
Integrative Constraint-Based Modeling and Proteomics Uncover Astrocytic Metabolic Adaptations to the Post-TBI Microenvironment


Abstract
1. Introduction
2. Results
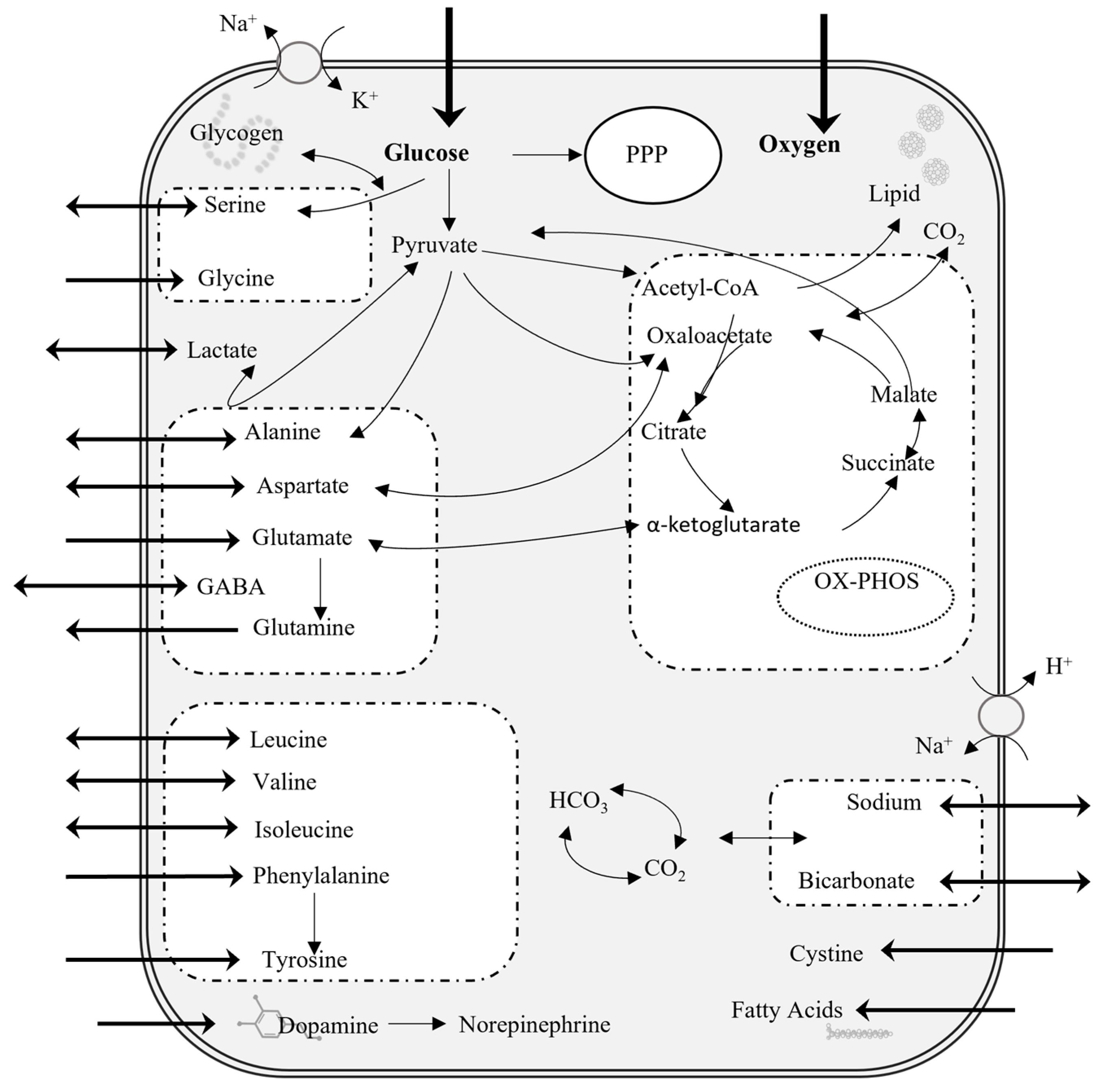
