hyperresponsiveness |
| BALF | bronchoalveolar lavage fluid |
| CCL | chemokine ligand |
| ELISA | enzyme-linked immunosorbent assay |
| Fl | flox |
| H and E | hematoxylin and eosin |
| Ig | immunoglobulin |
| IL | interleukin |
| i.n. | intranasal |
| i.p. | intraperitoneal |
| MCPT1 | mast cell protease 1 |
| Muc5a | mucin 5a |
| OVA | Ovalbumin |
| PAS | periodic acid-Schiff |
| wt | wildtype |
References
- Brightling, C.E.; Bradding, P.; Symon, F.A.; Holgate, S.T.; Wardlaw, A.J.; Pavord, I.D. Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 2002, 346, 1699–1705. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, T.; Samuchiwal, S.K.; Hallen, N.; Bankova, L.G.; Boyce, J.A.; Barrett, N.A.; Austen, K.F.; Dwyer, D.F. Lineage-specific regulation of inducible and constitutive mast cells in allergic airway inflammation. J. Exp. Med. 2021, 218, e20200321. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Ho, L.H.; Yu, M.; Monteforte, R.; Iikura, M.; Suto, H.; Galli, S.J. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J. Allergy Clin. Immunol. 2007, 120, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Lunderius, C.; Ho, L.H.; Schäfer, B.; Tsai, M.; Galli, S.J. TNF can contribute to multiple features of ovalbumin-induced allergic inflammation of the airways in mice. J. Allergy Clin. Immunol. 2007, 119, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E. Asthma: Defining of the persistent adult phenotypes. Lancet 2006, 368, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J. The Mast Cell-IgE Paradox: From Homeostasis to Anaphylaxis. Am. J. Pathol. 2016, 186, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Boyce, J.A. Mast cells: Beyond IgE. J. Allergy Clin. Immunol. 2003, 111, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Bradding, P.; Arthur, G. Mast cells in asthma–state of the art. Clin. Exp. Allergy 2016, 46, 194–263. [Google Scholar] [CrossRef] [PubMed]
- Banafea, G.H.; Bakhashab, S.; Alshaibi, H.F.; Pushparaj, P.N.; Rasool, M. The role of human mast cells in allergy and asthma. Bioengineered 2022, 13, 7049–7064. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, J.Y.; Wu, X.; Summer, S.; Whoriskey, J.; Saris, C.; Reagan, J.D. G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 2010, 86, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Aalberse, R.C.; Dieges, P.H.; Knul-Bretlova, V.; Vooren, P.; Aalbers, M.; van Leeuwen, J. IgG4 as A Blocking Antibody. Clin. Rev. Allergy 1983, 1, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Gehlhar, K.; Schlaak, M.; Becker, W.; Bufe, A. Monitoring allergen immunotherapy of pollen-allergic patients: The ratio of allergen-specific IgG4 to IgG1 correlates with clinical outcome. Clin. Exp. Allergy 1999, 29, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Akdis, C.A. Mechanisms of allergen-specific immunotherapy: Multiple suppressor factors at work in immune tolerance to allergens. J. Allergy Clin. Immunol. 2014, 133, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Platts-Mills, T.; Vaughan, J.; Squillace, S.; Woodfolk, J.; Sporik, R. Sensitisation, asthma, and a modified Th2 response in children exposed to cat allergen: A population-based cross-sectional study. Lancet 2001, 357, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Custovic, A.; Soderstrom, L.; Ahlstedt, S.; Sly, P.D.; Simpson, A.; Holt, P.G. Allergen-specific IgG antibody levels modify the relationship between allergen-specific IgE and wheezing in childhood. J. Allergy Clin. Immunol. 2011, 127, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.G.; Strickland, D.; Bosco, A.; Belgrave, D.; Hales, B.; Simpson, A.; Hollams, E.; Holt, B.; Kusel, M.; Ahlstedt, S.; et al. Distinguishing benign from pathologic TH2 immunity in atopic children. J. Allergy Clin. Immunol. 2016, 137, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, P.; Craemer, J.D.; Ruyck, N.D.; Rottey, S.; Hoon, J.d.; Hellings, P.W.; Volckaert, B.; Lesneuck, K.; Orengo, J.M.; Atanasio, A.; et al. Novel antibody cocktail targeting Bet v 1 rapidly and sustainably treats birch allergy symptoms in a phase 1 study. J. Allergy Clin. Immunol. 