
Comparative Transcriptomic Profiling in Patients Affected by Duchenne and Becker Muscular Dystrophies: A Focus on ECM Genes Dysregulation

 , , , and
, , , and
Abstract
1. Introduction
2. Results
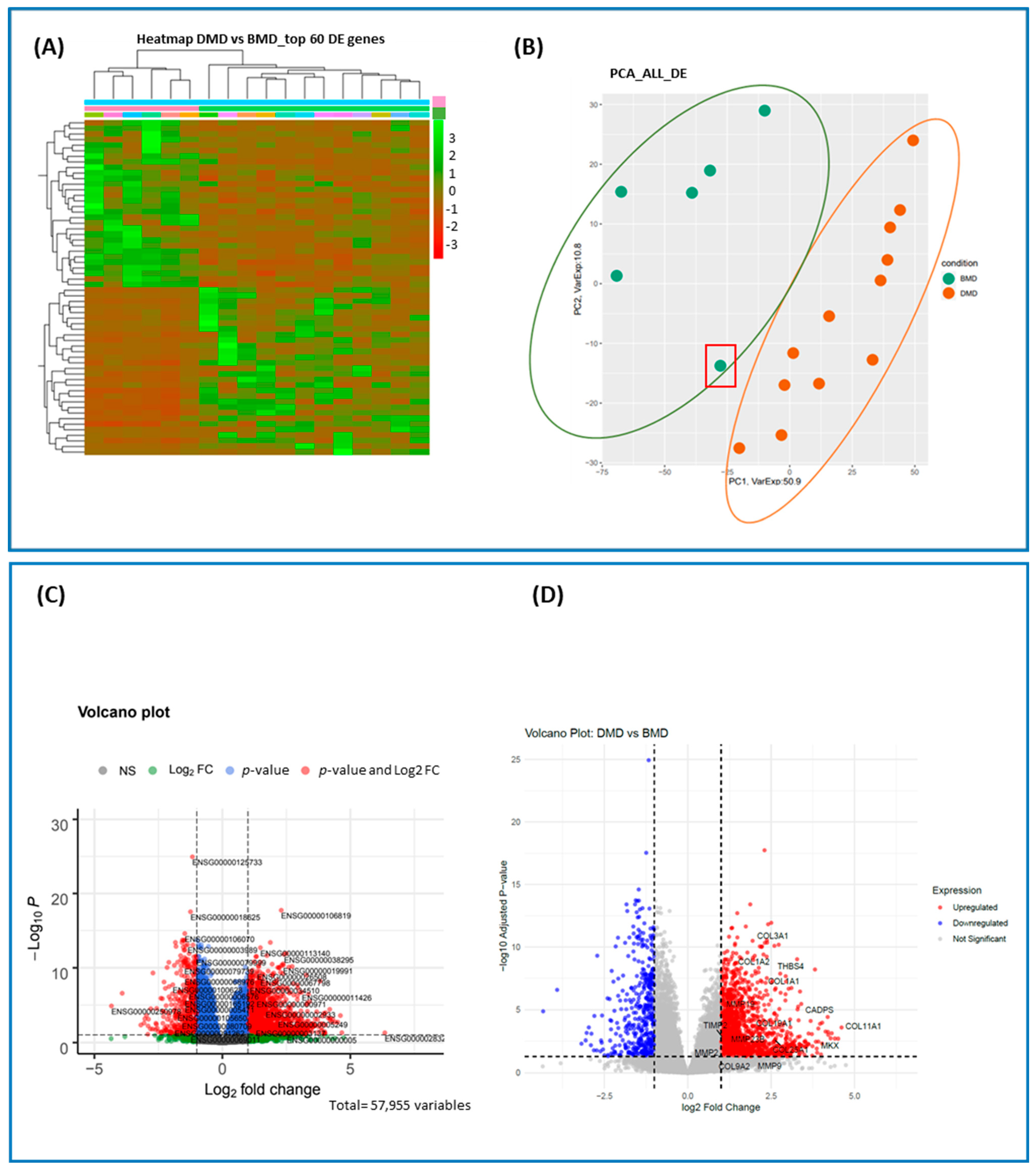
2.1. Overview of Differentially Expressed (DE) Genes
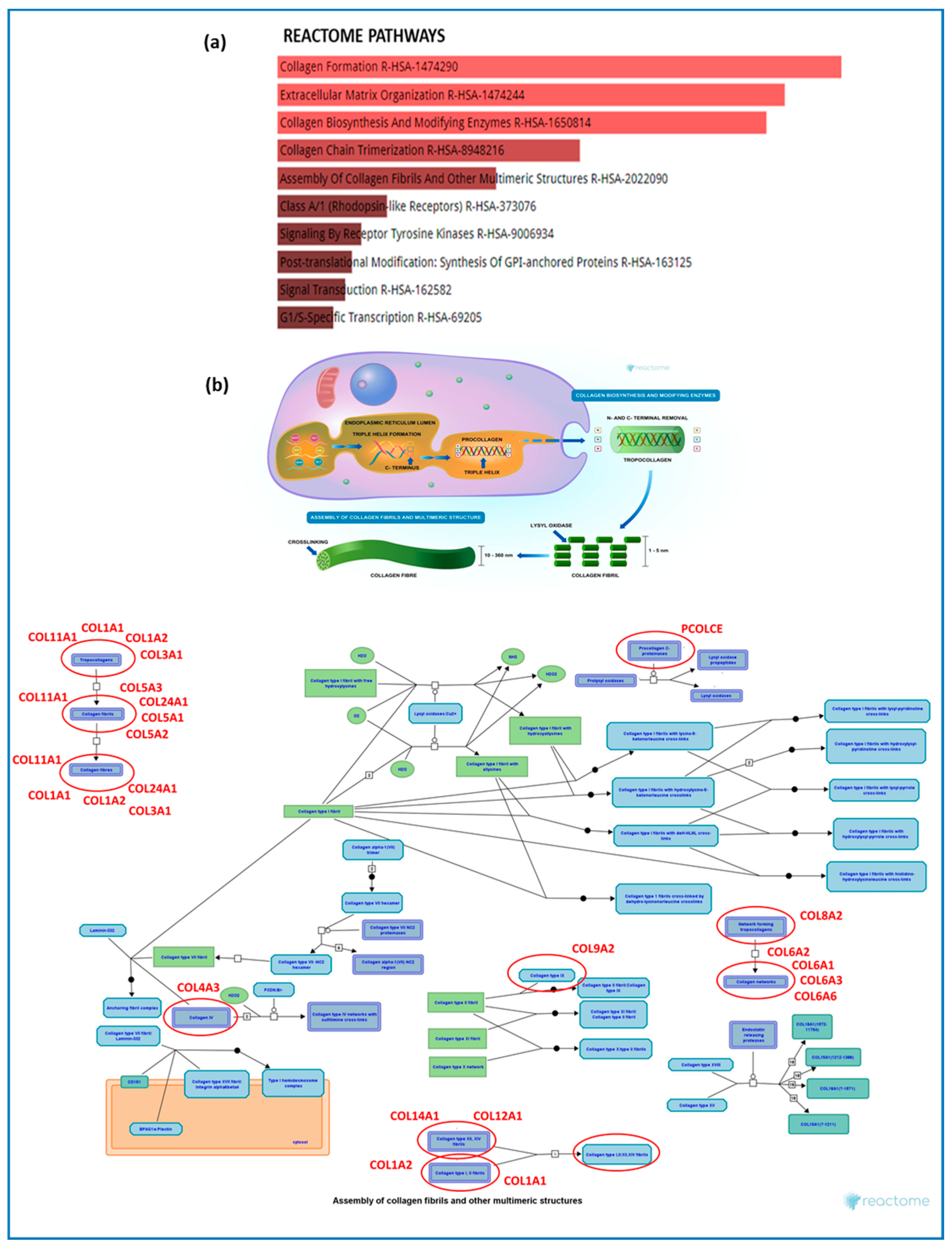
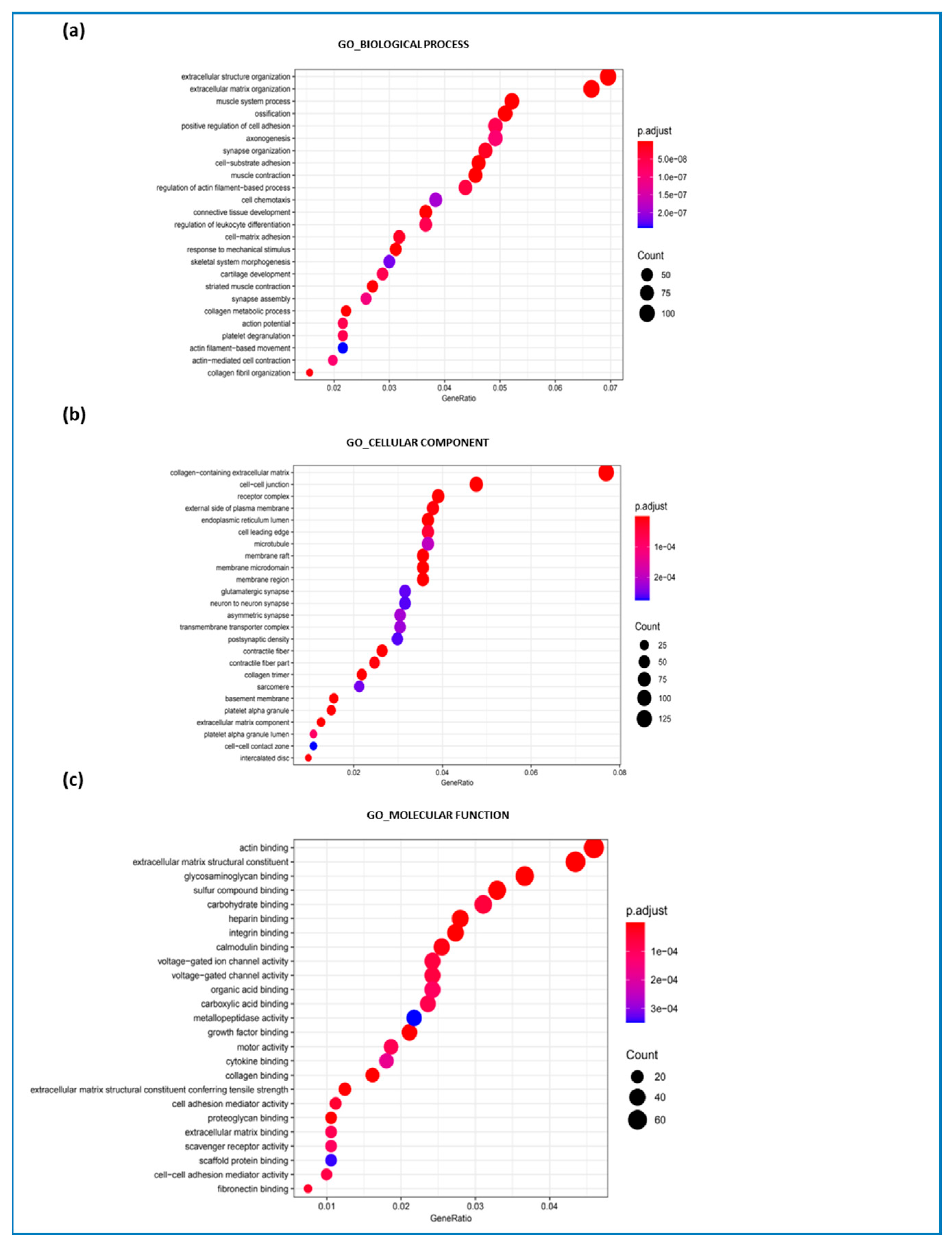
2.2. Gene Set Enrichment Analysis
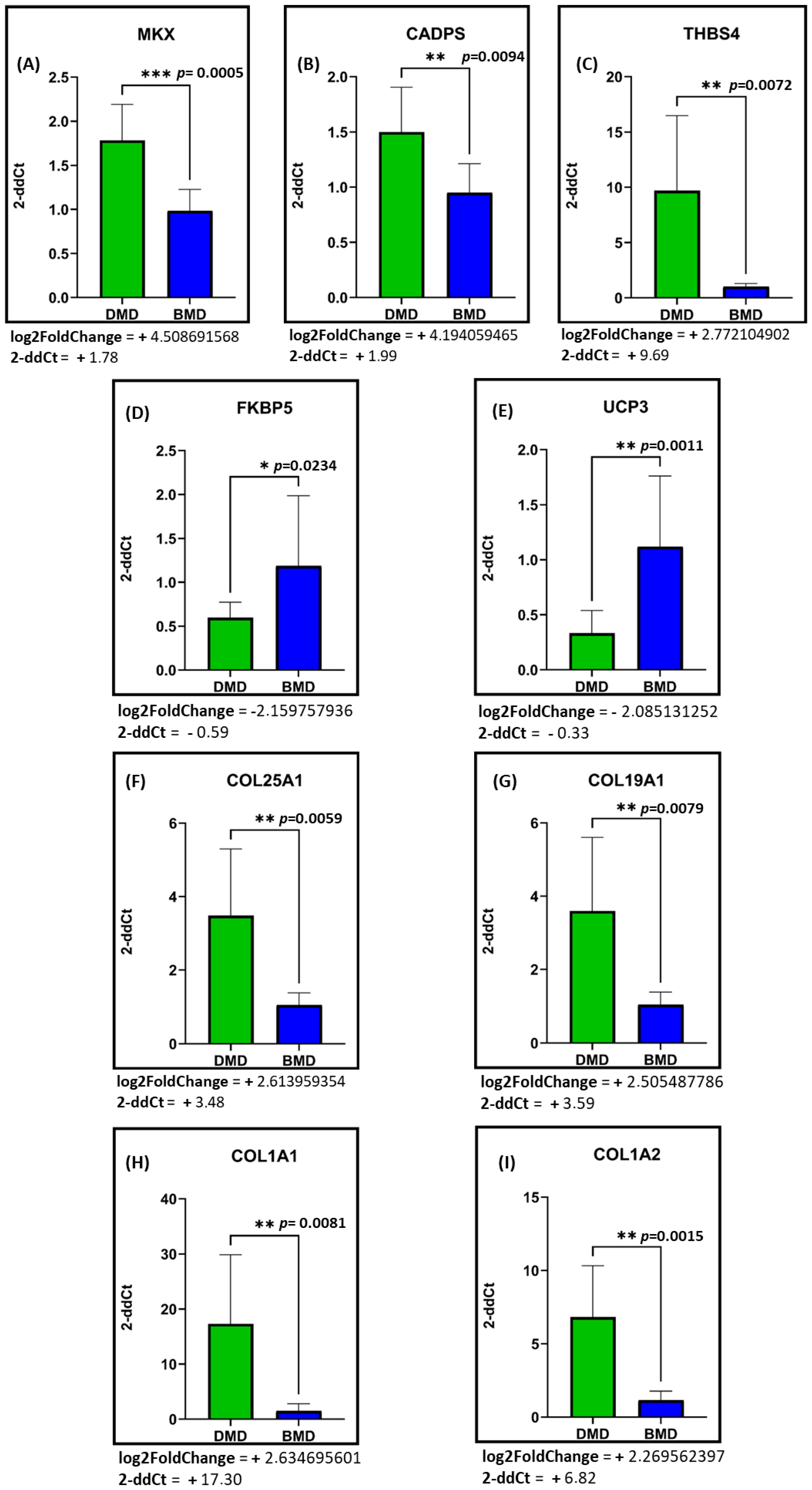
2.3. mRNA Expression Levels of the Most Differentially Expressed Genes Between DMD and BMD Patients
3. Discussion
3.1. Primary Deregulated Pathways: Collagen, ECM Component, and Fibrotic Processes
ECM Dysregulation Impact, Regeneration Failure, and Signaling Pathways Regulating ECM Genes
3.2. Secondary Deregulated Pathways: Synaptic Organization, Platelet Activation, and Inflammation
4. Materials and Methods
4.1. Patient Enrollment
4.2. Muscle Biopsy Isolation
4.3. Blood DNA Isolation, Muscle Sample Homogenization, and RNA Extraction
4.4. RNA-Seq Library Preparation and Bioinformatic Data Analysis
4.5. Pathway Analysis
4.6. Real-Time PCR
4.7. Statistical Analyses
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aartsma-Rus, A.; Janson, A.A.M.; Heemskerk, J.A.; DE Winter, C.L.; VAN Deutekom, J.C.T.; VAN Ommen, G.-J.B. Therapeutic modulation of DMD splicing by blocking exonic splicing enhancer sites with antisense oligonucleotides. Ann. N. Y. Acad. Sci. 2006, 1082, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, F.; Tonelli, L.; Farnè, M.; Selvatici, R.; Ferlini, A. DMD deletions underlining mild dystrophinopathies: Literature review highlights phenotype-related mutation clusters and provides insights about genetic mechanisms and prognosis. Front. Neurol. 2024, 14, 1288721. [Google Scholar] [CrossRef]
- Dabaj, I.; Ducatez, F.; Marret, S.; Bekri, S.; Tebani, A. Neuromuscular disorders in the omics era. Clin. Chim. Acta 2024, 553, 117691. [Google Scholar] [CrossRef]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, O.; Yokota, T. Advances in Genetic Characterization and Genotype-Phenotype Correlation of Duchenne and Becker Muscular Dystrophy in the Personalized Medicine Era. J. Pers. Med. 2020, 10, 111. [Google Scholar] [CrossRef]
