_Kim.png)
Yolk Sac Elements in Tumors Derived from Pluripotent Stem Cells: Borrowing Knowledge from Human Germ Cell Tumors

 , , ,
, , ,  and
and
Abstract
1. Introduction
1.1. Safety Concerns Regarding PSC-Derived Therapies
1.2. The In Vivo Teratoma Assay: The Current Method to Asses Malignant Potential of PSC
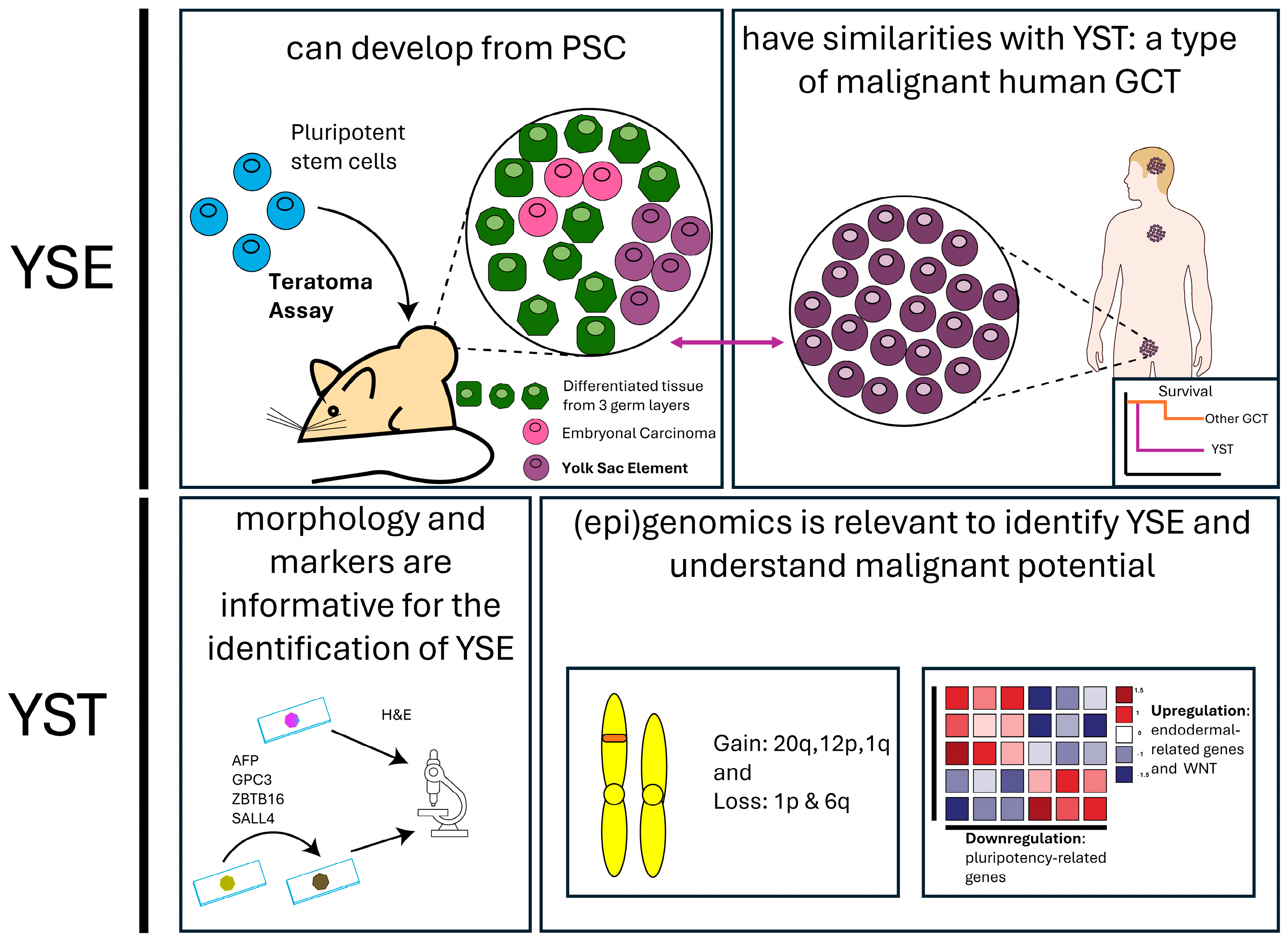
1.3. Borrowing Knowledge of Human Germ Cell Tumors for a Better Understanding of PSC-Derived Tumors
Prepubertal (Type I). GCTs predominantly occurring in prepubertal patients. Mostly with the histology of pure (benign) teratoma but can also occur as pure yolk sac tumor or in the combination of teratoma and/or yolk sac tumor (being malignant).
Post-pubertal (Type II). GCTs occurring in post-pubertal patients. These malignant tumors have an origin in a precursor lesion called germ cell neoplasia in situ and can occur in a more diverse array of histological subtypes, being subdivided into seminoma-like and nonseminoma (embryonal carcinoma, yolk sac tumor, choriocarcinoma, teratoma, and mixed).
