
Transsulfuration Pathway Products and H2S-Donors in Hyperhomocysteinemia: Potential Strategies Beyond Folic Acid

 , ,
, ,  , and
, and 
Abstract
1. Introduction
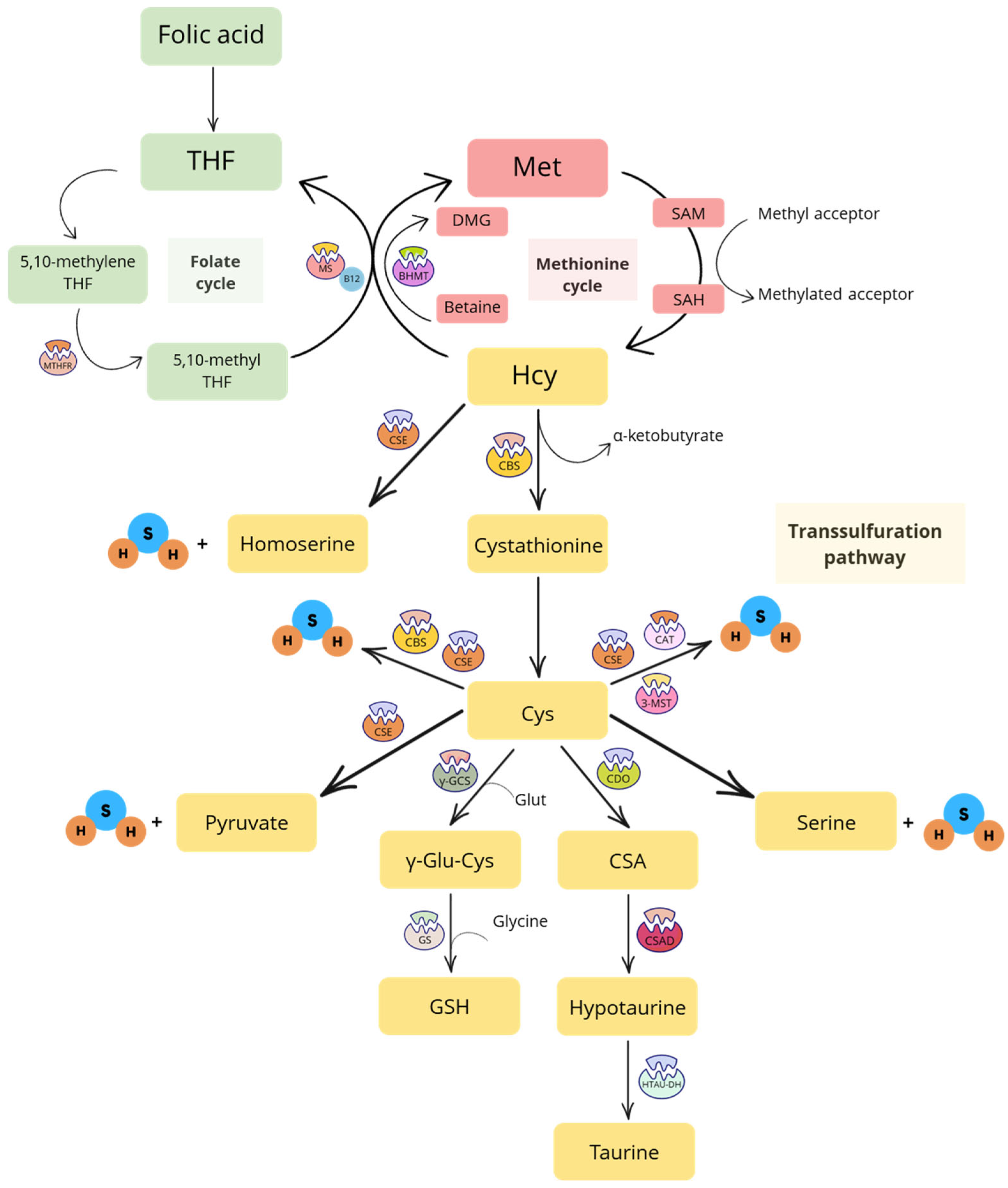
1.1. Transsulfuration Pathway
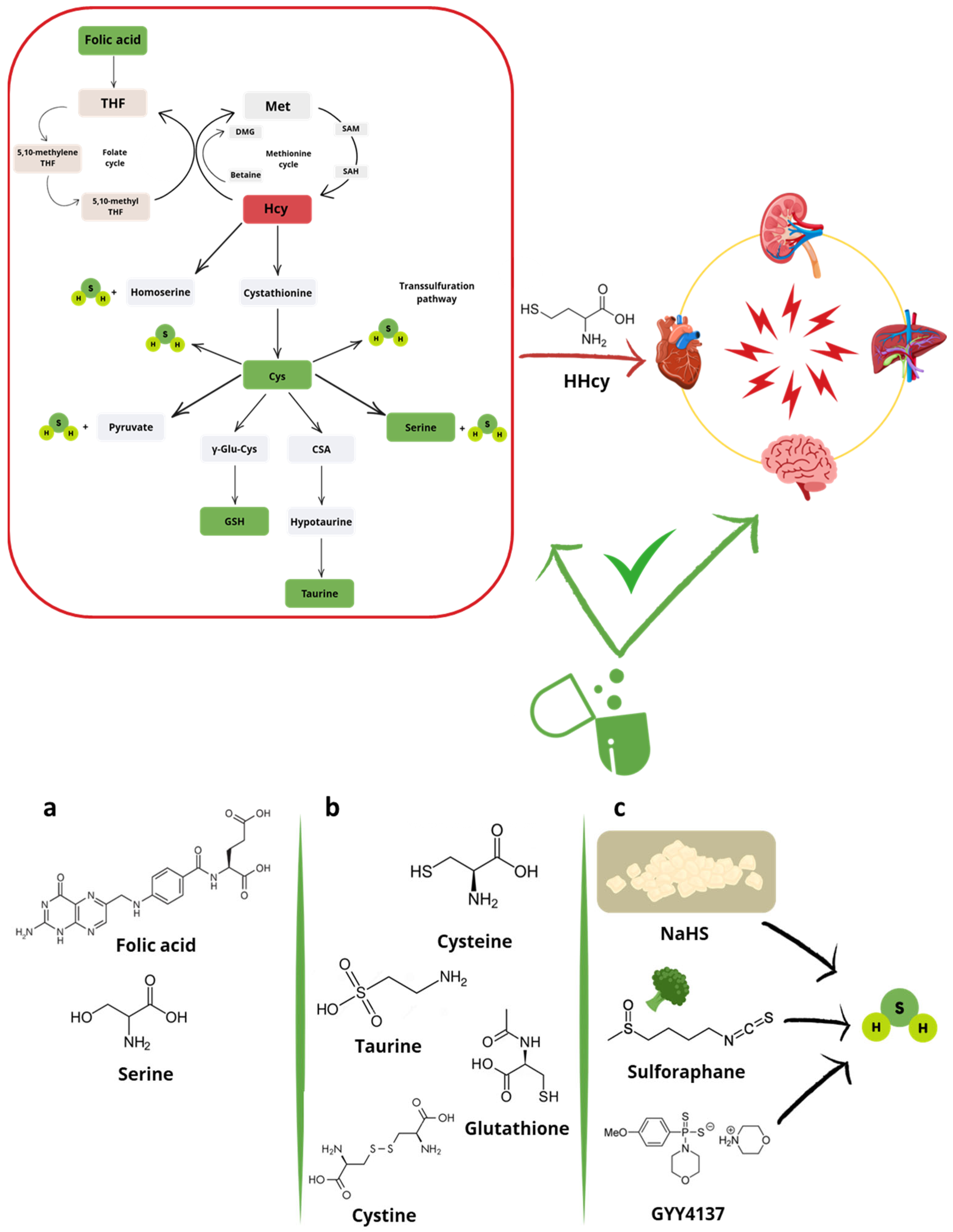
1.2. Hyperhomocysteinemia (HHcy)
2. Players of the Folate Cycle
3. Products of the Transsulfuration Pathway
3.1. Central Products (Cys, Cystine and Serine) and Synthetic Derivatives (NAC)
3.2. Final Products (Taurine)
4. H2S-Donors
4.1. Heart and Vessels
4.2. Brain
4.3. Kidney
4.4. Liver
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| ANP | Atrial-natriuretic peptide |
| ARE | Antioxidant response element |
| ATP | Adenosine triphosphate |
| BAX | BCL-2-like protein 4 |
| BCL2 | B-cell lymphoma 2 |
| BHMT | Betaine-homocysteine methyltransferase |
| BW | Body weight |
| CASP3 | Caspase-3 |
| CASP8 | Caspase-8 |
| CAT | Cysteine aminotransferase |
| CBS | Cystathionine β-synthase |
| CHF | Chronic heart failure |
| CDO | Cysteine dioxygenase |
| CO | Carbon monoxide |
| COX | Cyclooxygenase |
| CSAD | Cysteine sulfinic acid decarboxylase |
| CSE | Cystathionine γ-liase |
| CV | Cardiovascular |
| DATS | Diallytrisulfide |
| EC-SOD | Extracellular superoxide dismutase |
| eNOS | Endothelial nitric oxide synthase |
| ER | Endoplasmic reticulum |
| FMD | Flow-mediated dilation |
| GFR | Glomerular filtration rate |
| GOT1 | Glutamic oxaloacetic transaminase 1 |
| GPx | Glutathione peroxidase |
| GS | Glutathione synthase |
| HAECs | Human aortic endothelial cells |
| HCU | Homocystinuria |
| HcyT | Homocysteine thiolactone |
| HDL | High-density lipoprotein |
| HFpEF | Heart failure with preserved ejection fraction |
| HHcy | Hyperhomocysteinemia |
| HO-1 | Heme oxygenase-1 |
| HTAU-DH | Hypotaurine dehydrogenase |
| HUVECs | Human umbilical vein endothelial cells |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| KATP channels | ATP-sensitive potassium channels |
| Keap1 | Kelch-like ECH-associated protein 1 |
| L-NAME | N(ω)-nitro-L-arginine methyl ester |
| LDH | Lactate dehydrogenase |
| LDL | Low-density lipoprotein |
| MMPs | Matrix metalloproteinases |
| Mn-SOD | Mn-superoxide dismutase |
| MS | Methionine synthase |
| MTHFR | Methylenetetrahydrofolate reductase |
| NAC | N-acetylcysteine |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFS1 | Mitochondrial cysteine desulfurase |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OPA3 | Optic atrophy 3 |
| OTUB1 | OTU domain-containing ubiquitin aldehyde-binding protein 1 |
| PDI | Protein disulphide isomerase |
| PECAM | Platelet endothelial cell adhesion molecule |
| PON-1 | Paraoxonase-1 |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SGK1 | Serum and glucocorticoid-regulated kinase 1 |
| SHRs | Spontaneously hypertensive rats |
| SIRT1 | Sirtuin isoform 1 |
| SOD | Superoxide dismutase |
| SP1 | Specificity protein-1 |
| T2D | Type 2 diabetes |
| THF | Tetrahydrofolate |
| TNF-α | Tumor necrosis factor-α |
| TP53 | Tumor protein 53 |
| VSMCs | Vascular smooth muscle cells |
| Xc− | Cystine/glutamate antiporter |
| xCT | Cystine/glutamate antiporter light chain subunit |
| XO | Xanthine oxidase |
| β-MHC | β-myosin heavy chain |
| γ-GCS | γ-glutamyl cysteine synthetase |
| Glossary of Common Metabolite Terms | |
| BH4 | Tetrahydrobiopterin |
| Cys | Cysteine |
| DMG | Dimethylglycine |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| Hcy | Homocysteine |
| MDA | Malondialdehyde |
| Met | Methionine |
| SAH | S-adenosyl homocysteine |
| SAM | S-adenosyl methionine |
| THF | Tetrahydrofolate |
| γ-Glu-Cys | γ-glutamyl cysteine |
References
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef]
- Yuan, D.; Chu, J.; Lin, H.; Zhu, G.; Qian, J.; Yu, Y.; Yao, T.; Ping, F.; Chen, F.; Liu, X. Mechanism of homocysteine-mediated endothelial injury and its consequences for atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 1109445. [Google Scholar] [CrossRef]
- Wang, R. Roles of Hydrogen Sulfide in Hypertension Development and Its Complications: What, So What, Now What. Hypertension 2023, 80, 936–944. [Google Scholar] [CrossRef]
- Wang, J.; You, D.; Wang, H.; Yang, Y.; Zhang, D.; Lv, J.; Luo, S.; Liao, R.; Ma, L. Association between homocysteine and obesity: A meta-analysis. J. Evid. Based. Med. 2021, 14, 208–217. [Google Scholar] [CrossRef]
- Huang, T.; Ren, J.; Huang, J.; Li, D. Association of homocysteine with type 2 diabetes: A meta-analysis implementing Mendelian randomization approach. BMC Genom. 2013, 14, 867. [Google Scholar] [CrossRef]
- Liu, C.; Liu, L.; Wang, Y.; Chen, X.; Liu, J.; Peng, S.; Pi, J.; Zhang, Q.; Tomlinson, B.; Chan, P.; et al. Hyperhomocysteinemia Increases Risk of Metabolic Syndrome and Cardiovascular Death in an Elderly Chinese Community Population of a 7-Year Follow-Up Study. Front. Cardiovasc. Med. 2021, 8, 811670. [Google Scholar] [CrossRef]
- Gospodarczyk, A.; Marczewski, K.; Gospodarczyk, N.; Widuch, M.; Tkocz, M.; Zalejska-Fiolka, J. Homocysteine and cardiovascular disease—A current review. Wiad. Lek. 2022, 75, 2862–2866. [Google Scholar] [CrossRef]
- Qiu, J.; Yang, X.; Wang, Q.; Yang, X.; Ma, S.; Zhang, J.; Liu, W.; Li, X.; Chen, K.; Wang, K.; et al. Association of plasma homocysteine with cardiometabolic multimorbidity: A cross-sectional study in northwest China. Lipids Health Dis. 2024, 23, 370. [Google Scholar] [CrossRef]
- Zheng, Y.; Cantley, L.C. Toward a better understanding of folate metabolism in health and disease. J. Exp. Med. 2019, 216, 253–266. [Google Scholar] [CrossRef]
- Pajares, M.A.; Pérez-Sala, D. Betaine homocysteine S-methyltransferase: Just a regulator of homocysteine metabolism? Cell. Mol. Life Sci. 2006, 63, 2792–2803. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Holeček, M. Serine Metabolism in Health and Disease and as a Conditionally Essential Amino Acid. Nutrients 2022, 14, 1987. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef]