- Findlay, A.R.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. United Dystrophinopathy Project. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef]
- Cirak, S.; Feng, L.; Anthony, K.; Arechavala-Gomeza, V.; Torelli, S.; Sewry, C.; Morgan, J.E.; Muntoni, F. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 462–467. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Hendriksen, R.G.F.; Hoogland, G.; Schipper, S.; Hendriksen, J.G.M.; Vles, J.S.H.; Aalbers, M.W. A possible role of dystrophin in neuronal excitability: A review of the current literature. Neurosci. Biobehav. Rev. 2015, 51, 255–262. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Flanigan, K.M. Update in Duchenne and Becker muscular dystrophy. Curr. Opin. Neurol. 2019, 32, 722–727. [Google Scholar] [CrossRef]
- Elasbali, A.M.; Al-Soud, W.A.; Anwar, S.; Alhassan, H.H.; Adnan, M.; Hassan, I. A review on mechanistic insights into structure and function of dystrophin protein in pathophysiology and therapeutic targeting of Duchenne muscular dystrophy. Int. J. Biol. Macromol. 2024, 264 Pt 1, 130544. [Google Scholar] [CrossRef]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef]
- González-Jamett, A.; Vásquez, W.; Cifuentes-Riveros, G.; Martínez-Pando, R.; Sáez, J.C.; Cárdenas, A.M. Oxidative Stress, Inflammation and Connexin Hemichannels in Muscular Dystrophies. Biomedicines 2022, 10, 507. [Google Scholar] [CrossRef] [PubMed]
- Heret, M.; Saclier, M.; Messina, G.; Rossi, F.M.V. Macrophages in Skeletal Muscle Dystrophies, An Entangled Partner. J. Neuromuscul. Dis. 2022, 9, 1–23. [Google Scholar] [CrossRef]
- Rossi, G.; Taglietti, V.; Messina, G. Targeting Nfix to fix muscular dystrophies. Cell Stress 2017, 2, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Giovarelli, M.; Arnaboldi, F.; Zecchini, S.; Cornaghi, L.B.; Nava, A.; Sommariva, M.; Clementi, E.G.I.; Gagliano, N. Characterisation of Progressive Skeletal Muscle Fibrosis in the Mdx Mouse Model of Duchenne Muscular Dystrophy: An In Vivo and In Vitro Study. Int. J. Mol. Sci. 2022, 23, 8735. [Google Scholar] [CrossRef]
- Heezen, L.G.M.; Abdelaal, T.; Van Putten, M.; Aartsma-Rus, A.; Mahfouz, A.; Spitali, P. Spatial transcriptomics reveal markers of histopathological changes in Duchenne muscular dystrophy mouse models. Nat. Commun. 2023, 14, 4909. [Google Scholar] [CrossRef]