2. Histopathological Similarities Between GCT and Tumors Derived from PSC Relevant to Malignancy
2.1. Histopathology of Human GCT
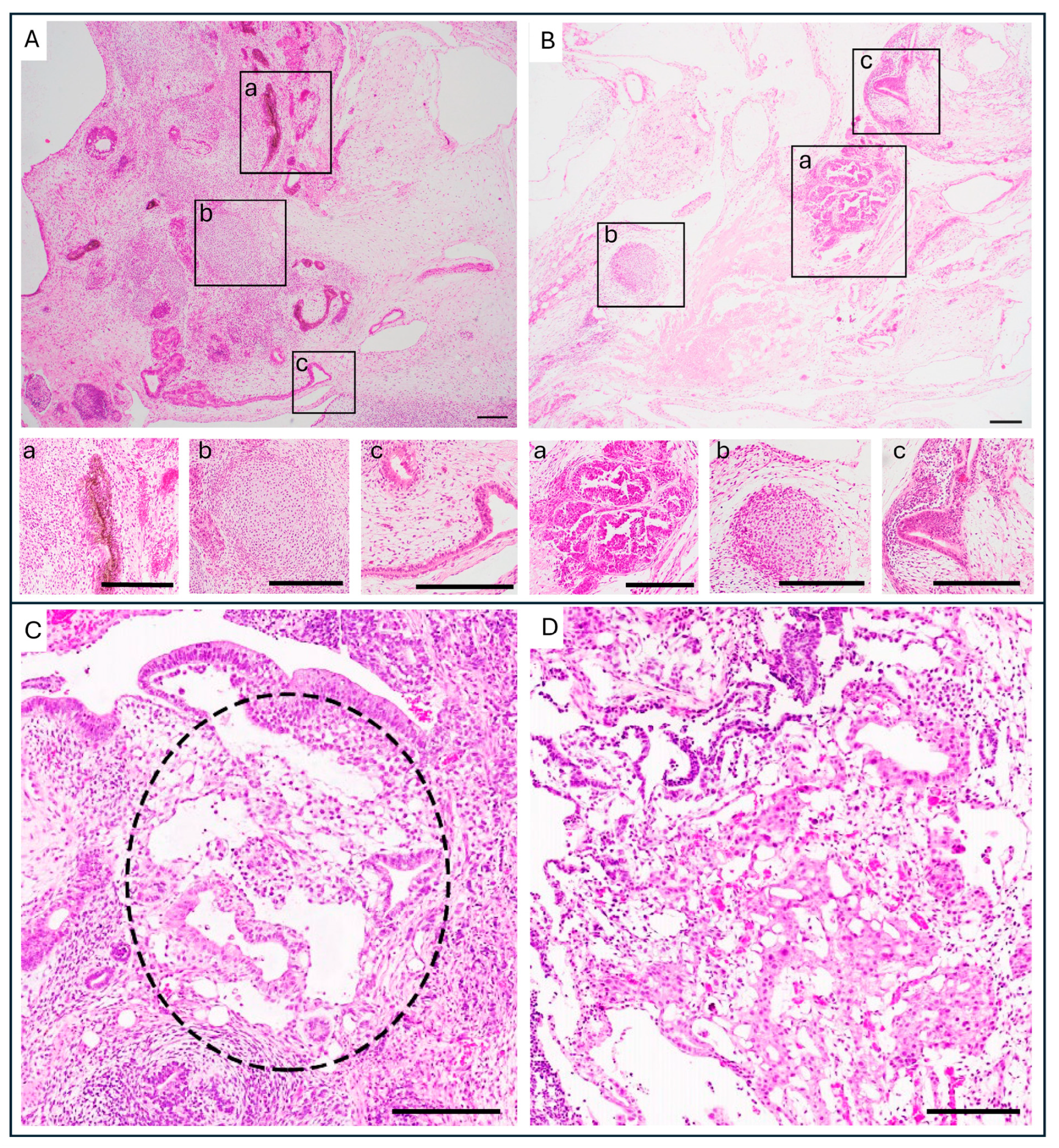
2.2. Histopathology of Tumors Derived from PSC Compared to GCT and the Risks YSE Pose
2.3. Immunohistochemistry of YST and YSE in Teratomas Derived from PSC
3. Genomics and Pathways of Malignancy Found in Yolk Sac Tumor
3.1. Chromosomal Aberrations Found in YST
3.2. YST Gene Expression
3.3. Mechanisms Related to Malignancy: YST and WNT Signaling
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PSC | Pluripotent Stem Cell |
| iPSC | Induced Pluripotent Stem Cell |
| GCT | Germ Cell Tumor |
| EC | Embryonal Carcinoma |
| YST | Yolk Sac Tumor |
| YSE | Yolk Sac Element |
| WHO | World Health Organization |
| AFP | Alpha-Fetoprotein |
| OCT4 | Octamer-binding Transcription factor 4 |
| GPC3 | Glypican-3 |
| ZBTB16 | Zinc finger and BTB domain-containing protein 16 |
| SALL4 | Sal-like protein 4 |
| HNF1β | Hepatocyte Nuclear Factor 1 homeobox B |
| FOXA2 | Forkhead Box Protein A2 |
| ISCI | International Stem Cell Initiative |
| BMP | Bone Morphogenetic Protein |
| APC | Adenomatous Polyposis Coli |
References
- Kirkeby, A.; Main, H.; Carpenter, M. Pluripotent Stem-Cell-Derived Therapies in Clinical Trial: A 2025 Update. Cell Stem Cell 2025, 32, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Barbaric, I.; Benvenisty, N.; Draper, J.S.; Ludwig, T.; Merkle, F.T.; Sato, Y.; Spits, C.; Stacey, G.N.; Wang, H.; et al. The Consequences of Recurrent Genetic and Epigenetic Variants in Human Pluripotent Stem Cells. Cell Stem Cell 2022, 29, 1624–1636. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Lezmi, E.; Benvenisty, N. The Tumorigenic Potential of Human Pluripotent Stem Cells. Stem Cells Transl. Med. 2022, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy Induces Profound Changes in Gene Expression, Proliferation and Tumorigenicity of Human Pluripotent Stem Cells. Nat. Commun. 2014, 5, 4825. [Google Scholar] [CrossRef]