2022, 149, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Blay, F.J.d.; Gherasim, A.; Domis, N.; Meier, P.; Shawki, F.; Wang, C.Q.; Orengo, J.M.; DeVeaux, M.; Ramesh, D.; Jalbert, J.J.; et al. REGN1908/1909 prevented cat allergen-induced early asthmatic responses in an environmental exposure unit. J. Allergy Clin. Immunol. 2022, 150, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Woodfolk, J.A. Benign TH2 immunity in children: A fresh perspective on control of the allergic response. J. Allergy Clin. Immunol. 2016, 137, 388–389. [Google Scholar] [CrossRef] [PubMed]
- Kanagaratham, C.; Derakhshan, T.; El Ansari, Y.S.; Furiness, K.N.; Hollers, E.; Keldsen, M.; Oettgen, H.C.; Dwyer, D.F. IgG:FcgammaRIIb signals block effector programs of IgE:FcepsilonRI-activated mast cells but spare survival pathways. J. Allergy Clin. Immunol. 2023, 152, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Engeroff, P.; Caviezel, F.; Mueller, D.; Thoms, F.; Bachmann, M.F.; Vogel, M. CD23 provides a noninflammatory pathway for IgE-allergen complexes. J. Allergy Clin. Immunol. 2020, 145, 301–311.e4. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Koyanagi, A.; Ago, H.; Yamamoto, M.; Kitaura, J.; Kasai, M.; Okumura, K. Allosteric inhibition of IgE-FcepsilonRI interactions by simultaneous targeting of IgE F(ab’)2 epitopes. Commun. Biol. 2024, 7, 1042. [Google Scholar] [CrossRef] [PubMed]
- Eggel, A.; Jardetzky, T.S. Structural and Functional Insights Into IgE Receptor Interactions and Disruptive Inhibition. Immunol. Rev. 2025, 331, e70031. [Google Scholar] [CrossRef] [PubMed]
- Burton, O.T.; Tamayo, J.M.; Stranks, A.J.; Koleoglou, K.J.; Oettgen, H.C. Allergen-specific IgG antibody signaling through FcgammaRIIb promotes food tolerance. J. Allergy Clin. Immunol. 2018, 141, 189–201.e3. [Google Scholar] [CrossRef] [PubMed]
- Burton, O.T.; Logsdon, S.L.; Zhou, J.S.; Medina-Tamayo, J.; Abdel-Gadir, A.; Rivas, M.N.; Koleoglou, K.J.; Chatila, T.A.; Schneider, L.C.; Rachid, R.; et al. Oral immunotherapy induces IgG antibodies that act through FcgammaRIIb to suppress IgE-mediated hypersensitivity. J. Allergy Clin. Immunol. 2014, 134, 1310–1317.e6. [Google Scholar] [CrossRef] [PubMed]
- Oettgen, H.C.; Martin, T.R.; Wynshaw-Boris, A.; Deng, C.; Drazen, J.M.; Leder, P. Active anaphylaxis in IgE-deficient mice. Nature 1994, 370, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Burton, O.T.; Rivas, M.N.; Zhou, J.S.; Logsdon, S.L.; Darling, A.R.; Koleoglou, K.J.; Roers, A.; Houshyar, H.; Crackower, M.A.; Chatila, T.A.; et al. Immunoglobulin E signal inhibition during allergen ingestion leads to reversal of established food allergy and induction of regulatory T cells. Immunity 2014, 41, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Amigorena, S.; Bonnerot, C.; Choquet, D.; Fridman, W.H.; Teillaud, J.L. Fc gamma RII expression in resting and activated B lymphocytes. Eur. J. Immunol. 1989, 19, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, R.; Sinclair, S.C. Regulation of the immune response. I. Reduction in ability of specific antibody to inhibit long-lasting IgG immunological priming after removal of the Fc fragment. J. Exp. Med. 1969, 129, 1183–1201. [Google Scholar] [PubMed]
- Suto, H.; Nakae, S.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast cell-associated TNF promotes dendritic cell migration. J. Immunol. 2006, 176, 4102–4112. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Kalesnikoff, J.; Grimbaldeston, M.A.; Piliponsky, A.M.; Williams, C.M.; Tsai, M. Mast cells as “tunable” effector and immunoregulatory cells: Recent advances. Annu. Rev. Immunol. 2005, 23, 749–786. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, T.; Dwyer, D.F. Detection and Isolation of Airway Mast Cell Subsets in Mouse and Human. Methods Mol. Biol. 2022, 2506, 223–235. [Google Scholar] [PubMed]