- Mázala, D.A.G.; Hindupur, R.; Moon, Y.J.; Shaikh, F.; Gamu, I.H.; Alladi, D.; Panci, G.; Weiss-Gayet, M.; Chazaud, B.; Partridge, T.A.; et al. Altered muscle niche contributes to myogenic deficit in the D2-mdx model of severe DMD. Cell Death Discov. 2023, 9, 224. [Google Scholar] [CrossRef]
- Coulis, G.; Jaime, D.; Guerrero-Juarez, C.; Kastenschmidt, J.M.; Farahat, P.K.; Nguyen, Q.; Pervolarakis, N.; McLinden, K.; Thurlow, L.; Movahedi, S.; et al. Single-cell and spatial transcriptomics identify a macrophage population associated with skeletal muscle fibrosis. Sci. Adv. 2023, 9, eadd9984. [Google Scholar] [CrossRef] [PubMed]
- Vitaliti, A.; Reggio, A.; Colletti, M.; Galardi, A.; Palma, A. Integration of single-cell datasets depicts profiles of macrophages and fibro/adipogenic progenitors in dystrophic muscle. Exp. Cell Res. 2024, 442, 114197. [Google Scholar] [CrossRef]
- Hinkle, E.R.; Wiedner, H.J.; Black, A.J.; Giudice, J. RNA processing in skeletal muscle biology and disease. Transcription 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Erriquez, D.; Perini, G.; Ferlini, A. Non-coding RNAs in muscle dystrophies. Int. J. Mol. Sci. 2013, 14, 19681–19704. [Google Scholar] [CrossRef]
- Nieves-Rodriguez, S.; Barthélémy, F.; Woods, J.D.; Douine, E.D.; Wang, R.T.; Scripture-Adams, D.D.; Chesmore, K.N.; Galasso, F.; Miceli, M.C.; Nelson, S.F. Transcriptomic analysis of paired healthy human skeletal muscles to identify modulators of disease severity in DMD. Front. Genet. 2023, 14, 1216066. [Google Scholar] [CrossRef]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Viganò, A.; Lochmüller, H.; Muntoni, F.; Ferlini, A.; Mora, M.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; Kuroda, K.; Rudnicki, M.A. Stem cell-based therapies to treat muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Loomis, T.; Smith, L.R. Thrown for a loop: Fibro-adipogenic progenitors in skeletal muscle fibrosis. Am. J. Physiol. Cell Physiol. 2023, 325, C895–C906. [Google Scholar] [CrossRef]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124 Pt 21, 3654–3664. [Google Scholar] [CrossRef]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Grand, F.L.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Ogura, Y.; Tajrishi, M.M.; Sato, S.; Hindi, S.M.; Kumar, A. Therapeutic potential of matrix metalloproteinases in Duchenne muscular dystrophy. Front. Cell Dev. Biol. 2014, 2, 11. [Google Scholar] [CrossRef]