- Hillenius, S.; Montilla-Rojo, J.; Eleveld, T.F.; Salvatori, D.C.F.; Looijenga, L.H.J. Safety Issues Related to Pluripotent Stem Cell-Based Therapies: Tumour Risk. In Pluripotent Stem Cell Therapy for Diabetes; Piemonti, L., Odorico, J., Kieffer, T.J., Sordi, V., de Koning, E., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 419–457. ISBN 978-3-031-41943-0. [Google Scholar]
- Krivec, N.; Ghosh, M.S.; Spits, C. Gains of 20q11.21 in Human Pluripotent Stem Cells: Insights from Cancer Research. Stem Cell Rep. 2024, 19, 11–27. [Google Scholar] [CrossRef]
- Stavish, D.; Price, C.J.; Gelezauskaite, G.; Alsehli, H.; Leonhard, K.A.; Taapken, S.M.; McIntire, E.M.; Laing, O.; James, B.M.; Riley, J.J.; et al. Feeder-Free Culture of Human Pluripotent Stem Cells Drives MDM4-Mediated Gain of Chromosome 1q. Stem Cell Rep. 2024, 19, 1217–1232. [Google Scholar] [CrossRef]
- Amir, H.; Touboul, T.; Sabatini, K.; Chhabra, D.; Garitaonandia, I.; Loring, J.F.; Morey, R.; Laurent, L.C. Spontaneous Single-Copy Loss of TP53 in Human Embryonic Stem Cells Markedly Increases Cell Proliferation and Survival. Stem Cells Dayt. Ohio 2017, 35, 872–885. [Google Scholar] [CrossRef]
- Garitaonandia, I.; Amir, H.; Boscolo, F.S.; Wambua, G.K.; Schultheisz, H.L.; Sabatini, K.; Morey, R.; Waltz, S.; Wang, Y.-C.; Tran, H.; et al. Increased Risk of Genetic and Epigenetic Instability in Human Embryonic Stem Cells Associated with Specific Culture Conditions. PLoS ONE 2015, 10, e0118307. [Google Scholar] [CrossRef]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human Pluripotent Stem Cells Recurrently Acquire and Expand Dominant Negative P53 Mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Montilla-Rojo, J.; Eleveld, T.F.; van Soest, M.; Hillenius, S.; Timmerman, D.M.; Gillis, A.J.M.; Roelen, B.A.J.; Mummery, C.L.; Looijenga, L.H.J.; Salvatori, D.C.F. Depletion of TP53 in Human Pluripotent Stem Cells Triggers Malignant-Like Behavior. Adv. Biol. 2025, 9, e2400538. [Google Scholar] [CrossRef] [PubMed]
- International Society for Stem Cell Research Standards for Human Stem Cell Use in Research. 2023. Available online: https://www.isscr.org/standards-document (accessed on 21 November 2023).