2.1. Flux Balance Analysis for Astrocytes at Steady State
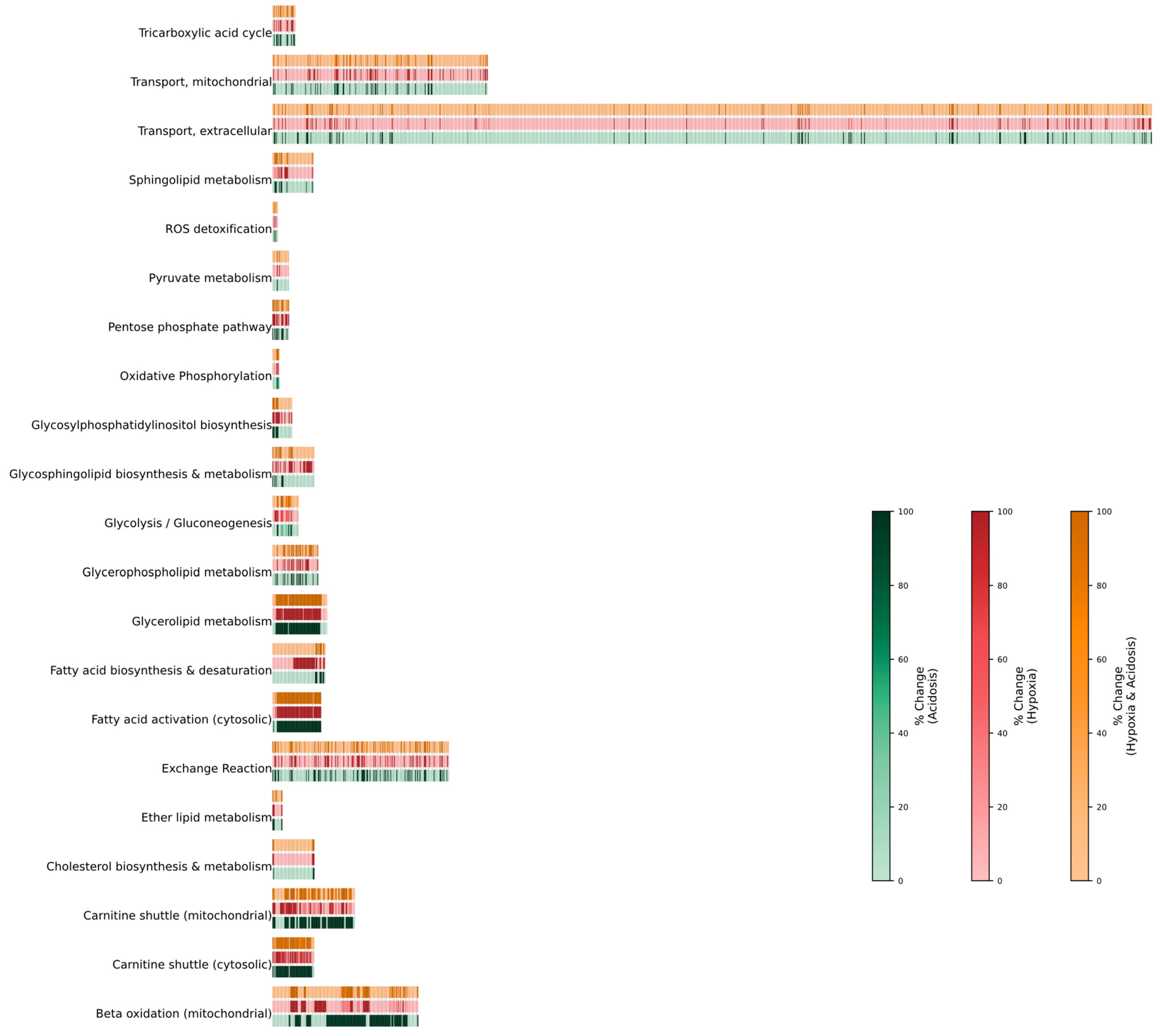
2.2. Change of Flux During Hypoxic, Acidic, or Combined Conditions
2.2.1. Hypoxia
2.2.2. Acidosis
2.2.3. Hypoxia + Acidosis
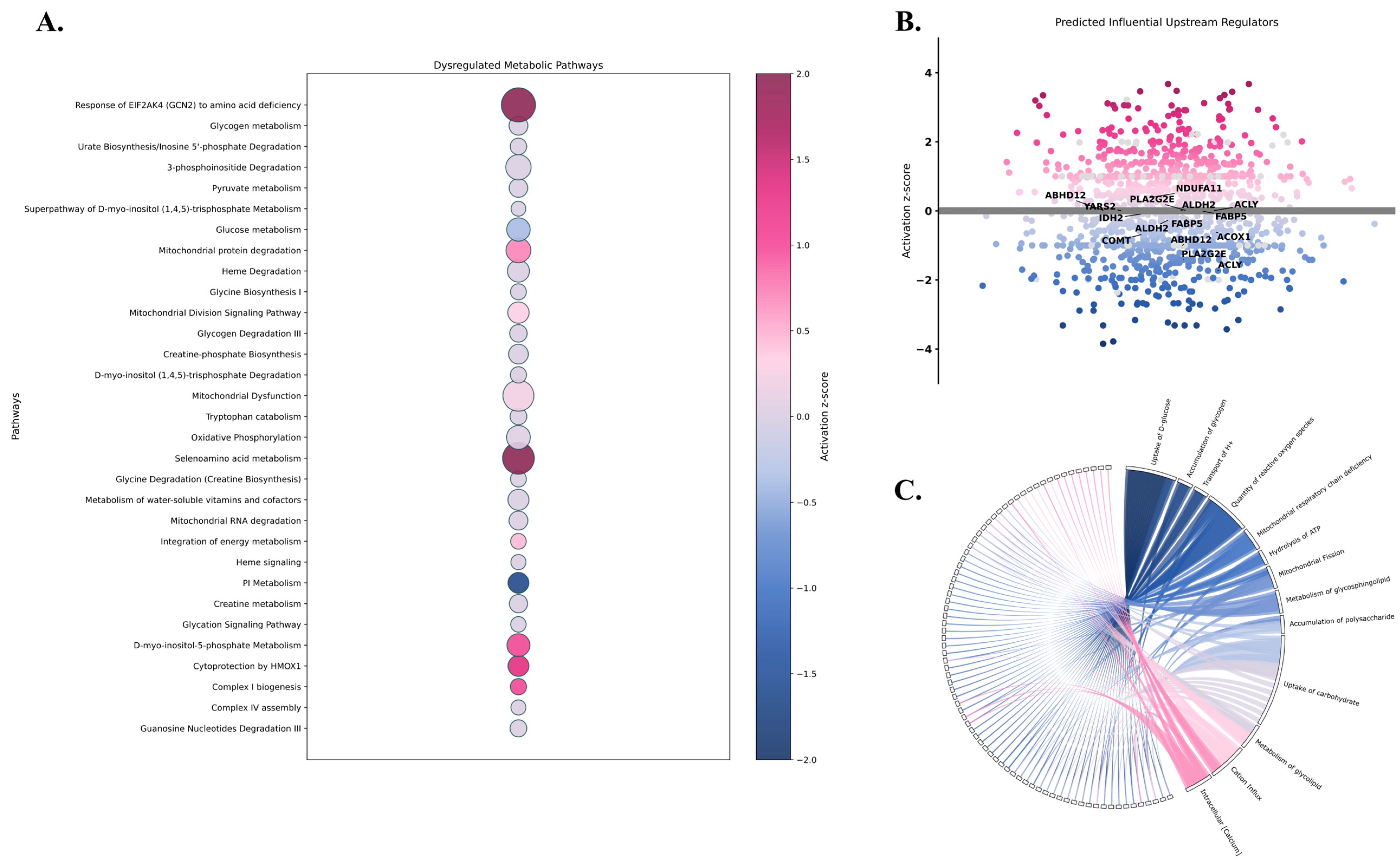