- Nadarajah, V.D.; Van Putten, M.; Chaouch, A.; Garrood, P.; Straub, V.; Lochmüller, H.; Ginjaar, H.B.; Aartsma-Rus, A.M.; Van Ommen, G.J.B.; Den Dunnen, J.T.; et al. Serum matrix metalloproteinase-9 (MMP-9) as a biomarker for monitoring disease progression in DMD. Neuromuscul. Disord. 2011, 21, 569–578. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New insights into fibrosis from the ECM degradation perspective: The macrophage-MMP-ECM interaction. Cell Biosci. 2022, 12, 138. [Google Scholar] [CrossRef]
- Zhao, H.; Dong, Y.; Tian, X.; Tan, T.K.; Liu, Z.; Zhao, Y.; Zhang, Y.; Harris, D.C.; Zheng, G. Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases. World J. Nephrol. 2013, 2, 84–89. [Google Scholar] [CrossRef]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. BioMed Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rosenthal, W.; Martinez, L.; Kaur, A.; Sparwasser, T.; Tidball, J.G.; Margeta, M.; Spencer, M.J.; Bluestone, J.A. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 2014, 6, 258ra142. [Google Scholar] [CrossRef]
- Evans, N.P.; Misyak, S.A.; Robertson, J.L.; Bassaganya-Riera, J.; Grange, R.W. Immune-mediated mechanisms potentially regulate the disease time-course of duchenne muscular dystrophy and provide targets for therapeutic intervention. PMR 2009, 1, 755–768. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.G.; Cabello-Verrugio, C.; Santander, C.; Cabrera, D.; Goldschmeding, R.; Brandan, E. CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J. Pathol. 2011, 225, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Vial, C.; Zúñiga, L.M.; Cabello-Verrugio, C.; Cañón, P.; Fadic, R.; Brandan, E. Skeletal muscle cells express the profibrotic cytokine connective tissue growth factor (CTGF/CCN2), which induces their dedifferentiation. J. Cell Physiol. 2008, 215, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix-What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef]