- Damjanov, I.; Andrews, P.W. Teratomas Produced from Human Pluripotent Stem Cells Xenografted into Immunodeficient Mice—A Histopathology Atlas. Int. J. Dev. Biol. 2016, 60, 337–419. [Google Scholar] [CrossRef] [PubMed]
- Gertow, K.; Przyborski, S.; Loring, J.F.; Auerbach, J.M.; Epifano, O.; Otonkoski, T.; Damjanov, I.; Ahrlund-Richter, L. Isolation of Human Embryonic Stem Cell-Derived Teratomas for the Assessment of Pluripotency. Curr. Protoc. Stem Cell Biol. 2007, 3, 1B. 4.1–1B. 4.29. [Google Scholar] [CrossRef]
- Damjanov, I.; Andrews, P.W. The Terminology of Teratocarcinomas and Teratomas. Nat. Biotechnol. 2007, 25, 1212. [Google Scholar] [CrossRef]
- Allison, T.F.; Andrews, P.W.; Avior, Y.; Barbaric, I.; Benvenisty, N.; Bock, C.; Brehm, J.; Brüstle, O.; Damjanov, I.; Elefanty, A.; et al. Assessment of Established Techniques to Determine Developmental and Malignant Potential of Human Pluripotent Stem Cells. Nat. Commun. 2018, 9, 1925. [Google Scholar] [CrossRef]
- Müller, F.-J.; Schuldt, B.M.; Williams, R.; Mason, D.; Altun, G.; Papapetrou, E.P.; Danner, S.; Goldmann, J.E.; Herbst, A.; Schmidt, N.O.; et al. A Bioinformatic Assay for Pluripotency in Human Cells. Nat. Methods 2011, 8, 315–317. [Google Scholar] [CrossRef]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of Human ES and IPS Cell Variation Enable High-Throughput Characterization of Pluripotent Cell Lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef]
- Tsankov, A.M.; Akopian, V.; Pop, R.; Chetty, S.; Gifford, C.A.; Daheron, L.; Tsankova, N.M.; Meissner, A. A QPCR ScoreCard Quantifies the Differentiation Potential of Human Pluripotent Stem Cells. Nat. Biotechnol. 2015, 33, 1182–1192. [Google Scholar] [CrossRef]
- Montilla-Rojo, J.; Bialecka, M.; Wever, K.E.; Mummery, C.L.; Looijenga, L.H.J.; Roelen, B.A.J.; Salvatori, D.C.F. Teratoma Assay for Testing Pluripotency and Malignancy of Stem Cells: Insufficient Reporting and Uptake of Animal-Free Methods—A Systematic Review. Int. J. Mol. Sci. 2023, 24, 3879. [Google Scholar] [CrossRef]
- Bouma, M.J.; Van Iterson, M.; Janssen, B.; Mummery, C.L.; Salvatori, D.C.F.; Freund, C. Differentiation-Defective Human Induced Pluripotent Stem Cells Reveal Strengths and Limitations of the Teratoma Assay and In Vitro Pluripotency Assays. Stem Cell Rep. 2017, 8, 1340–1353. [Google Scholar] [CrossRef]
- Kilic, I.; Idrees, M.T. The 2022 World Health Organization Classification of Germ Cell Tumors and Updates of American Joint Committee for Cancer Tumor Staging Classification. Diagn. Histopathol. 2023, 29, 259–268. [Google Scholar] [CrossRef]
- Organisation Mondiale de la Santé; Centre International de Recherche sur le Cancer (Eds.) Urinary and Male Genital Tumours. In World Health Organization Classification of Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2022; ISBN 978-92-832-4512-4. [Google Scholar]
- Nogales, F.F.; Jimenez, R.E. (Eds.) Pathology and Biology of Human Germ Cell Tumors; Springer: Berlin, Germany, 2017; ISBN 978-3-662-53773-2. [Google Scholar]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human Germ Cell Tumours from a Developmental Perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Matin, M.M.; Bahrami, A.R.; Damjanov, I.; Gokhale, P.; Draper, J.S. Embryonic Stem (ES) Cells and Embryonal Carcinoma (EC) Cells: Opposite Sides of the Same Coin. Biochem. Soc. Trans. 2005, 33, 1526–1530. [Google Scholar] [CrossRef]
- Solter, D. From Teratocarcinomas to Embryonic Stem Cells and beyond: A History of Embryonic Stem Cell Research. Nat. Rev. Genet. 2006, 7, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.J.; Ulbright, T.M.; Pera, M.F.; Looijenga, L.H.J. Lessons from Human Teratomas to Guide Development of Safe Stem Cell Therapies. Nat. Biotechnol. 2012, 30, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Ulbright, T.M. Germ Cell Tumors of the Gonads: A Selective Review Emphasizing Problems in Differential Diagnosis, Newly Appreciated, and Controversial Issues. Mod. Pathol. 2005, 18, S61–S79. [Google Scholar] [CrossRef]