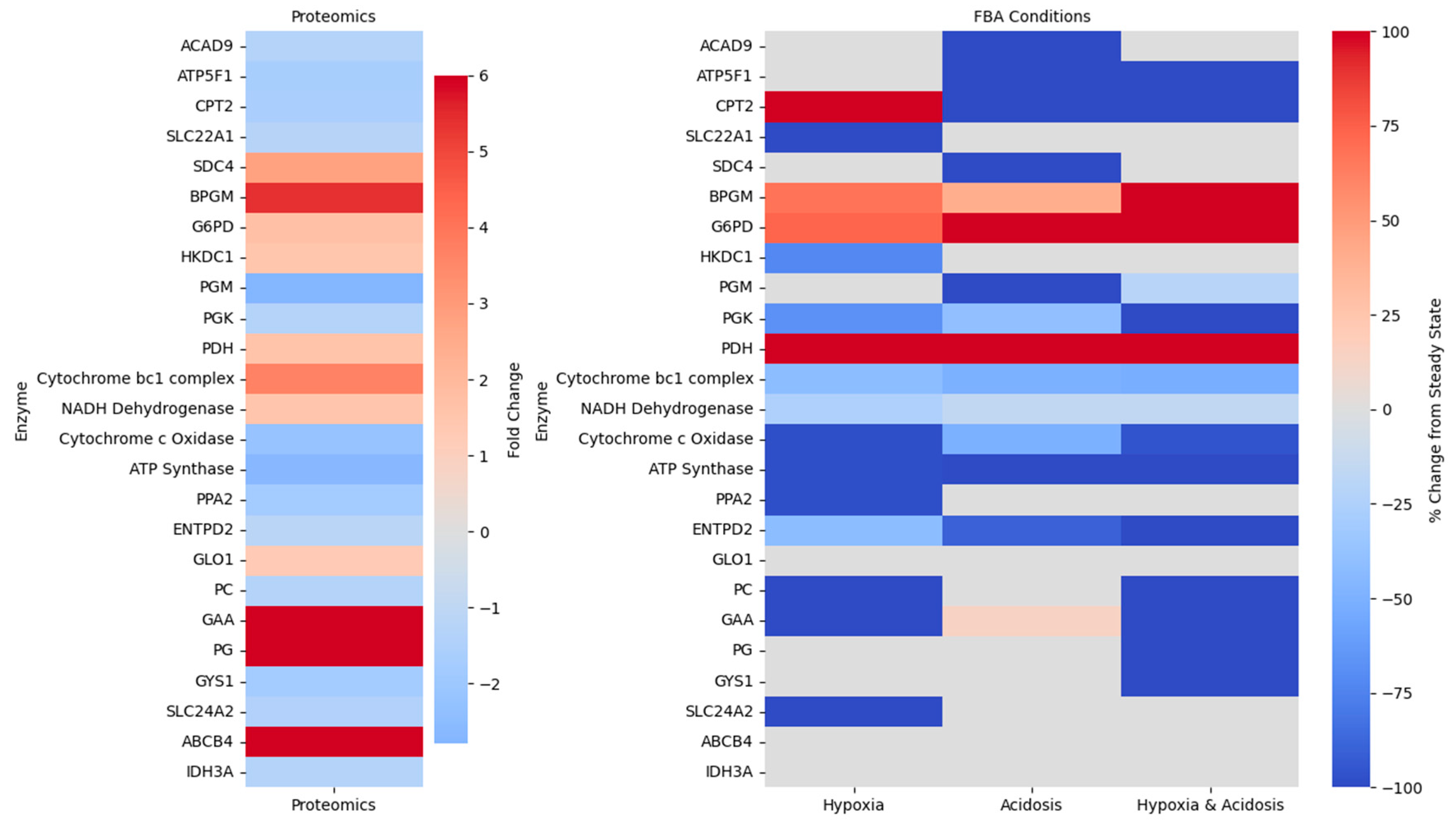
2.3. Proteomic Insights into Metabolic Pathophysiology Following mTBI
2.4. Bridging Predicted Metabolic Fluxes with Proteomic Alterations
3. Discussion
4. Materials and Methods
4.1. Flux Balance Analysis
- ATP Maintenance prioritizes ATP production to meet the energetic demands of the cell.
- 2.
- Glutamate input and glutamate output due to the essential role of astrocytes in neurotransmitter recycling.