- Levi, O.; Genin, O.; Angelini, C.; Halevy, O.; Pines, M. Inhibition of muscle fibrosis results in increases in both utrophin levels and the number of revertant myofibers in Duchenne muscular dystrophy. Oncotarget 2015, 6, 23249–23260. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef]
- Nelson, C.A.; Hunter, R.B.; Quigley, L.A.; Girgenrath, S.; Weber, W.D.; McCullough, J.A.; Dinardo, C.J.; Keefe, K.A.; Ceci, L.; Clayton, N.P.; et al. Inhibiting TGF-β activity improves respiratory function in mdx mice. Am. J. Pathol. 2011, 178, 2611–2621. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Riparini, G.; Simone, J.M.; Sartorelli, V. FACS-isolation and Culture of Fibro-Adipogenic Progenitors and Muscle Stem Cells from Unperturbed and Injured Mouse Skeletal Muscle. J. Vis. Exp. 2022, 184, e63983. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, L. The Many Roles of Macrophages in Skeletal Muscle Injury and Repair. Front. Cell Dev. Biol. 2022, 11, 952249. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68–69, 602–615. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, N.; Chu, H.Y.; Yu, Y.; Zhang, Z.-K.; Zhang, G.; Zhang, B.-T. Connective Tissue Growth Factor: From Molecular Understandings to Drug Discovery. Front. Cell Dev. Biol. 2020, 8, 593269. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’AMico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Lovering, R.M.; Iyer, S.R.; Edwards, B.; Davies, K.E. Alterations of neuromuscular junctions in Duchenne muscular dystrophy. Neurosci. Lett. 2020, 737, 135304. [Google Scholar] [CrossRef] [PubMed]
- Labarque, V.; Freson, K.; Thys, C.; Wittevrongel, C.; Hoylaerts, M.F.; De Vos, R.; Goemans, N.; Van Geet, C. Increased Gs signalling in platelets and impaired collagen activation, due to a defect in the dystrophin gene, result in increased blood loss during spinal surgery. Hum. Mol. Genet. 2008, 17, 357–366. [Google Scholar] [CrossRef]
- Shapiro, F.; Sethna, N. Blood loss in pediatric spine surgery. Eur. Spine J. 2004, 13 (Suppl. S1), 6–17. [Google Scholar] [CrossRef]
- Turturro, F.; Rocca, B.; Gumina, S.; De Cristofaro, R.; Mangiola, F.; Maggiano, N.; Evangelista, A.; Salsano, V.; Montanaro, A. Impaired primary hemostasis with normal platelet function in Duchenne muscular dystrophy during highly-invasive spinal surgery. Neuromuscul. Disord. 2005, 15, 532–540. [Google Scholar] [CrossRef]
- Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef]
- Burns, D.P.; Canavan, L.; Rowland, J.; O’Flaherty, R.; Brannock, M.; Drummond, S.E.; O’Malley, D.; Edge, D.; O’Halloran, K.D. Recovery of respiratory function in mdx mice co-treated with neutralizing interleukin-6 receptor antibodies and urocortin-2. J. Physiol. 2018, 596, 5175–5197. [Google Scholar]
- Madaro, L.; Bouché, M. From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: The role of lymphocytes. BioMed Res. Int. 2014, 2014, 438675. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding RNAs characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis patients. Sci. Rep. 2018, 8, 2378. [Google Scholar] [CrossRef]
- Zucca, S.; Gagliardi, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. RNA-Seq profiling in peripheral blood mononuclear cells of amyotrophic lateral sclerosis patients and controls. Sci. Data 2019, 6, 190006. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’aYan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Differentially Expressed Genes | ||
|---|---|---|
| Protein Coding | Non-Coding | |
| Upregulated | 1455 | 268 |
| Downregulated | 354 | 138 |
| Subtotal | 1809 | 406 |
| Total | 2215 | |
| Gene | Log2FC | p-Value | Adj p-Value | Protein | Function in Muscles |
|---|---|---|---|---|---|
| MYH1 | −1.700586668 | 0.000746513 | 0.004671804 | Myosin Heavy Chain 1 | Major contractile protein, it converts chemical energy into mechanical energy through the hydrolysis of ATP. |
| MYH14 | −1.122451416 | 1.52 × 10−9 | 0.000000088 | Myosin Heavy Chain 14 | Represents a conventional non-muscle myosin. It should not be confused with the unconventional myosin−14 (MYO14). |
| MYH11 | 1.028496227 | 0.0174439 | 0.059487717 | Myosin Heavy Chain 11 | Smooth muscle myosin. It functions as a major contractile protein, converting chemical energy into mechanical energy through the hydrolysis of ATP. |
| MYH8 | 2.268592775 | 0.001163423 | 0.006689683 | Myosin Heavy Chain 8 | Class II or conventional myosin heavy chains. Functions in skeletal muscle contraction. |
| MYH3 | 2.926975986 | 5.90 × 10−8 | 0.000001801 | Myosin Heavy Chain 3 | Major contractile protein actin-associated. |
