
Comparative Transcriptome Analysis in Tomato Fruit Reveals Genes, Pathways, and Processes Affected by the LEC1-LIKE4 Transcription Factor

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
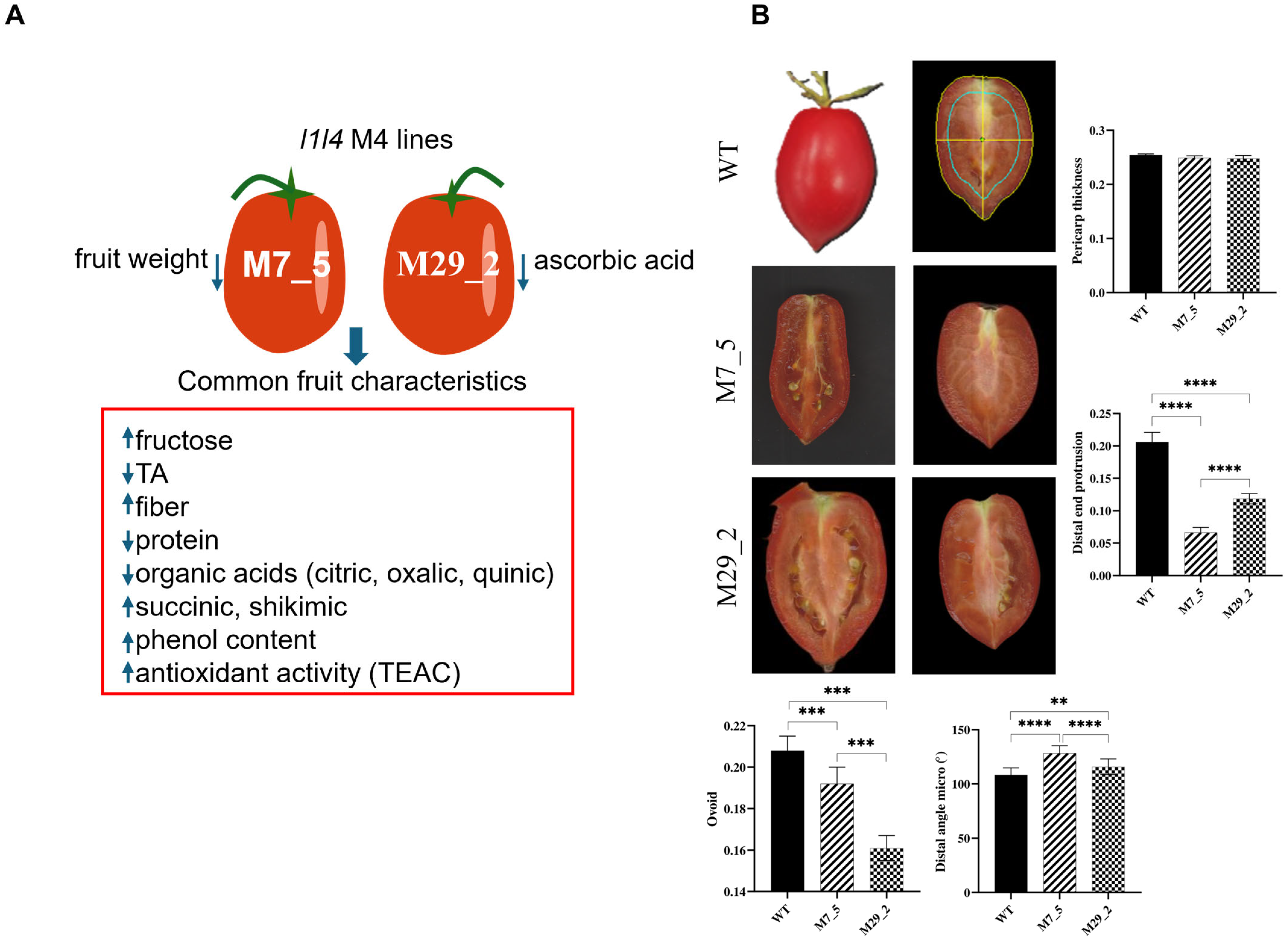
2.1. Fruit Morphometric Traits
2.2. Differentially Expressed Genes Among Samples and Groups
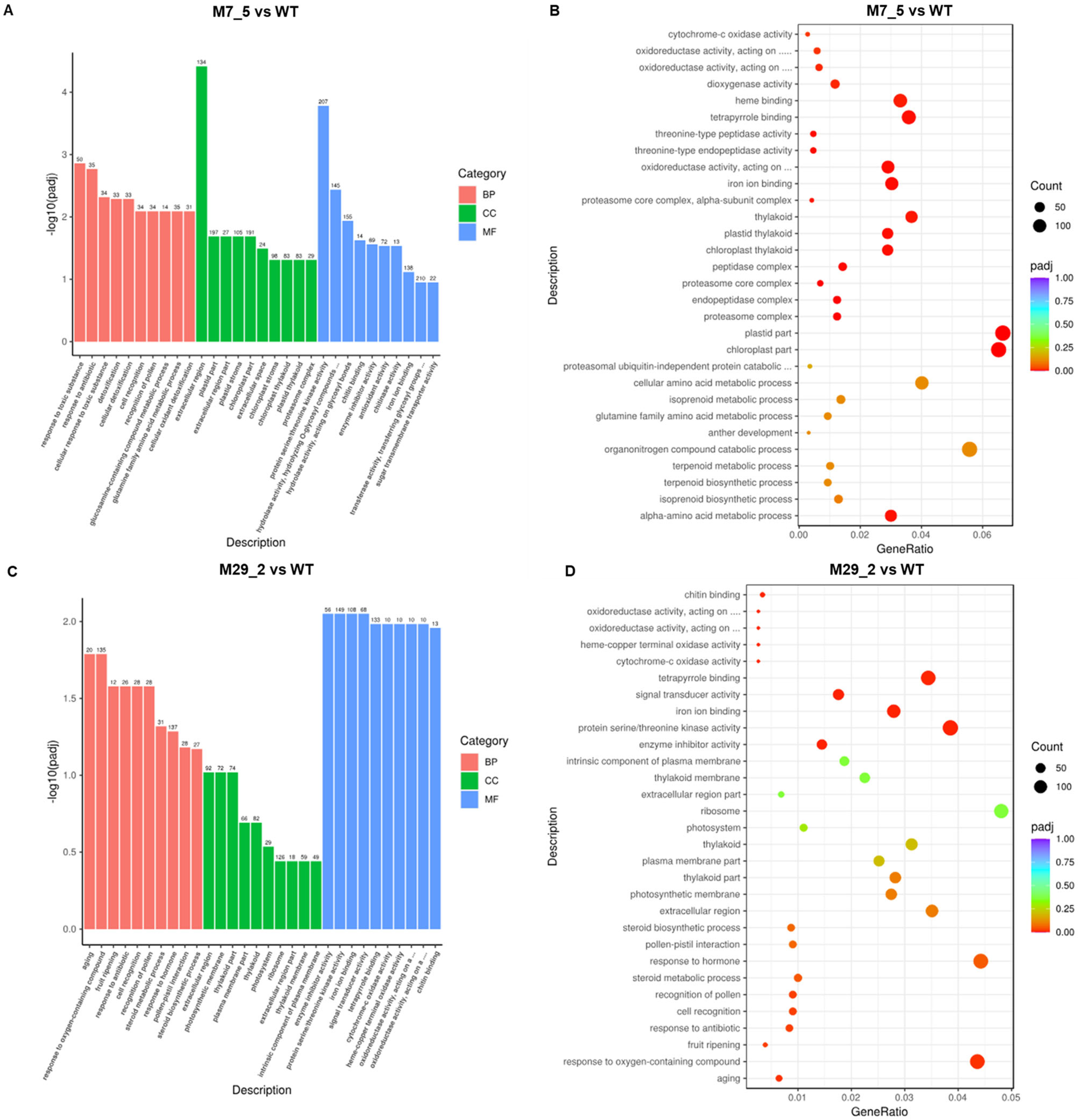
2.3. GO Term Analysis
2.4. KEGG Enrichment Analysis
2.5. Analysis of Histone-Related Genes
2.6. Analysis of TFs
2.7. Identification of Fruit Metabolism and Quality Genes
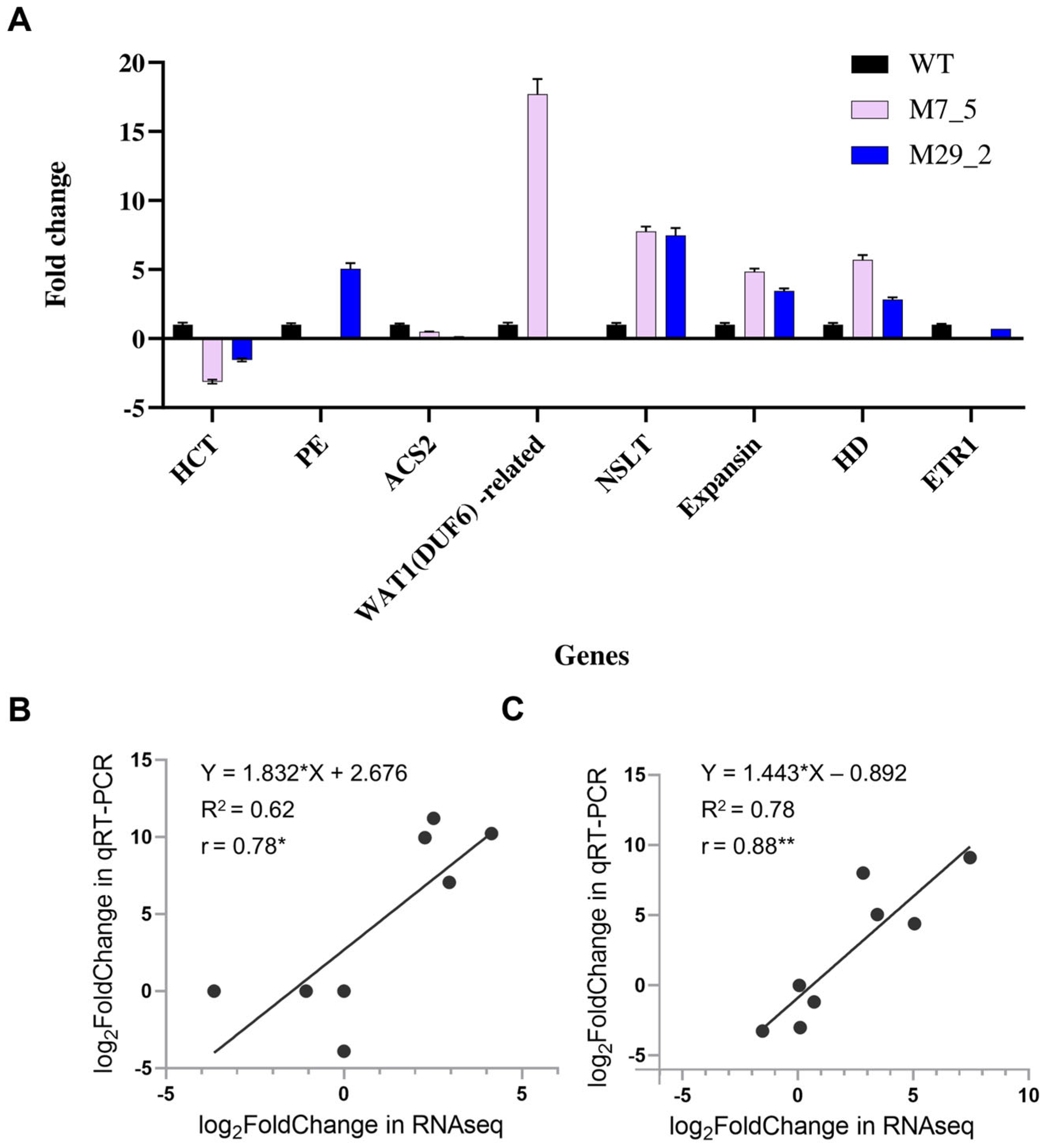
2.8. Validation of DEGs Genes with qRT-PCR Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Digital Fruit Phenotyping
4.3. RNA Extraction
4.4. Preparation of cDNA Libraries for Sequencing
4.5. Bioinformatics Analysis
4.6. Real-Time Quantitative PCR (qRT-PCR) Analysis
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACS | 1-aminocyclopropane-1-carboxylate synthase |
| ADH | alcohol dehydrogenase |
| BD | binding domain |
| BP | biological processes |
| CC | cellular components |
| CESA | cellulose synthase |
| CS | citrate synthase |
| DEG | differential gene expression |
| EF1-α | Elongation factor 1-α |
| ERFs | Ethylene Response Factors |
| FDR | False Discovery Rate |
| HD | histidine decarboxylase |
| HDACs | histone deacetylases |
| HKMT | Histone methyltransferase |
| FPKM | Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced |
| FRK | fructokinase |
| GGP | GDP-L-galactose phosphorylase |
| GMP | GDP-mannose pyrophosphorylase |
| GPP | L-galactose-1-phosphate phosphatase |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | KEGG Orthology |
| LEA | late embryogenesis abundant protein |
| L1L4 | LEAFY-COTYLEDON1-LIKE4 |
| MDH | malate dehydrogenase |
| MF | molecular functions |
| NF-Y | nuclear factor Y |
| NSLT | non-specific lipid transfer |
| padj | adjusted p-value |