- Jong, C.J.; Sandal, P.; Schaffer, S.W. The Role of Taurine in Mitochondria Health: More Than Just an Antioxidant. Molecules 2021, 26, 4913. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Zhang, D.; Du, J.; Tang, C.; Huang, Y.; Jin, H. H2S-Induced Sulfhydration: Biological Function and Detection Methodology. Front. Pharmacol. 2017, 8, 608. [Google Scholar] [CrossRef]
- Vandiver, M.; Snyder, S.H. Hydrogen sulfide: A gasotransmitter of clinical relevance. J. Mol. Med. 2012, 90, 255–263. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef]
- Spezzini, J.; Piragine, E.; d’Emmanuele di Villa Bianca, R.; Bucci, M.; Martelli, A.; Calderone, V. Hydrogen sulfide and epigenetics: Novel insights into the cardiovascular effects of this gasotransmitter. Br. J. Pharmacol. 2023, 180, 1793–1802. [Google Scholar] [CrossRef]
- Xi, C.; Pang, J.; Xue, W.; Cui, Y.; Jiang, N.; Zhi, W.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Transsulfuration pathway activation attenuates oxidative stress and ferroptosis in sickle primary erythroblasts and transgenic mice. Commun. Biol. 2025, 8, 15. [Google Scholar] [CrossRef]
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef]
- Mota-Martorell, N.; Jové, M.; Borrás, C.; Berdún, R.; Obis, È.; Sol, J.; Cabré, R.; Pradas, I.; Galo-Licona, J.D.; Puig, J.; et al. Methionine transsulfuration pathway is upregulated in long-lived humans. Free Radic. Biol. Med. 2021, 162, 38–52. [Google Scholar] [CrossRef] [PubMed]
- González-Lamuño, D.; Arrieta-Blanco, F.J.; Fuentes, E.D.; Forga-Visa, M.T.; Morales-Conejo, M.; Peña-Quintana, L.; Vitoria-Miñana, I. Hyperhomocysteinemia in Adult Patients: A Treatable Metabolic Condition. Nutrients 2023, 16, 135. [Google Scholar] [CrossRef]
- Tian, W.; Ju, J.; Guan, B.; Wang, T.; Zhang, J.; Song, L.; Xu, H. Role of hyperhomocysteinemia in atherosclerosis: From bench to bedside. Ann. Med. 2025, 57, 2457527. [Google Scholar] [CrossRef]
- Karatela, R.A.; Sainani, G.S. Plasma homocysteine in obese, overweight and normal weight hypertensives and normotensives. Indian Heart J. 2009, 61, 156–159. [Google Scholar] [PubMed]
- Karger, A.B.; Nomura, S.O.; Guan, W.; Garg, P.K.; Tison, G.H.; Szklo, M.; Budoff, M.J.; Tsai, M.Y. Association Between Elevated Total Homocysteine and Heart Failure Risk in the Multi-Ethnic Study of Atherosclerosis Cohort. J. Am. Heart Assoc. 2025, 14, e038168. [Google Scholar] [CrossRef]
- Rabelo, N.N.; Telles, J.P.M.; Pipek, L.Z.; Farias Vidigal Nascimento, R.; Gusmão, R.C.; Teixeira, M.J.; Figueiredo, E.G. Homocysteine is associated with higher risks of ischemic stroke: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0276087. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Hoshino, T.; Ishizuka, K.; Toi, S.; Takahashi, S.; Wako, S.; Arai, S.; Kitagawa, K. Hyperhomocysteinemia Increases Vascular Risk in Stroke Patients with Chronic Kidney Disease. J. Atheroscler. Thromb. 2023, 30, 1198–1209. [Google Scholar] [CrossRef]
- Cheng, C.K.; Wang, C.; Shang, W.; Lau, C.W.; Luo, J.Y.; Wang, L.; Huang, Y. A high methionine and low folate diet alters glucose homeostasis and gut microbiome. Biochem. Biophys. Rep. 2021, 25, 100921. [Google Scholar] [CrossRef]
- Ungvari, A.; Gulej, R.; Csik, B.; Mukli, P.; Negri, S.; Tarantini, S.; Yabluchanskiy, A.; Benyo, Z.; Csiszar, A.; Ungvari, Z. The Role of Methionine-Rich Diet in Unhealthy Cerebrovascular and Brain Aging: Mechanisms and Implications for Cognitive Impairment. Nutrients 2023, 15, 4662. [Google Scholar] [CrossRef] [PubMed]
- Al Mutairi, F. Hyperhomocysteinemia: Clinical Insights. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520962230. [Google Scholar] [CrossRef]
- Cui, X.; Navneet, S.; Wang, J.; Roon, P.; Chen, W.; Xian, M.; Smith, S.B. Analysis of MTHFR, CBS, Glutathione, Taurine, and Hydrogen Sulfide Levels in Retinas of Hyperhomocysteinemic Mice. Invest. Ophthalmol. Vis. Sci. 2017, 58, 1954–1963. [Google Scholar] [CrossRef]
- Morris, A.A.; Kožich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef]
- Malaviya, P.; Kowluru, R.A. Homocysteine and mitochondrial quality control in diabetic retinopathy. Eye Vis. 2024, 11, 5. [Google Scholar] [CrossRef]
- Kovalska, M.; Hnilicova, P.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Effect of Methionine Diet on Time-Related Metabolic and Histopathological Changes of Rat Hippocampus in the Model of Global Brain Ischemia. Biomolecules 2020, 10, 1128. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial dysfunction: The link between homocysteine and hydrogen sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.K.; Kan, M.Y. Homocysteine-Induced Endothelial Dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J.; Dole, V.S.; Loscalzo, J.; Handy, D.E.; Wagner, D.D. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef]
- Li, L.; Hasegawa, H.; Inaba, N.; Yoshioka, W.; Chang, D.; Liu, J.; Ichida, K. Diet-induced hyperhomocysteinemia impairs vasodilation in 5/6-nephrectomized rats. Amino Acids 2018, 50, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Piragine, E.; Gorica, E.; Citi, V.; Testai, L.; Pagnotta, E.; Matteo, R.; Pecchioni, N.; Montanaro, R.; Di Cesare Mannelli, L.; et al. Anti-Inflammatory Effect of the Natural H2S-Donor Erucin in Vascular Endothelium. Int. J. Mol. Sci. 2022, 23, 5593. [Google Scholar] [CrossRef] [PubMed]
- Faro, D.C.; Di Pino, F.L.; Monte, I.P. Inflammation, Oxidative Stress, and Endothelial Dysfunction in the Pathogenesis of Vascular Damage: Unraveling Novel Cardiovascular Risk Factors in Fabry Disease. Int. J. Mol. Sci. 2024, 25, 8273. [Google Scholar] [CrossRef]