| ACTA2 | 1.032325389 | 0.000988763 | 0.005874898 | Actin Alpha 2 | Smooth muscle actin that is involved in vascular contractility and blood pressure homeostasis. |
| ACTN1 | 1.147716074 | 1.29 × 10−5 | 0.000158151 | Actinin Alpha 1 | Skeletal muscle isoforms are localized to the Z-disc. They help anchor the myofibrillar actin filaments. |
| Code | Dob | Diagnosis | Age at Muscle Biopsy (Mnts) | Gene Variants | Age of Onset (Mnts) | Disease Duration (Yrs) | Ambulatory or Not | Age at Evaluation (Yrs) | NSS | 6MWT (cm) | FVC (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B1 | 1997 | Bmd | 29 | Ex. Del. 47–52 | 14 | 26 | Amb. | 17 | 34 | 609 | 1 |
| B2 | 1997 | Bmd | 32 | Ex. Del 48 | 18 | 25 | Amb. | 17 | 34 | 614 | 0.93 |
| B3 | 2000 | Bmd | 46 | Ex. Del 45–47 | 17 | 22 | Amb. | 17 | 29/34 | 475 | 1 |
| B4 | 2001 | Bmd | 122 | Ex. Del. 45–47 | 23 | 21 | Amb. | 16 | 33 | 614 | 1 |
| B5 | 2005 | Bmd | 92 | Ex 6: c.358G > Tp.120Vl > Phe | 72 | 13 | Amb. | 16 | 34 | 753 | 1 |
| B6 | 2006 | Bmd | 95 | Ex. Del. 48–50 | 22 | 17 | Amb. | 14 | 34 | 682 | 1 |
| D1 | 2002 | Dmd | 28 | Ex. Del 46–49 | 19 | 21 | Not amb. from 16 yrs | 15 | 21 | 232 | 1 |
| D2 | 1993 | Dmd | 136 | Ex. Dup. 19 | 49 | 27 | Amb. + deficit* | 17 | 21 | 360 | NA |
| D3 | 2006 | Dmd | 20 | Stop point mut. Ex. 70: c.10141C > Tp.Arg3381X | 13 | 18 | Amb. + deficit* | 17 | 21 | 449 | 0.78 |
| D4 | 2006 | Dmd | 17 | Ex. Del. 46–52 | 15 | 16 | Not amb. from 13 yrs | 12 | 2 | 246 | 0.77 |
| D5 | 2005 | Dmd | 36 | Ex. Dup. 1–9 | 19 | 17 | Not amb. from 16 yrs | 7 | 15 | 475 | NA |
| D6 | 2004 | Dmd | 56 | Ex. Del. 3–27 | 23 | 18 | Amb. + deficit* | 8 | 31 | 514 | NA |
| D7 | 2008 | Dmd | 17 | Ex. Del. 3–26 | 3 | 16 | Amb. + deficit* | 16 | 22 | 359 | 1 |
| D8 | 2003 | Dmd | 86 | Point mutation Int.5: c.358–1G > A | 29 | 19 | Not amb. from 14 yrs | 13 | 18 | 246 | 0.95 |
| D9 | 2008 | Dmd | 27 | Ex. Del. 8–12 | 25 | 14 | Not amb. from 15 yrs | 11 | 11 | 302 | NA |
| D10 | 2006 | Dmd | 57 | Ex. Del. 49–54 | 60 | 13 | Not amb. from 14 yrs | 13 | 12 | 247 | NA |
| D11 | 2008 | Dmd | 54 | Ex. Dup. 62–67 | 53 | 12 | Not amb. from 13 yrs | 12 | 13 | 186 | 1 |
| D12 | 2008 | Dmd | 56 | Ex. Del. 3–29 | 36 | 13 | Not amb. from 7 yrs | 5 | 14 | 201 | NA |