- Nogales, F.F.; Preda, O.; Nicolae, A. Yolk Sac Tumours Revisited. A Review of Their Many Faces and Names: Update on Nomenclature of Yolk Sac Tumours. Histopathology 2012, 60, 1023–1033. [Google Scholar] [CrossRef]
- Bialecka, M.; Montilla-Rojo, J.; Roelen, B.A.J.; Gillis, A.J.; Looijenga, L.H.J.; Salvatori, D.C.F. Humanised Mice and Immunodeficient Mice (NSG) Are Equally Sensitive for Prediction of Stem Cell Malignancy in the Teratoma Assay. Int. J. Mol. Sci. 2022, 23, 4680. [Google Scholar] [CrossRef]
- Griscelli, F.; Féraud, O.; Oudrhiri, N.; Gobbo, E.; Casal, I.; Chomel, J.-C.; Biéche, I.; Duvillard, P.; Opolon, P.; Turhan, A.G.; et al. Malignant Germ Cell–Like Tumors, Expressing Ki-1 Antigen (CD30), Are Revealed during In Vivo Differentiation of Partially Reprogrammed Human-Induced Pluripotent Stem Cells. Am. J. Pathol. 2012, 180, 2084–2096. [Google Scholar] [CrossRef]
- Salvatori, D.C.F.; Dorssers, L.C.J.; Gillis, A.J.M.; Perretta, G.; Van Agthoven, T.; Gomes Fernandes, M.; Stoop, H.; Prins, J.-B.; Oosterhuis, J.W.; Mummery, C.; et al. The MicroRNA-371 Family as Plasma Biomarkers for Monitoring Undifferentiated and Potentially Malignant Human Pluripotent Stem Cells in Teratoma Assays. Stem Cell Rep. 2018, 11, 1493–1505. [Google Scholar] [CrossRef]
- Bernbeck, B.; Schneider, D.T.; Bernbeck, B.; Koch, S.; Teske, C.; Lentrodt, J.; Harms, D.; Göbel, U.; Calaminus, G.; on Behalf of the MAKEI-Study Group. Germ Cell Tumors of the Head and Neck: Report from the MAKEI Study Group. Pediatr. Blood Cancer 2009, 52, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.-M.; Bilen, M.A.; Hess, K.R.; Broaddus, R.R.; Kopetz, S.; Wei, C.; Pagliaro, L.C.; Karam, J.A.; Ward, J.F.; Wood, C.G.; et al. Intratumoral Heterogeneity: Role of Differentiation in a Potentially Lethal Phenotype of Testicular Cancer. Cancer 2016, 122, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.B.; Srivastava, A.; Lanzotti, N.J.; Ellis, J.L.; Carlo, H.N.D.; Gearhart, J.P.; Bowen, D.K.; Gupta, M.; Picken, M.M.; Gupta, G.N.; et al. Variations in Germ Cell Tumor Histology by Age and Implications for Cancer-Specific Survival among Pediatric and Adult Males: A Population-Based Study. Urol. Oncol. Semin. Orig. Investig. 2024, 42, 292.e17–292.e26. [Google Scholar] [CrossRef]
- International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: A Prognostic Factor-Based Staging System for Metastatic Germ Cell Cancers. J. Clin. Oncol. 1997, 15, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, S.; Sauvé, N.; Collette, L.; Daugaard, G.; de Wit, R.; Albany, C.; Tryakin, A.; Fizazi, K.; Stahl, O.; Gietema, J.A.; et al. Predicting Outcomes in Men With Metastatic Nonseminomatous Germ Cell Tumors (NSGCT): Results From the IGCCCG Update Consortium. J. Clin. Oncol. 2021, 39, 1563–1574. [Google Scholar] [CrossRef]
- Looijenga, L.H.J.; Stoop, H.; de Leeuw, H.P.J.C.; de Gouveia Brazao, C.A.; Gillis, A.J.M.; van Roozendaal, K.E.P.; van Zoelen, E.J.J.; Weber, R.F.A.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) Identifies Cells with Pluripotent Potential in Human Germ Cell Tumors. Cancer Res. 2003, 63, 2244–2250. [Google Scholar]
- Ulbright, T.M.; Tickoo, S.K.; Berney, D.M.; Srigley, J.R. Best Practices Recommendations in the Application of Immunohistochemistry in Testicular Tumors: Report From the International Society of Urological Pathology Consensus Conference. Am. J. Surg. Pathol. 2014, 38, e50–e59. [Google Scholar] [CrossRef]
- Zynger, D.L.; Dimov, N.D.; Luan, C.; Teh, B.T.; Yang, X.J. Glypican 3: A Novel Marker in Testicular Germ Cell Tumors. Am. J. Surg. Pathol. 2006, 30, 1570–1575. [Google Scholar] [CrossRef]
- Xiao, G.-Q.; Li, F.; Unger, P.D.; Katerji, H.; Yang, Q.; McMahon, L.; Burstein, D.E. ZBTB16: A Novel Sensitive and Specific Biomarker for Yolk Sac Tumor. Mod. Pathol. 2016, 29, 591–598. [Google Scholar] [CrossRef]