| PCA | principal component analysis |
| PG | polygalacturonase |
| PME | pectin methylesterase |
| PSY | phytoene synthase |
| qRT-PCR | real-time quantitative PCR |
| SD | standard deviation |
| TCA | tricarboxylic acid |
| TF | transcription factor |
| WT | wild-type |
| ZFN | zinc-finger nuclease |
References
- Giovannoni, J.J. Genetic regulation of fruit development and ripening. Plant Cell 2004, 16 (Suppl. S1), S170–S180. [Google Scholar] [CrossRef]
- Li, S.; Li, K.; Ju, Z.; Cao, D.; Fu, D.; Zhu, H.; Zhu, B.; Luo, Y. Genome-Wide Analysis of Tomato NF-Y Factors and Their Role in Fruit Ripening. BMC Genom. 2016, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, R. The Molecular Biology of the CCAAT-Binding Factor NF-Y. Gene 1999, 239, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Myers, Z.A.; Holt, B.F. NUCLEAR FACTOR-Y: Still Complex after All These Years? Curr. Opin. Plant Biol. 2018, 45, 96–102. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Li, C.; Zhang, C.; Cui, L.; Ai, G.; Wang, X.; Zheng, F.; Zhang, D.; Zhang, J.; et al. NF-Y plays essential roles in flavonoid biosynthesis by modulating histone modifications in tomato. New Phytol. 2020, 229, 3237–3252. [Google Scholar] [CrossRef]
- Petrou, N.; Tsigarida, N.; Hilioti, Z. Genome Editing of the NF-YA8 Gene Modifies Tomato Plant Architecture and Fruit Traits. Plants 2025, in press. [CrossRef]
- Hilioti, Z.; Ganopoulos, I.; Ajith, S.; Bossis, I.; Tsaftaris, A. A novel arrangement of zinc finger nuclease system for in vivo targeted genome engineering: The tomato LEC1-LIKE4 gene case. Plant Cell Rep. 2016, 35, 2241–2255. [Google Scholar] [CrossRef] [PubMed]
- Hilioti, Z.; Ganopoulos, I.; Bossis, I.; Tsaftaris, A. LEC1-LIKE paralog transcription factor: How to survive extinction and fit in NF-Y protein complex. Gene 2014, 543, 220–233. [Google Scholar] [CrossRef]