- Yang, A.N.; Zhang, H.P.; Sun, Y.; Yang, X.L.; Wang, N.; Zhu, G.; Zhang, H.; Xu, H.; Ma, S.C.; Zhang, Y.; et al. High-methionine diets accelerate atherosclerosis by HHcy-mediated FABP4 gene demethylation pathway via DNMT1 in ApoE−/− mice. FEBS Lett. 2015, 589, 3998–4009. [Google Scholar] [CrossRef]
- Liao, D.; Yang, X.; Wang, H. Hyperhomocysteinemia and high-density lipoprotein metabolism in cardiovascular disease. Clin. Chem. Lab. Med. 2007, 45, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Taban-Shomal, O.; Hübner, U.; Böhm, M.; Herrmann, W. A review of homocysteine and heart failure. Eur. J. Heart Fail 2006, 8, 571–576. [Google Scholar] [CrossRef]
- Tyagi, S.C. A High-Fat Diet Induces Epigenetic 1-Carbon Metabolism, Homocystinuria, and Renal-Dependent HFpEF. Nutrients 2025, 17, 216. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Thomas, M.; Ghorpade, A.; Gendelman, H.E.; Banerjee, R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 2006, 281, 35785–35793. [Google Scholar] [CrossRef]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef]
- Shang, Y.; Siow, Y.L.; Isaak, C.K.; Karmin, O. Downregulation of Glutathione Biosynthesis Contributes to Oxidative Stress and Liver Dysfunction in Acute Kidney Injury. Oxidative Med. Cell. Longev. 2016, 2016, 9707292. [Google Scholar] [CrossRef]
- Zhang, W.; He, H.; Wang, H.; Wang, S.; Li, X.; Liu, Y.; Jiang, H.; Jiang, H.; Yan, Y.; Wang, Y.; et al. Activation of transsulfuration pathway by salvianolic acid a treatment: A homocysteine-lowering approach with beneficial effects on redox homeostasis in high-fat diet-induced hyperlipidemic rats. Nutr. Metab. 2013, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, A.; Fernández-Vivancos, E.; Montiel, C.; Vazquez, J.J.; Arnalich, F. Changes in the intracellular homocysteine and glutathione content associated with aging. Life Sci. 2000, 67, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef]
- Módis, K.; Coletta, C.; Asimakopoulou, A.; Szczesny, B.; Chao, C.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide 2014, 41, 146–156. [Google Scholar] [CrossRef]
- Ahn, C.S. Effect of taurine supplementation on plasma homocysteine levels of the middle-aged Korean women. Adv. Exp. Med. Biol. 2009, 643, 415–422. [Google Scholar] [CrossRef]
- Yang, Q.; He, G.W. Imbalance of Homocysteine and H2S: Significance, Mechanisms, and Therapeutic Promise in Vascular Injury. Oxidative Med. Cell. Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumar, S.; Kundu, S.; Sen, U. Hydrogen Sulfide Protects Hyperhomocysteinemia-Induced Renal Damage by Modulation of Caveolin and eNOS Interaction. Sci. Rep. 2019, 9, 2223. [Google Scholar] [CrossRef]
- Chang, L.; Geng, B.; Yu, F.; Zhao, J.; Jiang, H.; Du, J.; Tang, C. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids 2008, 34, 573–585. [Google Scholar] [CrossRef]
- Yakovlev, A.V.; Detterer, A.S.; Yakovleva, O.V.; Hermann, A.; Sitdikova, G.F. H2S prevents the disruption of the blood-brain barrier in rats with prenatal hyperhomocysteinemia. J. Pharmacol. Sci. 2024, 155, 131–139. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced blood-brain barrier permeability by inhibiting MMP-9. Int. J. Neurosci. 2022, 132, 1061–1071. [Google Scholar] [CrossRef]
- Nandi, S.S.; Mishra, P.K. H2S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 2017, 7, 3639. [Google Scholar] [CrossRef] [PubMed]
- Piragine, E.; Malanima, M.A.; Lucenteforte, E.; Martelli, A.; Calderone, V. Circulating Levels of Hydrogen Sulfide (H2S) in Patients with Age-Related Diseases: A Systematic Review and Meta-Analysis. Biomolecules 2023, 13, 1023. [Google Scholar] [CrossRef] [PubMed]
- Bearden, S.E.; Beard, R.S., Jr.; Pfau, J.C. Extracellular transsulfuration generates hydrogen sulfide from homocysteine and protects endothelium from redox stress. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1568–H1576. [Google Scholar] [CrossRef]
- Huang, X.; Bao, H.; Ding, C.; Li, J.; Cao, T.; Liu, L.; Wei, Y.; Zhou, Z.; Zhang, N.; Song, Y.; et al. Optimal folic acid dosage in lowering homocysteine: Precision Folic Acid Trial to lower homocysteine (PFAT-Hcy). Eur. J. Nutr. 2024, 63, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Asbaghi, O.; Ghanavati, M.; Ashtary-Larky, D.; Bagheri, R.; Rezaei Kelishadi, M.; Nazarian, B.; Nordvall, M.; Wong, A.; Dutheil, F.; Suzuki, K.; et al. Effects of Folic Acid Supplementation on Oxidative Stress Markers: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Antioxidants 2021, 10, 871. [Google Scholar] [CrossRef]
- Surai, P.F.; Earle-Payne, K.; Kidd, M.T. Taurine as a Natural Antioxidant: From Direct Antioxidant Effects to Protective Action in Various Toxicological Models. Antioxidants 2021, 10, 1876. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Munteanu, C.; Turnea, M.A.; Rotariu, M. Hydrogen Sulfide: An Emerging Regulator of Oxidative Stress and Cellular Homeostasis-A Comprehensive One-Year Review. Antioxidants 2023, 12, 1737. [Google Scholar] [CrossRef]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Du, C.; Lin, X.; Xu, W.; Zheng, F.; Cai, J.; Yang, J.; Cui, Q.; Tang, C.; Cai, J.; Xu, G.; et al. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid. Redox Signal 2019, 30, 184–197. [Google Scholar] [CrossRef]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Z.; Tao, Y.; He, L.; Zhan, F.; Li, J. Linking homocysteine and ferroptosis in cardiovascular disease: Insights and implications. Apoptosis 2024, 29, 1944–1958. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Liu, R.; Chen, L. Ferroptosis inhibitor ferrostatin-1 alleviates homocysteine-induced ovarian granulosa cell injury by regulating TET activity and DNA methylation. Mol. Med. Rep. 2022, 25, 12645. [Google Scholar] [CrossRef]