- Cao, D.; Li, J.; Guo, C.C.; Allan, R.W.; Humphrey, P.A. SALL4 Is a Novel Diagnostic Marker for Testicular Germ Cell Tumors. Am. J. Surg. Pathol. 2009, 33, 1065–1077. [Google Scholar] [CrossRef]
- Nogales, F.F.; Quiñonez, E.; López-Marín, L.; Dulcey, I.; Preda, O. A Diagnostic Immunohistochemical Panel for Yolk Sac (Primitive Endodermal) Tumours Based on an Immunohistochemical Comparison with the Human Yolk Sac. Histopathology 2014, 65, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Luo, R.; Sun, B.; Zhao, J.; Xu, Q.; Feng, S.; Chen, X.; Wang, C. SALL4 Is a Useful Marker for Pediatric Yolk Sac Tumors. Pediatr. Surg. Int. 2020, 36, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Bing, Z.; Pasha, T.; Tomaszewski, J.E.; Zhang, P. CDX2 Expression in Yolk Sac Component of Testicular Germ Cell Tumors. Int. J. Surg. Pathol. 2009, 17, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Fankhauser, C.; Hermanns, T.; Beyer, J.; Christiansen, A.; Moch, H.; Bode, P.K. HNF1β Is a Sensitive and Specific Novel Marker for Yolk Sac Tumor: A Tissue Microarray Analysis of 601 Testicular Germ Cell Tumors. Mod. Pathol. 2020, 33, 2354–2360. [Google Scholar] [CrossRef]
- Ricci, C.; Ambrosi, F.; Franceschini, T.; Giunchi, F.; Di Filippo, G.; Franchini, E.; Massari, F.; Mollica, V.; Tateo, V.; Bianchi, F.M.; et al. FoxA2 Is a Reliable Marker for the Diagnosis of Yolk Sac Tumour Postpubertal-type. Histopathology 2023, 83, 465–476. [Google Scholar] [CrossRef]
- Wruck, W.; Bremmer, F.; Kotthoff, M.; Fichtner, A.; Skowron, M.A.; Schönberger, S.; Calaminus, G.; Vokuhl, C.; Pfister, D.; Heidenreich, A.; et al. The Pioneer and Differentiation Factor FOXA2 Is a Key Driver of Yolk-sac Tumour Formation and a New Biomarker for Paediatric and Adult Yolk-sac Tumours. J. Cell. Mol. Med. 2021, 25, 1394–1405. [Google Scholar] [CrossRef]
- Xiao, G.-Q.; Priemer, D.S.; Wei, C.; Aron, M.; Yang, Q.; Idrees, M.T. ZBTB16 Is a Sensitive and Specific Marker in Detection of Metastatic and Extragonadal Yolk Sac Tumour. Histopathology 2017, 71, 562–569. [Google Scholar] [CrossRef]
- Osman, H.; Cheng, L.; Ulbright, T.M.; Idrees, M.T. The Utility of CDX2, GATA3, and DOG1 in the Diagnosis of Testicular Neoplasms: An Immunohistochemical Study of 109 Cases. Hum. Pathol. 2016, 48, 18–24. [Google Scholar] [CrossRef]
- Zynger, D.L.; McCallum, J.C.; Luan, C.; Chou, P.M.; Yang, X.J. Glypican 3 Has a Higher Sensitivity than Alpha-Fetoprotein for Testicular and Ovarian Yolk Sac Tumour: Immunohistochemical Investigation with Analysis of Histological Growth Patterns. Histopathology 2010, 56, 750–757. [Google Scholar] [CrossRef]
- Looijenga, L.H.J.; Rosenberg, C.; van Gurp, R.J.H.L.M.; Geelen, E.; van Echten-Arends, J.; de Jong, B.; Mostert, M.; Wolter Oosterhuis, J. Comparative Genomic Hybridization of Microdissected Samples from Different Stages in the Development of a Seminoma and a Non-Seminoma. J. Pathol. 2000, 191, 187–192. [Google Scholar] [CrossRef]
- Palmer, R.D.; Foster, N.A.; Vowler, S.L.; Roberts, I.; Thornton, C.M.; Hale, J.P.; Schneider, D.T.; Nicholson, J.C.; Coleman, N. Malignant Germ Cell Tumours of Childhood: New Associations of Genomic Imbalance. Br. J. Cancer 2007, 96, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, D.M.; Eleveld, T.F.; Sriram, S.; Dorssers, L.C.J.; Gillis, A.J.M.; Schmidtova, S.; Kalavska, K.; van de Werken, H.J.G.; Oing, C.; Honecker, F.; et al. Chromosome 3p25.3 Gain Is Associated With Cisplatin Resistance and Is an Independent Predictor of Poor Outcome in Male Malignant Germ Cell Tumors. J. Clin. Oncol. 2022, 40, 3077–3087. [Google Scholar] [CrossRef]
- Takami, H.; Satomi, K.; Fukuoka, K.; Nakamura, T.; Tanaka, S.; Mukasa, A.; Saito, N.; Suzuki, T.; Yanagisawa, T.; Sugiyama, K.; et al. Distinct Patterns of Copy Number Alterations May Predict Poor Outcome in Central Nervous System Germ Cell Tumors. Sci. Rep. 2023, 13, 15760. [Google Scholar] [CrossRef]
- Eakin, G.S.; Hadjantonakis, A.-K.; Papaioannou, V.E.; Behringer, R.R. Developmental Potential and Behavior of Tetraploid Cells in the Mouse Embryo. Dev. Biol. 2005, 288, 150–159. [Google Scholar] [CrossRef]