4.2. Experimental Proteomic Data Acquisition
4.3. Ingenuity Pathway Analysis of Differentially Expressed Proteins
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TBI | Traumatic brain injury |
| mTBI | Mild traumatic brain injury |
| CNS | Central nervous system |
| TCA | Tricarboxylic acid |
| PPP | Pentose Phosphate Pathway |
| OxPhos | Oxidative Phosphorylation |
| ATP | Adenosine triphosphate |
| ETC | Electron transport chain |
| CO2 | Carbon dioxide |
| FBA | Flux balance analysis |
| DEPs | Differentially expressed proteins |
| FC | Fold change |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| IPA | Ingenuity Pathway Analysis |
References
- Joannides, A.; Korhonen, T.K.; Clark, D.; Gnanakumar, S.; Venturini, S.; Mohan, M.; Bashford, T.; Baticulon, R.; Bhagavatula, I.D.; Esene, I.; et al. An international, prospective observational study on traumatic brain injury epidemiology study protocol: GEO-TBI: Incidence. NIHR Open Res. 2023, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Anderson, D.B.; Chen, L.; Feng, S.; Zhou, H. Global, regional and national burden of traumatic brain injury and spinal cord injury, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. BMJ Open 2023, 13, e075049. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.A. Traumatic brain injury–related emergency department visits, hospitalizations, and deaths—United States, 2007 and 2013. MMWR. Surveill. Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Agoston, D.V. Modeling the Long-Term Consequences of Repeated Blast-Induced Mild Traumatic Brain Injuries. J. Neurotrauma 2017, 34, S44–S52. [Google Scholar] [CrossRef]
- Chapman, J.C.; Diaz-Arrastia, R. Military traumatic brain injury: A review. Alzheimer’s Dement. 2014, 10 (Suppl. S3), S97–S104. [Google Scholar] [CrossRef]
- Kashdan, T.B.; Julian, T.; Merritt, K.; Uswatte, G. Social anxiety and posttraumatic stress in combat veterans: Relations to well-being and character strengths. Behav. Res. Ther. 2006, 44, 561–583. [Google Scholar] [CrossRef]
- Kraus, J.F.; Nourjah, P. The epidemiology of mild, uncomplicated brain injury. J. Trauma Acute Care Surg. 1988, 28, 1637–1643. [Google Scholar] [CrossRef]
- Bower, J.H.; Maraganore, D.M.; Peterson, B.J.; McDonnell, S.K.; Ahlskog, J.E.; Rocca, W.A. Head trauma preceding PD: A case-control study. Neurology 2003, 60, 1610–1615. [Google Scholar] [CrossRef]
- Clausen, T.; Khaldi, A.; Zauner, A.; Reinert, M.; Doppenberg, E.; Menzel, M.; Soukup, J.; Alves, O.L.; Bullock, M.R. Cerebral acid-base homeostasis after severe traumatic brain injury. J. Neurosurg. 2005, 103, 597–607. [Google Scholar] [CrossRef]
- Timofeev, I.; Nortje, J.; Al-Rawi, P.G.; Hutchinson, P.J.; Gupta, A.K. Extracellular brain pH with or without hypoxia is a marker of profound metabolic derangement and increased mortality after traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 422–427. [Google Scholar] [CrossRef]
- Veenith, T.V.; Carter, E.L.; Geeraerts, T.; Grossac, J.; Newcombe, V.F.; Outtrim, J.; Gee, G.S.; Lupson, V.; Smith, R.; Aigbirhio, F.I.; et al. Pathophysiologic Mechanisms of Cerebral Ischemia and Diffusion Hypoxia in Traumatic Brain Injury. JAMA Neurol. 2016, 73, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Hillered, L.; Smith, M.L.; Siesjo, B.K. Lactic acidosis and recovery of mitochondrial function following forebrain ischemia in the rat. J. Cereb. Blood Flow Metab. 1985, 5, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Rehncrona, S. Brain acidosis. Ann. Emerg. Med. 1985, 14, 770–776. [Google Scholar] [CrossRef]