- Shi, J.; Chen, D.; Wang, Z.; Li, S.; Zhang, S. Homocysteine induces ferroptosis in endothelial cells through the systemXc(-)/GPX4 signaling pathway. BMC Cardiovasc. Disord 2023, 23, 316. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 2020, 160, 552–565. [Google Scholar] [CrossRef]
- Chen, S.; Bu, D.; Zhu, J.; Yue, T.; Guo, S.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. Endogenous hydrogen sulfide regulates xCT stability through persulfidation of OTUB1 at cysteine 91 in colon cancer cells. Neoplasia 2021, 23, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ying, X.; Wang, Y.; Zou, Z.; Yuan, A.; Xiao, Z.; Geng, N.; Qiao, Z.; Li, W.; Lu, X.; et al. Hydrogen sulfide alleviates mitochondrial damage and ferroptosis by regulating OPA3-NFS1 axis in doxorubicin-induced cardiotoxicity. Cell. Signal. 2023, 107, 110655. [Google Scholar] [CrossRef]
- Poltorack, C.D.; Dixon, S.J. Understanding the role of cysteine in ferroptosis: Progress & paradoxes. FEBS J. 2022, 289, 374–385. [Google Scholar] [CrossRef]
- Shi, Z.; Naowarojna, N.; Pan, Z.; Zou, Y. Multifaceted mechanisms mediating cystine starvation-induced ferroptosis. Nat. Commun. 2021, 12, 4792. [Google Scholar] [CrossRef]
- Kaye, A.D.; Jeha, G.M.; Pham, A.D.; Fuller, M.C.; Lerner, Z.I.; Sibley, G.T.; Cornett, E.M.; Urits, I.; Viswanath, O.; Kevil, C.G. Folic Acid Supplementation in Patients with Elevated Homocysteine Levels. Adv. Ther. 2020, 37, 4149–4164. [Google Scholar] [CrossRef]
- Cui, S.; Li, W.; Wang, P.; Lv, X.; Gao, Y.; Huang, G. Folic acid inhibits homocysteine-induced cell apoptosis in human umbilical vein endothelial cells. Mol. Cell. Biochem. 2018, 444, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wen, J.; Wang, X.; Xiao, C. High-dose folic acid improves endothelial function by increasing tetrahydrobiopterin and decreasing homocysteine levels. Mol. Med. Rep. 2014, 10, 1609–1613. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.M.; Kim, H.J.; Lee, I.; Chang, N. Folic acid supplementation can reduce the endothelial damage in rat brain microvasculature due to hyperhomocysteinemia. J. Nutr. 2005, 135, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Zhang, W.; Wang, G.; Zhao, X.; Gao, N.; Liu, Z.; Xu, R. Low Dose of Folic Acid Can Ameliorate Hyperhomocysteinemia-Induced Cardiac Fibrosis and Diastolic Dysfunction in Spontaneously Hypertensive Rats. Int. Heart J. 2021, 62, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Collaboration, H.L.T. Lowering blood homocysteine with folic acid based supplements: Meta-analysis of randomised trials. Homocysteine Lowering Trialists’ Collaboration. BMJ 1998, 316, 894–898. [Google Scholar] [CrossRef]
- Jayedi, A.; Zargar, M.S. Intake of vitamin B6, folate, and vitamin B12 and risk of coronary heart disease: A systematic review and dose-response meta-analysis of prospective cohort studies. Crit. Rev. Food Sci. Nutr. 2019, 59, 2697–2707. [Google Scholar] [CrossRef]
- Jeon, J.; Park, K. Dietary Vitamin B6 Intake Associated with a Decreased Risk of Cardiovascular Disease: A Prospective Cohort Study. Nutrients 2019, 11, 1484. [Google Scholar] [CrossRef]
- Cui, R.; Iso, H.; Date, C.; Kikuchi, S.; Tamakoshi, A. Dietary folate and vitamin b6 and B12 intake in relation to mortality from cardiovascular diseases: Japan collaborative cohort study. Stroke 2010, 41, 1285–1289. [Google Scholar] [CrossRef]
- Vianna, A.C.; Mocelin, A.J.; Matsuo, T.; Morais-Filho, D.; Largura, A.; Delfino, V.A.; Soares, A.E.; Matni, A.M. Uremic hyperhomocysteinemia: A randomized trial of folate treatment for the prevention of cardiovascular events. Hemodial. Int. 2007, 11, 210–216. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.; Zheng, Y.; Muka, T.; Troup, J.; Hu, F.B. Folic Acid Supplementation and the Risk of Cardiovascular Diseases: A Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2016, 5, 3768. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Morita, T.; Sugiyama, K. Cysteine supplementation decreases plasma homocysteine concentration in rats fed on a low-casein diet in rats. Biosci. Biotechnol. Biochem. 2007, 71, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Stabler, S.P.; Allen, R.H.; Abman, S.H.; Maclean, K.N. Altered hepatic sulfur metabolism in cystathionine β-synthase-deficient homocystinuria: Regulatory role of taurine on competing cysteine oxidation pathways. FASEB J. 2014, 28, 4044–4054. [Google Scholar] [CrossRef]
- Fukada, S.; Shimada, Y.; Morita, T.; Sugiyama, K. Suppression of methionine-induced hyperhomocysteinemia by glycine and serine in rats. Biosci. Biotechnol. Biochem. 2006, 70, 2403–2409. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, P.; Steenge, G.R.; Boelsma, E.; van Vliet, T.; Olthof, M.R.; Katan, M.B. Dietary serine and cystine attenuate the homocysteine-raising effect of dietary methionine: A randomized crossover trial in humans. Am. J. Clin. Nutr. 2004, 80, 674–679. [Google Scholar] [CrossRef]
- Kondakçı, G.; Aydın, A.F.; Doğru-Abbasoğlu, S.; Uysal, M. The effect of N-acetylcysteine supplementation on serum homocysteine levels and hepatic and renal oxidative stress in homocysteine thiolactone-treated rats. Arch. Physiol. Biochem. 2017, 123, 128–133. [Google Scholar] [CrossRef]
- Wu, C.; Li, Y.; Liu, S.; Wang, L.; Wang, X. Catalpol inhibits HHcy-induced EndMT in endothelial cells by modulating ROS/NF-κB signaling. BMC Cardiovasc. Disord. 2024, 24, 431. [Google Scholar] [CrossRef]
- Hildebrandt, W.; Sauer, R.; Bonaterra, G.; Dugi, K.A.; Edler, L.; Kinscherf, R. Oral N-acetylcysteine reduces plasma homocysteine concentrations regardless of lipid or smoking status. Am. J. Clin. Nutr. 2015, 102, 1014–1024. [Google Scholar] [CrossRef]
- Thaha, M.; Yogiantoro, M.; Tomino, Y. Intravenous N-acetylcysteine during haemodialysis reduces the plasma concentration of homocysteine in patients with end-stage renal disease. Clin. Drug Investig. 2006, 26, 195–202. [Google Scholar] [CrossRef]