- Gago, C.; Drosou, V.; Paschalidis, K.; Guerreiro, A.; Miguel, G.; Antunes, D.; Hilioti, Z. Targeted Gene Disruption Coupled with Metabolic Screen Approach to Uncover the LEAFY COTYLEDON1-LIKE4 (L1L4) Function in Tomato Fruit Metabolism. Plant Cell Rep. 2017, 36, 1065–1082. [Google Scholar] [CrossRef]
- Rodríguez, G.R.; Muños, S.; Anderson, C.; Sim, S.-C.; Michel, A.; Causse, M.; Gardener, B.B.M.; Francis, D.; van der Knaap, E. Distribution ofSUN, OVATE, LC, and FASin the Tomato Germplasm and the Relationship to Fruit Shape Diversity. Plant Physiol. 2011, 156, 275–285. [Google Scholar] [CrossRef]
- Gillaspy, G.; Ben-David, H.; Gruissem, W. Fruits: A Developmental Perspective. Plant Cell 1993, 5, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-Base Resolution Methylomes of Tomato Fruit Development Reveal Epigenome Modifications Associated with Ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical Roles of DNA Demethylation in the Activation of Ripening-Induced Genes and Inhibition of Ripening-Repressed Genes in Tomato Fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, X.; Liu, X.; Li, Y.; Wu, K.; Hou, X. Arabidopsis NF-YCs Mediate the Light-Controlled Hypocotyl Elongation via Modulating Histone Acetylation. Mol. Plant 2017, 10, 260–273. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.-W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of Chromatin Structure by Site-Specific Histone H3 Methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Karlić, R.; Chung, H.-R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone Modification Levels Are Predictive for Gene Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef]