| GENE SYMBOL | PRIMER FORWARD | PRIMER REVERSE | AMPLICON LENGTHS |
|---|---|---|---|
| MKX | CCTTACAGGCATGAAGGGGG | GTGGTGCTTTCCAACAGTGC | 73 bp |
| CADPS | TGCAGAAAATGTAGGCCGGT | CGTGGTGCTCCTCATTTTGC | 107 bp |
| THBS4 | AACCCAGAGCTGAACCCTTG | ACACACATGTCACATCCCCC | 73 bp |
| FKBP5 | GGACTGGACAGTGCCAATGA | GGCACATGGAGATCTGCAGT | 151 bp |
| UCP3 | AGCCCCCTCGACTGTATGAT | CCCAAACGCAAAAAGGAGGG | 89 bp |
| COL25A1 | CCAAAATCGCCTCTCCCGAT | TTGGCACAGATTGTCCCAGT | 55 bp |
| COL19A1 | CCTTACAGGCATGAAGGGGG | TCCCATGGAGCCCTTGTTTC | 64 bp |
| COL1A1 | CCTGGGGCAAGACAGTGATT | TCGAAGCCGAATTCCTGGTC | 109 bp |
| COL1A2 | CTGGTAGTCGTGGTGCAAGT | AGGACCTTCTTTTCCAGCGG | 143 bp |
| GAPDH (HG *) | AGGAGTAAGACCCCTGGACC | GGGGAGATTCAGTGTGGTGG | 113 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzo, B.; Dragoni, F.; Dainesi, M.I.; Di Gerlando, R.; Minucchi, E.; Berardinelli, A.L.; Gagliardi, S. Comparative Transcriptomic Profiling in Patients Affected by Duchenne and Becker Muscular Dystrophies: A Focus on ECM Genes Dysregulation. Int. J. Mol. Sci. 2025, 26, 6594. https://doi.org/10.3390/ijms26146594
Rizzo B, Dragoni F, Dainesi MI, Di Gerlando R, Minucchi E, Berardinelli AL, Gagliardi S. Comparative Transcriptomic Profiling in Patients Affected by Duchenne and Becker Muscular Dystrophies: A Focus on ECM Genes Dysregulation. International Journal of Molecular Sciences. 2025; 26(14):6594. https://doi.org/10.3390/ijms26146594
Chicago/Turabian StyleRizzo, Bartolo, Francesca Dragoni, Maria Irene Dainesi, Rosalinda Di Gerlando, Evelyne Minucchi, Angela Lucia Berardinelli, and Stella Gagliardi. 2025. "Comparative Transcriptomic Profiling in Patients Affected by Duchenne and Becker Muscular Dystrophies: A Focus on ECM Genes Dysregulation" International Journal of Molecular Sciences 26, no. 14: 6594. https://doi.org/10.3390/ijms26146594
APA StyleRizzo, B., Dragoni, F., Dainesi, M. I., Di Gerlando, R., Minucchi, E., Berardinelli, A. L., & Gagliardi, S. (2025). Comparative Transcriptomic Profiling in Patients Affected by Duchenne and Becker Muscular Dystrophies: A Focus on ECM Genes Dysregulation. International Journal of Molecular Sciences, 26(14), 6594. https://doi.org/10.3390/ijms26146594