- Scholze, A.; Rinder, C.; Beige, J.; Riezler, R.; Zidek, W.; Tepel, M. Acetylcysteine reduces plasma homocysteine concentration and improves pulse pressure and endothelial function in patients with end-stage renal failure. Circulation 2004, 109, 369–374. [Google Scholar] [CrossRef]
- Nonaka, H.; Tsujino, T.; Watari, Y.; Emoto, N.; Yokoyama, M. Taurine prevents the decrease in expression and secretion of extracellular superoxide dismutase induced by homocysteine: Amelioration of homocysteine-induced endoplasmic reticulum stress by taurine. Circulation 2001, 104, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Xu, J.X.; Zhao, J.; Pang, Y.Z.; Tang, C.S.; Qi, Y.F. Taurine antagonized oxidative stress injury induced by homocysteine in rat vascular smooth muscle cells. Acta Pharmacol. Sin. 2004, 25, 341–346. [Google Scholar]
- Zhang, Z.; Zhao, L.; Zhou, Y.; Lu, X.; Wang, Z.; Wang, J.; Li, W. Taurine ameliorated homocysteine-induced H9C2 cardiomyocyte apoptosis by modulating endoplasmic reticulum stress. Apoptosis 2017, 22, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhao, J.; Xu, J.; Jiang, W.; Tang, C.S.; Qi, Y.F. Effects of taurine and homocysteine on calcium homeostasis and hydrogen peroxide and superoxide anions in rat myocardial mitochondria. Clin. Exp. Pharmacol. Physiol. 2004, 31, 237–243. [Google Scholar] [CrossRef]
- Chang, L.; Xu, J.; Yu, F.; Zhao, J.; Tang, X.; Tang, C. Taurine protected myocardial mitochondria injury induced by hyperhomocysteinemia in rats. Amino Acids 2004, 27, 37–48. [Google Scholar] [CrossRef]
- Yalçinkaya, S.; Unlüçerçi, Y.; Giriş, M.; Olgaç, V.; Doğru-Abbasoğlu, S.; Uysal, M. Oxidative and nitrosative stress and apoptosis in the liver of rats fed on high methionine diet: Protective effect of taurine. Nutrition 2009, 25, 436–444. [Google Scholar] [CrossRef]
- Maclean, K.N.; Jiang, H.; Phinney, W.N.; McLagan, B.M.; Roede, J.R.; Stabler, S.P. Derangement of hepatic polyamine, folate, and methionine cycle metabolism in cystathionine beta-synthase-deficient homocystinuria in the presence and absence of treatment: Possible implications for pathogenesis. Mol. Genet. Metab. 2021, 132, 128–138. [Google Scholar] [CrossRef]
- Van Hove, J.L.K.; Freehauf, C.L.; Ficicioglu, C.; Pena, L.D.M.; Moreau, K.L.; Henthorn, T.K.; Christians, U.; Jiang, H.; Cowan, T.M.; Young, S.P.; et al. Biomarkers of oxidative stress, inflammation, and vascular dysfunction in inherited cystathionine β-synthase deficient homocystinuria and the impact of taurine treatment in a phase 1/2 human clinical trial. J. Inherit. Metab. Dis. 2019, 42, 424–437. [Google Scholar] [CrossRef]
- Martelli, A.; Piragine, E.; Gorica, E.; Citi, V.; Testai, L.; Pagnotta, E.; Lazzeri, L.; Pecchioni, N.; Ciccone, V.; Montanaro, R.; et al. The H2S-Donor Erucin Exhibits Protective Effects against Vascular Inflammation in Human Endothelial and Smooth Muscle Cells. Antioxidants 2021, 10, 961. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Piragine, E.; Citi, V.; Testai, L.; Pagnotta, E.; Ugolini, L.; Lazzeri, L.; Di Cesare Mannelli, L.; Manzo, O.L.; Bucci, M.; et al. Erucin exhibits vasorelaxing effects and antihypertensive activity by H2S-releasing properties. Br. J. Pharmacol. 2020, 177, 824–835. [Google Scholar] [CrossRef]
- Barresi, E.; Nesi, G.; Citi, V.; Piragine, E.; Piano, I.; Taliani, S.; Da Settimo, F.; Rapposelli, S.; Testai, L.; Breschi, M.C.; et al. Iminothioethers as Hydrogen Sulfide Donors: From the Gasotransmitter Release to the Vascular Effects. J. Med. Chem. 2017, 60, 7512–7523. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Testai, L.; Marino, A.; Breschi, M.C.; Calderone, V. Hydrogen sulfide releasing capacity of natural isothiocyanates: Is it a reliable explanation for the multiple biological effects of Brassicaceae? Planta Med. 2014, 80, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.W.; Zhou, J.; Chen, C.S.; Zhao, Y.; Tan, C.H.; Li, L.; Moore, P.K.; Deng, L.W. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.L.; Fang, F.; Qiao, P.F.; Yan, N.; Gao, D.; Yan, Y. AP39, a Mitochondria-Targeted Hydrogen Sulfide Donor, Supports Cellular Bioenergetics and Protects against Alzheimer’s Disease by Preserving Mitochondrial Function in APP/PS1 Mice and Neurons. Oxidative Med. Cell. Longev. 2016, 2016, 8360738. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, X.; Lu, Y.; Liang, D.; Huang, D. Isothiocyanates as H2S Donors Triggered by Cysteine: Reaction Mechanism and Structure and Activity Relationship. Org. Lett. 2019, 21, 5977–5980. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Hormesis: A fundamental concept in biology. Microb. Cell. 2014, 1, 145–149. [Google Scholar] [CrossRef]
- Calderone, V.; Martelli, A.; Testai, L.; Citi, V.; Breschi, M.C. Using hydrogen sulfide to design and develop drugs. Expert. Opin. Drug Discov. 2016, 11, 163–175. [Google Scholar] [CrossRef]
- Spezzini, J.; Piragine, E.; Flori, L.; Calderone, V.; Martelli, A. Natural H2S-donors: A new pharmacological opportunity for the management of overweight and obesity. Phytother. Res. 2024, 38, 2388–2405. [Google Scholar] [CrossRef]
- Citi, V.; Barresi, E.; Piragine, E.; Spezzini, J.; Testai, L.; Da Settimo, F.; Martelli, A.; Taliani, S.; Calderone, V. Anti-Proliferative Properties of the Novel Hybrid Drug Met-ITC, Composed of the Native Drug Metformin with the Addition of an Isothiocyanate H2S Donor Moiety, in Different Cancer Cell Lines. Int. J. Mol. Sci. 2023, 24, 6131. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Bucci, M.; Piragine, E.; Testai, L.; Vellecco, V.; Cirino, G.; Calderone, V. Searching for novel hydrogen sulfide donors: The vascular effects of two thiourea derivatives. Pharmacol. Res. 2020, 159, 105039. [Google Scholar] [CrossRef]