- Brandt, E.B.; Strait, R.T.; Hershko, D.; Wang, Q.; Muntel, E.E.; Scribner, T.A.; Zimmermann, N.; Finkelman, F.D.; Rothenberg, M.E. Mast cells are required for experimental oral allergen-induced diarrhea. J. Clin. Investig. 2003, 112, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, R.; Osterfeld, H.; Wu, D.; Chen, C.Y.; Arumugam, M.; Groschwitz, K.; Strait, R.; Wang, Y.H.; Finkelman, F.D.; Hogan, S.P. Intestinal mast cell levels control severity of oral antigen-induced anaphylaxis in mice. Am. J. Pathol. 2012, 180, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.E.; Ownbey, R.; Mehlhop, P.D.; Loiselle, P.M.; Rijn, M.v.d.; Bonventre, J.V.; Oettgen, H.C.; Leder, P.; Luster, A.D. Eotaxin triggers eosinophil-selective chemotaxis and calcium flux via a distinct receptor and induces pulmonary eosinophilia in the presence of interleukin 5 in mice. Mol. Med. 1996, 2, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Menzies-Gow, A.; Ying, S.; Sabroe, I.; Stubbs, V.L.; Soler, D.; Williams, T.J.; Kay, A.B. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J. Immunol. 2002, 169, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Rihar, M.; Bahri, R.; Forstnerič, V.; Bulfone-Paus, S.; Korošec, P. CCL2/C-C chemokine receptor type 2-mediated interactions among mast cells, basophils, and endothelial cells. Clin. Transl. Allergy 2025, 15, e70044. [Google Scholar] [CrossRef] [PubMed]
- Shamji, M.H.; Singh, I.; Layhadi, J.A.; Ito, C.; Karamani, A.; Kouser, L.; Sharif, H.; Tang, J.; Handijiev, S.; Parkin, R.V.; et al. Passive Prophylactic Administration with a Single Dose of Anti-Fel d 1 Monoclonal Antibodies REGN1908–1909 in Cat Allergen-induced Allergic Rhinitis: A Randomized, Double-Blind, Placebo-controlled Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Atanasio, A.; Franklin, M.C.; Kamat, V.; Hernandez, A.R.; Badithe, A.; Ben, L.H.; Jones, J.; Bautista, J.; Yancopoulos, G.D.; Olson, W.; et al. Targeting immunodominant Bet v 1 epitopes with monoclonal antibodies prevents the birch allergic response. J. Allergy Clin. Immunol. 2022, 149, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Scholten, J.; Hartmann, K.; Gerbaulet, A.; Krieg, T.; Müller, W.; Testa, G.; Roers, A. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 2008, 17, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Breukel, C.; van Loo, P.F.; Kaa, J.v.d.; Claassens, J.W.; Bujard, H.; Schönig, K.; Verbeek, J.S. Highly B lymphocyte-specific tamoxifen inducible transgene expression of CreER T2 by using the LC-1 locus BAC vector. Genesis 2009, 47, 729–735. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | FcγRIIb | Sensitization Allergen | OVA-IgG | Challenge Allergen |
|---|---|---|---|---|
| MCPT5Cre+ FcgRIIbwt/wt | Present | PBS | − | OVA |
| OVA | − | OVA | ||
| OVA | + | OVA | ||
| MCPT5Cre+ FcgRIIbfl/fl | Absent | PBS | − | OVA |
| OVA | − | OVA | ||
| OVA | + | OVA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanagaratham, C.; El Ansari, Y.S.; Furiness, K.N.; Oettgen, H.C. IgG:FcγRIIb Signaling on Mast Cells Blocks Allergic Airway Inflammation. Int. J. Mol. Sci. 2025, 26, 6779. https://doi.org/10.3390/ijms26146779
Kanagaratham C, El Ansari YS, Furiness KN, Oettgen HC. IgG:FcγRIIb Signaling on Mast Cells Blocks Allergic Airway Inflammation. International Journal of Molecular Sciences. 2025; 26(14):6779. https://doi.org/10.3390/ijms26146779
Chicago/Turabian StyleKanagaratham, Cynthia, Yasmeen S. El Ansari, Kameryn N. Furiness, and Hans C. Oettgen. 2025. "IgG:FcγRIIb Signaling on Mast Cells Blocks Allergic Airway Inflammation" International Journal of Molecular Sciences 26, no. 14: 6779. https://doi.org/10.3390/ijms26146779
APA StyleKanagaratham, C., El Ansari, Y. S., Furiness, K. N., & Oettgen, H. C. (2025). IgG:FcγRIIb Signaling on Mast Cells Blocks Allergic Airway Inflammation. International Journal of Molecular Sciences, 26(14), 6779. https://doi.org/10.3390/ijms26146779