- Lü, P.; Yu, S.; Zhu, N.; Chen, Y.-R.; Zhou, B.; Pan, Y.; Tzeng, D.; Fabi, J.P.; Argyris, J.; Garcia-Mas, J.; et al. Genome Encode Analyses Reveal the Basis of Convergent Evolution of Fleshy Fruit Ripening. Nat. Plants 2018, 4, 784–791. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Wang, L.; Tian, Y.; Jia, N.; Chen, S.; Shi, N.; Huang, X.; Zhou, C.; Yu, Y.; et al. Regulation of Ethylene-Responsive SlWRKYs Involved in Color Change during Tomato Fruit Ripening. Sci. Rep. 2017, 7, 16674. [Google Scholar] [CrossRef]
- Allan, A.C.; Hellens, R.P.; Laing, W.A. MYB Transcription Factors That Colour Our Fruit. Trends Plant Sci. 2008, 13, 99–102. [Google Scholar] [CrossRef]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and Comparative Analysis of MYB and BHLH Plant Transcription Factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef] [PubMed]
- Loreti, E.; Betti, F.; Ladera-Carmona, M.J.; Fontana, F.; Novi, G.; Valeri, M.C.; Perata, P. ARGONAUTE1 and ARGONAUTE4 Regulate Gene Expression and Hypoxia Tolerance. Plant Physiol. 2019, 182, 287–300. [Google Scholar] [CrossRef]
- Brummell, D.A. Cell Wall Metabolism during Maturation, Ripening and Senescence of Peach Fruit. J. Exp. Bot. 2004, 55, 2029–2039. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, K.A.; Bennett, A.B. Polygalacturonases: Many Genes in Search of a Function. Plant Physiol. 1998, 117, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Loosening of Plant Cell Walls by Expansins. Nature 2000, 407, 321–326. [Google Scholar] [CrossRef]
- Osorio, S.; Alba, R.; Damasceno, C.M.B.; Lopez-Casado, G.; Lohse, M.; Zanor, M.I.; Tohge, T.; Usadel, B.; Rose, J.K.C.; Fei, Z.; et al. Systems Biology of Tomato Fruit Development: Combined Transcript, Protein, and Metabolite Analysis of Tomato Transcription Factor (Nor, Rin) and Ethylene Receptor (Nr) Mutants Reveals Novel Regulatory Interactions. Plant Physiol. 2011, 157, 405–425. [Google Scholar] [CrossRef]
- Smirnoff, N.; Wheeler, G.L. Ascorbic Acid in Plants: Biosynthesis and Function. Crit. Rev. Plant Sci. 2000, 19, 267–290. [Google Scholar] [CrossRef]