- Zaorska, E.; Hutsch, T.; Gawryś-Kopczyńska, M.; Ostaszewski, R.; Ufnal, M.; Koszelewski, D. Evaluation of thioamides, thiolactams and thioureas as hydrogen sulfide (H2S) donors for lowering blood pressure. Bioorg. Chem. 2019, 88, 102941. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Testai, L.; Citi, V.; Marino, A.; Bellagambi, F.G.; Ghimenti, S.; Breschi, M.C.; Calderone, V. Pharmacological characterization of the vascular effects of aryl isothiocyanates: Is hydrogen sulfide the real player? Vascul. Pharmacol. 2014, 60, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Citi, V.; Corvino, A.; Fiorino, F.; Frecentese, F.; Magli, E.; Perissutti, E.; Santagada, V.; Brogi, S.; Flori, L.; Gorica, E.; et al. Structure-activity relationships study of isothiocyanates for H2S releasing properties: 3-Pyridyl-isothiocyanate as a new promising cardioprotective agent. J. Adv. Res. 2021, 27, 41–53. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Wen, S.; Huang, K.; Huang, J.; Chu, X.; Wang, F.; Pang, L. Novel controlled and targeted releasing hydrogen sulfide system exerts combinational cerebral and myocardial protection after cardiac arrest. J. Nanobiotechnol. 2021, 19, 40. [Google Scholar] [CrossRef]
- Oh, C.; Lee, W.; Park, J.; Choi, J.; Lee, S.; Li, S.; Jung, H.N.; Lee, J.S.; Hwang, J.E.; Park, J.; et al. Development of Spleen Targeting H2S Donor Loaded Liposome for the Effective Systemic Immunomodulation and Treatment of Inflammatory Bowel Disease. ACS Nano 2023, 17, 4327–4345. [Google Scholar] [CrossRef]
- Ali, H.; Opere, C.; Singh, S. In vitro-controlled release delivery system for hydrogen sulfide donor. AAPS PharmSciTech 2014, 15, 910–919. [Google Scholar] [CrossRef]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal 2011, 15, 363–372. [Google Scholar] [CrossRef]
- Kesherwani, V.; Nandi, S.S.; Sharawat, S.K.; Shahshahan, H.R.; Mishra, P.K. Hydrogen sulfide mitigates homocysteine-mediated pathological remodeling by inducing miR-133a in cardiomyocytes. Mol. Cell. Biochem. 2015, 404, 241–250. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, S.; Dong, S.; Wu, J.; Song, M.; Zhong, X.; Liu, Y. Sodium hydrosulfide attenuates hyperhomocysteinemia rat myocardial injury through cardiac mitochondrial protection. Mol. Cell. Biochem. 2015, 399, 189–200. [Google Scholar] [CrossRef]
- Fan, J.; Zheng, F.; Li, S.; Cui, C.; Jiang, S.; Zhang, J.; Cai, J.; Cui, Q.; Yang, J.; Tang, X.; et al. Hydrogen sulfide lowers hyperhomocysteinemia dependent on cystathionine γ lyase S-sulfhydration in ApoE-knockout atherosclerotic mice. Br. J. Pharmacol. 2019, 176, 3180–3192. [Google Scholar] [CrossRef]
- Jiang, S.; Xu, W.; Chen, Z.; Cui, C.; Fan, X.; Cai, J.; Gong, Y.; Geng, B. Hydrogen sulphide reduces hyperhomocysteinaemia-induced endothelial ER stress by sulfhydrating protein disulphide isomerase to attenuate atherosclerosis. J. Cell. Mol. Med. 2021, 25, 3437–3448. [Google Scholar] [CrossRef]
- Piragine, E.; Citi, V.; Lawson, K.; Calderone, V.; Martelli, A. Regulation of blood pressure by natural sulfur compounds: Focus on their mechanisms of action. Biochem. Pharmacol. 2022, 206, 115302. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Shen, X.; Jiang, X.; Shan, H.; Cimini, M.; Fang, P.; Ji, Y.; Park, J.Y.; Drosatos, K.; Yang, X.; et al. Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: Role of insufficient hydrogen sulfide. Redox. Biol. 2018, 16, 215–225. [Google Scholar] [CrossRef]
- Piragine, E.; Citi, V.; Lawson, K.; Calderone, V.; Martelli, A. Potential Effects of Natural H2S-Donors in Hypertension Management. Biomolecules 2022, 12, 581. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous K(ATP) channel opener. Embo J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef]
- Yakovleva, O.V.; Ziganshina, A.R.; Dmitrieva, S.A.; Arslanova, A.N.; Yakovlev, A.V.; Minibayeva, F.V.; Khaertdinov, N.N.; Ziyatdinova, G.K.; Giniatullin, R.A.; Sitdikova, G.F. Hydrogen Sulfide Ameliorates Developmental Impairments of Rat Offspring with Prenatal Hyperhomocysteinemia. Oxidative Med. Cell. Longev. 2018, 2018, 2746873. [Google Scholar] [CrossRef]
- Yakovleva, O.; Bogatova, K.; Mukhtarova, R.; Yakovlev, A.; Shakhmatova, V.; Gerasimova, E.; Ziyatdinova, G.; Hermann, A.; Sitdikova, G. Hydrogen Sulfide Alleviates Anxiety, Motor, and Cognitive Dysfunctions in Rats with Maternal Hyperhomocysteinemia via Mitigation of Oxidative Stress. Biomolecules 2020, 10, 995. [Google Scholar] [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced mitochondrial dysfunctions in brain. Mitochondrion 2020, 50, 158–169. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Hydrogen sulfide suppresses homocysteine-induced glial activation and inflammatory response. Nitric Oxide 2019, 90, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ray, R.S.; Sandhir, R. Hydrogen sulfide attenuates homocysteine-induced neurotoxicity by preventing mitochondrial dysfunctions and oxidative damage: In vitro and in vivo studies. Neurochem. Int. 2018, 120, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Sandhir, R. Neuroprotective Effect of Hydrogen Sulfide in Hyperhomocysteinemia Is Mediated Through Antioxidant Action Involving Nrf2. Neuromolecular. Med. 2018, 20, 475–490. [Google Scholar] [CrossRef]
- Nath, N.; Prasad, H.K.; Kumar, M. Cerebroprotective effects of hydrogen sulfide in homocysteine-induced neurovascular permeability: Involvement of oxidative stress, arginase, and matrix metalloproteinase-9. J. Cell. Physiol. 2019, 234, 3007–3019. [Google Scholar] [CrossRef]
- Tang, X.Q.; Chen, R.Q.; Dong, L.; Ren, Y.K.; Del Soldato, P.; Sparatore, A.; Liao, D.F. Role of paraoxonase-1 in the protection of hydrogen sulfide-donating sildenafil (ACS6) against homocysteine-induced neurotoxicity. J. Mol. Neurosci. 2013, 50, 70–77. [Google Scholar] [CrossRef]