- Eakin, G.S.; Behringer, R.R. Tetraploid Development in the Mouse. Dev. Dyn. 2003, 228, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Xu, D.; Zhang, M.; Chen, Z.; Cheng, L.; Du, S.; Lu, M.; Zhou, T.; Li, R.; Bai, F.; et al. Aneuploid Embryonic Stem Cells Drive Teratoma Metastasis. Nat. Commun. 2024, 15, 1087. [Google Scholar] [CrossRef]
- Montilla-Rojo, J.; Hillenius, S.; Eleveld, T.F.; Salvatori, D.C.F.; Looijenga, L.H.J. Chromosome-Specific Aberrations, Rather than General Aneuploidy, May Drive Mouse Embryonic Stem Cell-Derived Teratoma Metastasis. Stem Cell Res. Ther. 2025, 16, 181. [Google Scholar] [CrossRef]
- Korkola, J.E.; Houldsworth, J.; Dobrzynski, D.; Olshen, A.B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.K. Gene Expression-Based Classification of Nonseminomatous Male Germ Cell Tumors. Oncogene 2005, 24, 5101–5107. [Google Scholar] [CrossRef]
- Kubota, Y.; Seki, M.; Kawai, T.; Isobe, T.; Yoshida, M.; Sekiguchi, M.; Kimura, S.; Watanabe, K.; Sato-Otsubo, A.; Yoshida, K.; et al. Comprehensive Genetic Analysis of Pediatric Germ Cell Tumors Identifies Potential Drug Targets. Commun. Biol. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Killian, J.K.; Dorssers, L.C.J.; Trabert, B.; Gillis, A.J.M.; Cook, M.B.; Wang, Y.; Waterfall, J.J.; Stevenson, H.; Smith, W.I.; Noyes, N.; et al. Imprints and DPPA3 Are Bypassed during Pluripotency- and Differentiation-Coupled Methylation Reprogramming in Testicular Germ Cell Tumors. Genome Res. 2016, 26, 1490–1504. [Google Scholar] [CrossRef]
- Xu, L.; Pierce, J.L.; Sanchez, A.; Chen, K.S.; Shukla, A.A.; Fustino, N.J.; Stuart, S.H.; Bagrodia, A.; Xiao, X.; Guo, L.; et al. Integrated Genomic Analysis Reveals Aberrations in WNT Signaling in Germ Cell Tumors of Childhood and Adolescence. Nat. Commun. 2023, 14, 2636. [Google Scholar] [CrossRef]
- Fritsch, M.K.; Schneider, D.T.; Schuster, A.E.; Murdoch, F.E.; Perlman, E.J. Activation of Wnt/β-Catenin Signaling in Distinct Histologic Subtypes of Human Germ Cell Tumors. Pediatr. Dev. Pathol. 2006, 9, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.-D.; Diao, L.-T.; Xu, H.; Xiao, Z.-D.; Li, J.-H.; Zhou, H.; Qu, L.-H. β-Catenin/LEF1 Transactivates the MicroRNA-371-373 Cluster That Modulates the Wnt/β-Catenin-Signaling Pathway. Oncogene 2012, 31, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.M.; Stoop, H.; Nagel, R.; Liu, Y.-P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates MiRNA-372 and MiRNA-373 As Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef]
- Gillis, A.; Stoop, H.; Hersmus, R.; Oosterhuis, J.; Sun, Y.; Chen, C.; Guenther, S.; Sherlock, J.; Veltman, I.; Baeten, J.; et al. High-Throughput MicroRNAome Analysis in Human Germ Cell Tumours. J. Pathol. 2007, 213, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Looijenga, L.H.J.; Gillis, A.J.M.; Stoop, H.; Hersmus, R.; Oosterhuis, J.W. Relevance of MicroRNAs in Normal and Malignant Development, Including Human Testicular Germ Cell Tumours. Int. J. Androl. 2007, 30, 304–315. [Google Scholar] [CrossRef]
- Murray, M.J.; Halsall, D.J.; Hook, C.E.; Williams, D.M.; Nicholson, J.C.; Coleman, N. Identification of MicroRNAs from the MiR-371∼373 and MiR-302 Clusters as Potential Serum Biomarkers of Malignant Germ Cell Tumors. Am. J. Clin. Pathol. 2011, 135, 119–125. [Google Scholar] [CrossRef]
- Dieckmann, K.-P.; Radtke, A.; Geczi, L.; Matthies, C.; Anheuser, P.; Eckardt, U.; Sommer, J.; Zengerling, F.; Trenti, E.; Pichler, R.; et al. Serum Levels of MicroRNA-371a-3p (M371 Test) as a New Biomarker of Testicular Germ Cell Tumors: Results of a Prospective Multicentric Study. J. Clin. Oncol. 2019, 37, 1412–1423. [Google Scholar] [CrossRef]
- Schönberger, S.; Mohseni, M.M.; Ellinger, J.; Tran, G.V.Q.; Becker, M.; Claviez, A.; Classen, C.-F.; Hermes, B.; Driever, P.H.; Jorch, N.; et al. MicroRNA-Profiling of MiR-371~373- and MiR-302/367-Clusters in Serum and Cerebrospinal Fluid Identify Patients with Intracranial Germ Cell Tumors. J. Cancer Res. Clin. Oncol. 2023, 149, 791–802. [Google Scholar] [CrossRef]