- Kalimo, H.; Rehncrona, S.; Soderfeldt, B.; Olsson, Y.; Siesjo, B.K. Brain lactic acidosis and ischemic cell damage: 2. Histopathology. J. Cereb. Blood Flow Metab. 1981, 1, 313–327. [Google Scholar] [CrossRef]
- Ying, W.; Han, S.K.; Miller, J.W.; Swanson, R.A. Acidosis potentiates oxidative neuronal death by multiple mechanisms. J. Neurochem. 1999, 73, 1549–1556. [Google Scholar] [CrossRef]
- Chi, J.H.; Knudson, M.M.; Vassar, M.J.; McCarthy, M.C.; Shapiro, M.B.; Mallet, S.; Holcroft, J.J.; Moncrief, H.; Noble, J.; Wisner, D.; et al. Prehospital hypoxia affects outcome in patients with traumatic brain injury: A prospective multicenter study. J. Trauma Acute Care Surg. 2006, 61, 1134–1141. [Google Scholar] [CrossRef]
- Yan, E.B.; Satgunaseelan, L.; Paul, E.; Bye, N.; Nguyen, P.; Agyapomaa, D.; Kossmann, T.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Post-traumatic hypoxia is associated with prolonged cerebral cytokine production, higher serum biomarker levels, and poor outcome in patients with severe traumatic brain injury. J. Neurotrauma 2014, 31, 618–629. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Glenn, T.C.; Shik, V.; Boscardin, J.; Mahamid, E.; Zaaroor, M. Monitoring of cerebral blood flow and metabolism in traumatic brain injury. J. Neurotrauma 2005, 22, 955–965. [Google Scholar] [CrossRef]
- Bouma, G.J.; Muizelaar, J.P.; Stringer, W.A.; Choi, S.C.; Fatouros, P.; Young, H.F. Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J. Neurosurg. 1992, 77, 360–368. [Google Scholar] [CrossRef]
- Menon, D.K.; Coles, J.P.; Gupta, A.K.; Fryer, T.D.; Smielewski, P.; Chatfield, D.A.; Aigbirhio, F.; Skepper, J.N.; Minhas, P.S.; Hutchinson, P.J.; et al. Diffusion limited oxygen delivery following head injury. Crit. Care Med. 2004, 32, 1384–1390. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Qi, Y.B.; Gao, Y.N.; Chen, W.G.; Zhou, T.; Zang, Y.; Li, J. Astrocyte metabolism and signaling pathways in the CNS. Front. Neurosci. 2023, 17, 1217451. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 6568. [Google Scholar] [CrossRef]
- Guilhaume-Correa, F.; Pickrell, A.M.; VandeVord, P.J. The Imbalance of Astrocytic Mitochondrial Dynamics Following Blast-Induced Traumatic Brain Injury. Biomedicines 2023, 11, 329. [Google Scholar] [CrossRef]
- Rahman, M.H.; Suk, K. Mitochondrial Dynamics and Bioenergetic Alteration During Inflammatory Activation of Astrocytes. Front. Aging Neurosci. 2020, 12, 614410. [Google Scholar] [CrossRef]
- Jiang, T.; Cadenas, E. Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell 2014, 13, 1059–1067. [Google Scholar] [CrossRef]
- Martin-Jimenez, C.A.; Salazar-Barreto, D.; Barreto, G.E.; Gonzalez, J. Genome-Scale Reconstruction of the Human Astrocyte Metabolic Network. Front. Aging Neurosci. 2017, 9, 23. [Google Scholar] [CrossRef]
- Mangia, S.; Simpson, I.A.; Vannucci, S.J.; Carruthers, A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: Evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009, 109 (Suppl. S1), 55–62. [Google Scholar] [CrossRef]
- Sharma, B.; Schmidt, L.; Nguyen, C.; Kiernan, S.; Dexter-Meldrum, J.; Kuschner, Z.; Ellis, S.; Bhatia, N.D.; Agriantonis, G.; Whittington, J. The effect of L-carnitine on critical illnesses such as traumatic brain injury (TBI), acute kidney injury (AKI), and hyperammonemia (HA). Metabolites 2024, 14, 363. [Google Scholar] [CrossRef]