- Mackness, M.; Mackness, B. Human paraoxonase-1 (PON1): Gene structure and expression, promiscuous activities and multiple physiological roles. Gene 2015, 567, 12–21. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Mallamaci, F.; Zoccali, C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Sen, U.; Basu, P.; Abe, O.A.; Givvimani, S.; Tyagi, N.; Metreveli, N.; Shah, K.S.; Passmore, J.C.; Tyagi, S.C. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am. J. Physiol. Renal. Physiol. 2009, 297, F410–F419. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Ren, L.; Pushpakumar, S.; Sen, U. Hydrogen sulphide mitigates homocysteine-induced apoptosis and matrix remodelling in mesangial cells through Akt/FOXO1 signalling cascade. Cell. Signal. 2019, 61, 66–77. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Li, B.; Song, W.; Ding, Z.; Wang, S.; Shan, Y. Sulforaphane attenuates homocysteine-induced endoplasmic reticulum stress through Nrf-2-driven enzymes in immortalized human hepatocytes. J. Agric. Food. Chem. 2014, 62, 7477–7485. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cao, Y.; Chen, S.; Liu, Q.; Chai, J.; Wang, W. Insufficient S-sulfhydration of serum and glucocorticoid-regulated kinase 1 participates in hyperhomocysteinemia-induced liver injury. Free Radic. Biol. Med. 2024, 225, 517–527. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| First Author, Year | Preclinical Model/Subjects | Treatment (Concentration/Dose) | Duration | Main Findings |
|---|---|---|---|---|
| Preclinical studies | ||||
| Cao, 2021 [85] | SHRs with Hcy-induced HHcy | Folic acid (0.4 mg/kg/day, orally) | 6 weeks | ↓ Interstitial and perivascular collagen deposition in cardiomyocytes ↓ Diastolic dysfunction |
| Chang, 2004 [102] | VSMCs exposed to HHcy | Taurine (5, 10, and 20 mM) | 12 h | ↓ LDH and ROS production ↑ Mn-SOD and catalase activity |
| Chang, 2004 [104] | Rat mitochondria exposed to HHcy | Taurine (5,10, and 20 µM) | - | ↓ Ca2+ uptake inhibition |
| Chang, 2004 [105] | Rats with DL-Met-induced HHcy | L-Taurine (1% in the chow) | 6 weeks | ↓ ROS production ↑ Ca2+ fluxes in myocardial mitochondria |
| Cui, 2018 [82] | HUVECs exposed to HHcy | Folic acid (0-1 µM) | 48 h | ↑ Cell viability ↓ Apoptosis |
| Fukada, 2006 [94] | Rats with Met-induced HHcy | Serine (0.5%, 1%, or 2% in the chow; 100, 200, 300, or 500 mg/kg/day, i.p.) | 10 days | ↓ Hcy levels |
| Jiang, 2014 [93] | CBS-deficient HCU mice | Cys (1.5 mg/mL in drinking water) | 1 week | Normalization of hepatic CDO levels |
| Kondakçı, 2017 [96] | Rats with HcyT-induced HHcy | NAC (1 g/kg/day, unknown route of administration) | 6 weeks | ↓ Serum Hcy ↓ Hepatic and renal ROS |
| Lee, 2005 [84] | Rats with Hcy-induced HHcy | Folic acid (8 mg per kg of chow) | 8 weeks | ↓ Vascular damage ↓ Serum Hcy levels |
| Maclean, 2021 [107] | CBS-deficient HCU mice | Taurine (20 mg/mL in drinking water) | 1 week | Normalization of hepatic expression levels of CDO, CSAD, and GOT1 |
| Nonaka, 2009 [101] | VSMCs exposed to HHcy | Taurine (10 mM) | 30 min | ↓ Oxidative stress in the vascular wall |
| Okawa, 2007 [92] | Rats with HHcy induced by low-protein diet | Cys (0.3% or 0.6% in the chow) | 14 days | ↓ HHcy |
| Wu, 2024 [97] | HUVECs exposed to HHcy | NAC (5 mM) | 48 h | ↓ Morphofunctional alterations in the vascular endothelium |
| Yalçinkaya, 2009 [106] | Rats with HHcy induced by high-Met diet | Taurine (1.5% w/v in drinking water) | 6 months | ↓ Oxidative stress ↓ Nitrosative stress ↓ Apoptosis ↓ Hepatic necrosis |
| Zhang, 2014 [83] | HUVECs exposed to HHcy | Folic acid (5–10 nM) | 12 h | ↑ NO production ↑ BH4 levels |
| Zhang, 2017 [103] | Rat cardiomyoblasts exposed to HHcy | Taurine (40 mM) | 60 min | ↓ Apoptosis |
| Clinical Studies | ||||
| Hildebrandt, 2015 [98] | Hyperlipidemic and normolipidemic men | NAC (1.8 g/day, orally) | 4 weeks | ↓ Plasma Hcy concentrations ↓ Systolic blood pressure |
| Scholze, 2004 [100] | Patients with end-stage renal failure | NAC (5 g, i.v.) | During hemodialysis (4 h) | ↓ Plasma Hcy levels ↑ Endothelial function |
| Thaha, 2006 [99] | Patients with end-stage renal failure | NAC (5 g, i.v.) | During hemodialysis (4 h) | ↓ Plasma Hcy levels ↓ Blood pressure ↓ Pulse pressure |
| Van Hove, 2019 [108] | Patients with inherited CBS-deficient HCU | Taurine (75 mg/kg, orally) | 4 days | ↑ FMD |
| Verhoef, 2004 [95] | Healthy men who ingested low-protein diet supplemented with Met (once, at breakfast) | Serine (60.6 mg/kg/BW) and cystine (12.3 mg/kg/BW), both in the diet | Once (at breakfast) | ↓ Plasma Hcy levels |
| Vianna, 2007 [90] | Patients with end-stage renal disease | Folic acid (10 mg, orally) | 2 years | ↓ Blood Hcy levels ↓ Intima-media wall thickness |
| First Author, Year | Preclinical Model | Treatment (Concentration/Dose) | Duration | Main Findings |
|---|---|---|---|---|
| Chang, 2008 [58] | Rats with DL-Hcy-induced HHcy | Unknown source of H2S (2.8 or 14 µmol/kg/day, i.p.) | 3 weeks | ↑ H2S levels in myocardium ↓ Plasma Hcy levels ↓ Mn-SOD and COX activity |
| Cheng, 2018 [136] | Small mesenteric arteries of db/db mice with high-Met diet-induced HHcy | DATS (5 µM) or NaHS (10–60 µM) | 30 min | ↓ Oxidative stress ↑ Vasorelaxation |
| Fan, 2019 [132] | ApoE−/− atherosclerotic mice with L-Met-induced HHcy | NaHS (5.6 mg/kg twice a day, i.p.) or GYY4137 (3.6 mg/kg twice a day, i.p.) | 16 weeks | ↓ Blood lipid levels ↓ Serum Hcy ↓ Atherosclerotic lesions in aortic tissue ↑ CSE and 3-MST expression * in aortic tissue ↑ H2S levels in aortic tissue |
| He, 2014 [153] | Human hepatocytes exposed to HHcy | Sulforaphane (1–20 µM) | 24 h | ↓ ROS production |