- Foyer, C.H.; Noctor, G. Ascorbate and Glutathione: The Heart of the Redox Hub. Plant Physiol. 2011, 155, 2–18. [Google Scholar] [CrossRef]
- Mu, J.; Tan, H.; Zheng, Q.; Fu, F.; Liang, Y.; Zhang, J.; Yang, X.; Wang, T.; Chong, K.; Wang, X.-J.; et al. LEAFY COTYLEDON1 Is a Key Regulator of Fatty Acid Biosynthesis in Arabidopsis. Plant Physiol. 2008, 148, 1042–1054. [Google Scholar] [CrossRef]
- Manan, S.; Alabbosh, K.F.; Al-Andal, A.; Ahmad, W.; Khan, K.A.; Zhao, J. Soybean LEAFY COTYLEDON 1: A Key Target for Genetic Enhancement of Oil Biosynthesis. Agronomy 2023, 13, 2810. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koidou, V.; Valasiadis, D.; Petrou, N.; Emmanouilidou, C.; Hilioti, Z. Comparative Transcriptome Analysis in Tomato Fruit Reveals Genes, Pathways, and Processes Affected by the LEC1-LIKE4 Transcription Factor. Int. J. Mol. Sci. 2025, 26, 6728. https://doi.org/10.3390/ijms26146728
Koidou V, Valasiadis D, Petrou N, Emmanouilidou C, Hilioti Z. Comparative Transcriptome Analysis in Tomato Fruit Reveals Genes, Pathways, and Processes Affected by the LEC1-LIKE4 Transcription Factor. International Journal of Molecular Sciences. 2025; 26(14):6728. https://doi.org/10.3390/ijms26146728
Chicago/Turabian StyleKoidou, Venetia, Dimitrios Valasiadis, Nestor Petrou, Christina Emmanouilidou, and Zoe Hilioti. 2025. "Comparative Transcriptome Analysis in Tomato Fruit Reveals Genes, Pathways, and Processes Affected by the LEC1-LIKE4 Transcription Factor" International Journal of Molecular Sciences 26, no. 14: 6728. https://doi.org/10.3390/ijms26146728
APA StyleKoidou, V., Valasiadis, D., Petrou, N., Emmanouilidou, C., & Hilioti, Z. (2025). Comparative Transcriptome Analysis in Tomato Fruit Reveals Genes, Pathways, and Processes Affected by the LEC1-LIKE4 Transcription Factor. International Journal of Molecular Sciences, 26(14), 6728. https://doi.org/10.3390/ijms26146728