- Capurro, M.I.; Xiang, Y.-Y.; Lobe, C.; Filmus, J. Glypican-3 Promotes the Growth of Hepatocellular Carcinoma by Stimulating Canonical Wnt Signaling. Cancer Res. 2005, 65, 6245–6254. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Kim, H.; Feng, M.; Phung, Y.; Xavier, C.P.; Rubin, J.S.; Ho, M. Inactivation of Wnt Signaling by a Human Antibody That Recognizes the Heparan Sulfate Chains of Glypican-3 for Liver Cancer Therapy. Hepatol. Baltim. Md 2014, 60, 576–587. [Google Scholar] [CrossRef]
- Liu, Q.; Song, Q.; Luo, C.; Wei, J.; Xu, Y.; Zhao, L.; Wang, Y. A Novel Bispecific Antibody as an Immunotherapeutic Agent in Hepatocellular Carcinoma. Mol. Immunol. 2023, 162, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Yang, T.; Li, H.; Li, S.; Li, T.; Ye, H.; Lu, M.; Chu, X.; Zhong, G.; Zhou, J.; et al. Frequently Expressed Glypican-3 as a Promising Novel Therapeutic Target for Osteosarcomas. Cancer Sci. 2022, 113, 3618–3632. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, X.; Lei, Y.; Wang, G.; Liu, M. Glypican-3: A Novel and Promising Target for the Treatment of Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 824208. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, N.; Zhang, Y.-F.; Fu, H.; Feng, M.; Schneider, D.; Su, L.; Wu, X.; Zhou, J.; Mackay, S.; et al. Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice. Gastroenterology 2020, 158, 2250–2265. [Google Scholar] [CrossRef]
- Okpanyi, V.; Schneider, D.T.; Zahn, S.; Sievers, S.; Calaminus, G.; Nicholson, J.C.; Palmer, R.D.; Leuschner, I.; Borkhardt, A.; Schönberger, S. Analysis of the Adenomatous Polyposis Coli (APC) Gene in Childhood and Adolescent Germ Cell Tumors. Pediatr. Blood Cancer 2011, 56, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of Pluripotency in Human and Mouse Embryonic Stem Cells through Activation of Wnt Signaling by a Pharmacological GSK-3-Specific Inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef]
- Sokol, S.Y. Maintaining Embryonic Stem Cell Pluripotency with Wnt Signaling. Dev. Camb. Engl. 2011, 138, 4341–4350. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Marker | Cellular Localization | Expression in YSE | Expression in YST |
|---|---|---|---|
| AFP | Cytoplasmic | Reported [15,32,34] | [31,42,45,46,47,49,50,51,52] |
| CDX2 | Nuclear | Not reported | [46,48,53] |
| FOXA2 | Nuclear | Not reported | [50,51] |
| GPC3 | Cell membrane/cytoplasmic | Reported [15,35] | [42,43,45,46,47,50,51,52,54] |
| HNF1β | Nuclear | Not reported | [49] |
| SALL4 | Nuclear | Reported [18] | [42,45,47,51] |
| ZBTB16 | Nuclear | Reported [18] | [44,52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Soest, M.; Montilla-Rojo, J.; Eleveld, T.F.; Looijenga, L.H.J.; Salvatori, D.C.F. Yolk Sac Elements in Tumors Derived from Pluripotent Stem Cells: Borrowing Knowledge from Human Germ Cell Tumors. Int. J. Mol. Sci. 2025, 26, 6464. https://doi.org/10.3390/ijms26136464
van Soest M, Montilla-Rojo J, Eleveld TF, Looijenga LHJ, Salvatori DCF. Yolk Sac Elements in Tumors Derived from Pluripotent Stem Cells: Borrowing Knowledge from Human Germ Cell Tumors. International Journal of Molecular Sciences. 2025; 26(13):6464. https://doi.org/10.3390/ijms26136464
Chicago/Turabian Stylevan Soest, Marnix, Joaquin Montilla-Rojo, Thomas F. Eleveld, Leendert H. J. Looijenga, and Daniela C. F. Salvatori. 2025. "Yolk Sac Elements in Tumors Derived from Pluripotent Stem Cells: Borrowing Knowledge from Human Germ Cell Tumors" International Journal of Molecular Sciences 26, no. 13: 6464. https://doi.org/10.3390/ijms26136464
APA Stylevan Soest, M., Montilla-Rojo, J., Eleveld, T. F., Looijenga, L. H. J., & Salvatori, D. C. F. (2025). Yolk Sac Elements in Tumors Derived from Pluripotent Stem Cells: Borrowing Knowledge from Human Germ Cell Tumors. International Journal of Molecular Sciences, 26(13), 6464. https://doi.org/10.3390/ijms26136464