- Gessner, P.; Lum, J.; Frenguelli, B.G. The mammalian purine salvage pathway as an exploitable route for cerebral bioenergetic support after brain injury. Neuropharmacology 2023, 224, 109370. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhao, Y.; Haidacher, S.J.; Wang, E.; Parsley, M.O.; Gao, J.; Sadygov, R.G.; Starkey, J.M.; Luxon, B.A.; Spratt, H.; et al. Detection of Structural and Metabolic Changes in Traumatically Injured Hippocampus by Quantitative Differential Proteomics. J. Neurotrauma 2012, 30, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.H.; Shin, D.; Kim, Y.; Kim, Y.S.; Seong, M.Y.; Lee, J.J.; Seo, H.G.; Cho, W.S.; Ro, Y.S.; et al. Multiplexed Quantitative Proteomics Reveals Proteomic Alterations in Two Rodent Traumatic Brain Injury Models. J. Proteome Res. 2024, 23, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Ojo, J.O.; Crynen, G.; Algamal, M.; Vallabhaneni, P.; Leary, P.; Mouzon, B.; Reed, J.M.; Mullan, M.; Crawford, F. Unbiased Proteomic Approach Identifies Pathobiological Profiles in the Brains of Preclinical Models of Repetitive Mild Traumatic Brain Injury, Tauopathy, and Amyloidosis. ASN Neuro 2020, 12, 1759091420914768. [Google Scholar] [CrossRef]
- Chateil, J.-F.; Biran, M.; Thiaudière, E.; Canioni, P.; Merle, M. Metabolism of [1-13C] glucose and [2-13C] acetate in the hypoxic rat brain. Neurochem. Int. 2001, 38, 399–407. [Google Scholar] [CrossRef]
- Lebon, V.; Petersen, K.F.; Cline, G.W.; Shen, J.; Mason, G.F.; Dufour, S.; Behar, K.L.; Shulman, G.I.; Rothman, D.L. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: Elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J. Neurosci. 2002, 22, 1523–1531. [Google Scholar] [CrossRef]
- Verleysdonk, S.; Martin, H.; Willker, W.; Leibfritz, D.; Hamprecht, B. Rapid uptake and degradation of glycine by astroglial cells in culture: Synthesis and release of serine and lactate. Glia 1999, 27, 239–248. [Google Scholar] [CrossRef]
- Westergaard, N.; Varming, T.; Peng, L.; Sonnewald, U.; Hertz, L.; Schousboe, A. Uptake, release and metabolism of alaume in neurons and astrocytes in primary cultures. J. Neurosci. Res. 1993, 35, 540–545. [Google Scholar] [CrossRef]
- Aureli, T.; Di Cocco, M.E.; Calvani, M.; Conti, F. The entry of [1-13C] glucose into biochemical pathways reveals a complex compartmentation and metabolite trafficking between glia and neurons: A study by 13C-NMR spectroscopy. Brain Res. 1997, 765, 218–227. [Google Scholar] [CrossRef]
- Shen, J.; Petersen, K.F.; Behar, K.L.; Brown, P.; Nixon, T.W.; Mason, G.F.; Petroff, O.A.; Shulman, G.I.; Shulman, R.G.; Rothman, D.L. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc. Natl. Acad. Sci. USA 1999, 96, 8235–8240. [Google Scholar] [CrossRef]
- Murin, R.; Mohammadi, G.; Leibfritz, D.; Hamprecht, B. Glial metabolism of isoleucine. Neurochem. Res. 2009, 34, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Lanz, B.; Gruetter, R.; Duarte, J.M. Metabolic Flux and Compartmentation Analysis in the Brain In vivo. Front. Endocrinol. 2013, 4, 156. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.; Cerdan, S. Quantitative 13C NMR studies of metabolic compartmentation in the adult mammalian brain. NMR Biomed. 1999, 12, 451–462. [Google Scholar] [CrossRef]
- Amaral, A.I.; Teixeira, A.P.; Håkonsen, B.I.; Sonnewald, U.; Alves, P.M. A comprehensive metabolic profile of cultured astrocytes using isotopic transient metabolic flux analysis and 13C-labeled glucose. Front. Neuroenergetics 2011, 3, 5. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001, 902, 156–163. [Google Scholar] [CrossRef]