| Jiang, 2021 [133] | ApoE−/− atherosclerotic mice with L-Met-induced HHcy | NaHS (5.6 mg/kg twice a day, i.p.) or GYY4137 (3.6 mg/kg twice a day, i.p.) | 16 weeks | ↓ ER stress in aortic plaques |
| Jiang, 2021 [133] | HAECs exposed to HHcy | NaHS (1 mM) or GYY4137 (1 mM) | 2 h | ↓ PDI expression |
| Kesherwani, 2015 [130] | Murine cardiomyocytes exposed to HHcy | Na2S (30 µM) | 24 h | ↓ Cardiac hypertrophy |
| Kumar, 2018a [144] | N2a cells exposed to HHcy | NaHS (250 µM) | 24 h | Prevention of cytotoxicity, ROS production, alterations in mitochondrial membrane potential, apoptosis, and changes in cell cycle |
| Kumar, 2018a [144] | Rats with Hcy-induced HHcy | NaHS (30 µmol/kg/day, i.p.) | 30 days | ↓ DNA fragmentation in the cortex and hippocampus ↑ Memory and cognitive deficits |
| Kumar, 2018b [145] | Rats with Hcy-induced HHcy | NaHS (30 µmol/kg/day, i.p.) | 30 days | Reverse of acetylcholinesterase activity ↓ Chromatin condensation ↓ Oxidative stress in the cortex and hippocampus |
| Kumar, 2019 [143] | Rats with Hcy-induced HHcy | NaHS (30 µmol/kg/day, i.p.) | 30 days | ↑ CBS and CSE activity Normalization of H2S and polysulfides levels in the brain ↓ Pro-inflammatory mediators ↓ iNOS expression ↓ PECAM expression in cortical microvessels Reversal of eNOS expression* in the cortex |
| Kumar, 2020 [142] | Rats with Hcy-induced HHcy | NaHS (30 µmol/kg/day, i.p.) | 30 days | Prevention of apoptosis and ROS production ↓ Mitochondrial swelling in the brain ↓ Nitrite levels ↓ Mn-SOD activity |
| Kumar, 2022 [60] | Rats with Hcy-induced HHcy | NaHS (30 µmol/kg/day, i.p.) | 30 days | Maintenance of blood–brain barrier integrity via inhibition of MMPs |
| Majumder, 2019 [152] | Mesangial cells exposed to HHcy | GYY4137 (250 µM) | 24 h | ↓ MMPs activity ↓ Apoptosis ↓ Oxidative stress ↓ Mitochondrial dysfunction Prevention of collagen I and fibronectin expression |
| Malaviya, 2024 [35] | Human retinal endothelial cells exposed to HHcy | GYY4137 (150 µM) | 96 h | ↑ H2S intracellular levels ↓ ROS mitochondrial levels ↓ DNA damage ↓ Mitophagy |
| Nath, 2019 [146] | Mice with DL-Hcy-induced HHcy | NaHS (20 µM/day, orally) | 10 weeks | Restoration of H2S physiological concentrations in the brain ↑ Antioxidant enzymes activity ↑ GSH levels ↓ MMPs ↓ Nitrite levels Maintenance of blood–brain barrier integrity |
| Nandi, 2017 [61] | Murine cardiomyocytes exposed to HHcy | Na2S (5–100 µM) or GYY4137 (5–100 µM) | 24 h | ↓ CSE expression* ↑ CBS expression ↓ SP1 activity |
| Pushpakumar, 2019 [57] | CBS+/− mice | NaHS (30 µM/day, orally) | 8 weeks | Restoration of renal cortical vascularity Prevention of the increase in MMPs expression and collagen deposition in the kidney |
| Pushpakumar, 2019 [57] | VSMCs exposed to HHcy | NaHS (30 µM, orally) | 48 h | ↓ Proliferation |
| Sen, 2009 [151] | CBS+/− mice | NaHS (30 µM/day orally) | 8 weeks | Prevention of the increase in plasma Hcy ↑ H2S bioavailability ↓ Glomerular oxidative stress ↓ Cell death ↑ GSH/GSSG in cortical tissue Normalization of MMPs activity in the kidney |
| Tang, 2013 [147] | PC12 rat cells exposed to HHcy | Sildenafil (16 µM) | 30 min | Prevention of apoptosis and ROS production |
| Wang, 2015 [131] | Rats with L-Met-induced HHcy | NaHS (80 µM/day, i.p.) | 12 weeks | ↓ Cardiac impairment ↓ Mitochondrial alterations ↓ Cardiomyocytes apoptosis |
| Wang, 2015 [131] | Neonatal rat cardiomyocytes exposed to HHcy | NaHS (80 µM) | 3 days | ↓ ROS production |
| Yakovleva, 2018 [139] | Rat offspring with prenatal HHcy induced by Met | NaHS (3 mg/kg, subcutaneously, alternating 7 days of injections with 7 days of adaptation) | 3 weeks prior to and during pregnancy | Improvement in developmental impairments |
| Yakovleva, 2020 [140] | Rat offspring with prenatal HHcy induced by Met | NaHS (3 mg/kg, subcutaneously, alternating 7 days of injections with 7 days of adaptation) | 3 weeks prior to and during pregnancy | Alleviation of motor and cognitive dysfunctions ↑ SOD and GPx activity in the brain |
| Yakovlev, 2024 [59] | Rat offspring with prenatal HHcy induced by Met | NaHS (3 mg/kg, subcutaneously, alternating 7 days of injections with 7 days of adaptation) | 3 weeks prior to and during pregnancy | ↓ Hcy levels Restoration of H2S levels in the brain Preservation of blood–brain barrier integrity, ↑ mitochondrial activity, and ↓ brain levels of nitrites and pro-inflammatory cytokines |
| Zhu, 2024 [154] | Mice with HHcy induced by high-Met diet | NaHS (10 mg/kg/day, i.p.) | 17 weeks | ↓ Liver injury |
| Zhu, 2024 [154] | HepG2 hepatic cells exposed to HHcy | NaHS (200 µM) | 24 h | ↓ Autophagy ↓ Oxidative stress ↓ Lipid deposition |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flori, L.; Veneziano, S.; Martelli, A.; Piragine, E.; Calderone, V. Transsulfuration Pathway Products and H2S-Donors in Hyperhomocysteinemia: Potential Strategies Beyond Folic Acid. Int. J. Mol. Sci. 2025, 26, 6430. https://doi.org/10.3390/ijms26136430
Flori L, Veneziano S, Martelli A, Piragine E, Calderone V. Transsulfuration Pathway Products and H2S-Donors in Hyperhomocysteinemia: Potential Strategies Beyond Folic Acid. International Journal of Molecular Sciences. 2025; 26(13):6430. https://doi.org/10.3390/ijms26136430
Chicago/Turabian StyleFlori, Lorenzo, Sara Veneziano, Alma Martelli, Eugenia Piragine, and Vincenzo Calderone. 2025. "Transsulfuration Pathway Products and H2S-Donors in Hyperhomocysteinemia: Potential Strategies Beyond Folic Acid" International Journal of Molecular Sciences 26, no. 13: 6430. https://doi.org/10.3390/ijms26136430
APA StyleFlori, L., Veneziano, S., Martelli, A., Piragine, E., & Calderone, V. (2025). Transsulfuration Pathway Products and H2S-Donors in Hyperhomocysteinemia: Potential Strategies Beyond Folic Acid. International Journal of Molecular Sciences, 26(13), 6430. https://doi.org/10.3390/ijms26136430