- Hannuniemi, R.; Oja, S. Uptake of leucine, lysine, aspartic acid, and glycine into isolated neurons and astrocytes. Neurochem. Res. 1981, 6, 873–884. [Google Scholar] [CrossRef]
- Zwingmann, C.; Richter-Landsberg, C.; Brand, A.; Leibfritz, D. NMR spectroscopic study on the metabolic fate of [3-13C] alanine in astrocytes, neurons, and cocultures: Implications for glia-neuron interactions in neurotransmitter metabolism. Glia 2000, 32, 286–303. [Google Scholar] [CrossRef]
- Zwingmann, C.; Leibfritz, D. Regulation of glial metabolism studied by 13C-NMR. NMR Biomed. Int. J. Devoted Dev. Appl. Magn. Reson. In Vivo 2003, 16, 370–399. [Google Scholar] [CrossRef]
- Gruetter, R.; Seaquist, E.R.; Ugurbil, K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E100–E112. [Google Scholar] [CrossRef]
- Talty, C.-E.; Murphy, S.; VandeVord, P. Mild traumatic brain injury gives rise to chronic depression-like behavior and associated alterations in glutamatergic protein expression. Neuroscience 2024, 560, 198–210. [Google Scholar] [CrossRef]
- Swanger, S.A.; Vance, K.M.; Pare, J.-F.; Sotty, F.; Fog, K.; Smith, Y.; Traynelis, S.F. NMDA receptors containing the GluN2D subunit control neuronal function in the subthalamic nucleus. J. Neurosci. 2015, 35, 15971–15983. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.L.; Wilson, K.A.; Shandra, O.; Wei, X.; Mahmutovic, D.; Maharathi, B.; Robel, S.; VandeVord, P.J.; Olsen, M.L. Applying Proteomics and Computational Approaches to Identify Novel Targets in Blast-Associated Post-Traumatic Epilepsy. Int. J. Mol. Sci. 2024, 25, 2880. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Metabolic Rate µmol/g/min | References |
|---|---|---|
| Acetoacetate | 0.012 | [35] |
| Glutamine | 0.500 | |
| Asparagine | 0.0037 | [36] |
| Ornithine | 0.0031 | [37] |
| Valine | 0.0018 | [38] |
| Serine | 0.0016 | [37] |
| Linoleate | 0.0011 | [39] |
| Threonine | 0.0008 | [40] |
| Isoleucine | 0.0004 | [41] |
| Methionine | 0.0017 | [42] |
| Tyrosine | 0.0017 | [36] |
| Arginine | 0.0025 | [43] |
| Histidine | 0.0025 | [44] |
| Cystine | 0.0045 | [45] |
| Lysine | 0.011 | [46] |
| Leucine | 0.0145 | [46] |
| Glutamate | 0.232 | [47] |
| Acetate | 0.0013 | |
| Glycine | 0.0053 | [47] |
| Proline | 0.0066 | [48] |
| Alanine | 0.0079 | |
| Glucose | 0.19 | [40,49] |
| CMR_O2 | 0.530 | [48] |
| CMR_CO2 | 0.515 | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, K.A.; Talty, C.-E.; Parker, B.C.; VandeVord, P.J. Integrative Constraint-Based Modeling and Proteomics Uncover Astrocytic Metabolic Adaptations to the Post-TBI Microenvironment. Int. J. Mol. Sci. 2025, 26, 6456. https://doi.org/10.3390/ijms26136456
Wilson KA, Talty C-E, Parker BC, VandeVord PJ. Integrative Constraint-Based Modeling and Proteomics Uncover Astrocytic Metabolic Adaptations to the Post-TBI Microenvironment. International Journal of Molecular Sciences. 2025; 26(13):6456. https://doi.org/10.3390/ijms26136456
Chicago/Turabian StyleWilson, Kelsey A., Caiti-Erin Talty, Brian C. Parker, and Pamela J. VandeVord. 2025. "Integrative Constraint-Based Modeling and Proteomics Uncover Astrocytic Metabolic Adaptations to the Post-TBI Microenvironment" International Journal of Molecular Sciences 26, no. 13: 6456. https://doi.org/10.3390/ijms26136456
APA StyleWilson, K. A., Talty, C.-E., Parker, B. C., & VandeVord, P. J. (2025). Integrative Constraint-Based Modeling and Proteomics Uncover Astrocytic Metabolic Adaptations to the Post-TBI Microenvironment. International Journal of Molecular Sciences, 26(13), 6456. https://doi.org/10.3390/ijms26136456