
Transgenerational Epigenetic and Phenotypic Inheritance Across Five Generations in Sheep

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Transgenerational Effects of Methionine Supplementation on Phenotypes
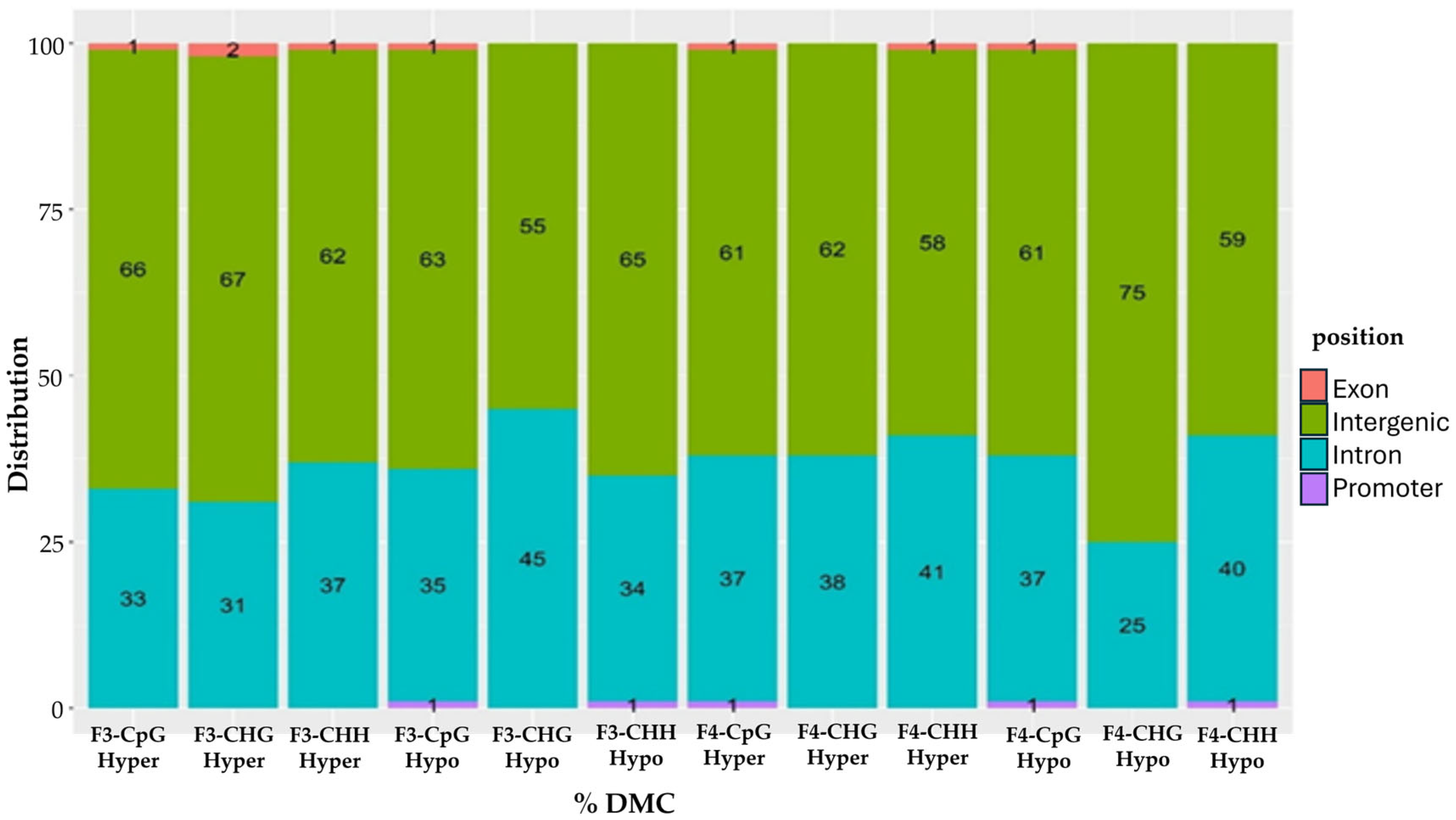
2.2. Overlapping and De Novo Differentially Methylated Cytosines (DMCs) Between Treatment vs. Control in F3 and F4
2.3. Differentially Methylated Genes (DMGs) Between Treatment vs. Control in F3 and F4 Generations
2.4. Transgenerationally Inherited Differentially Methylated Genes (TEI-DMGs)
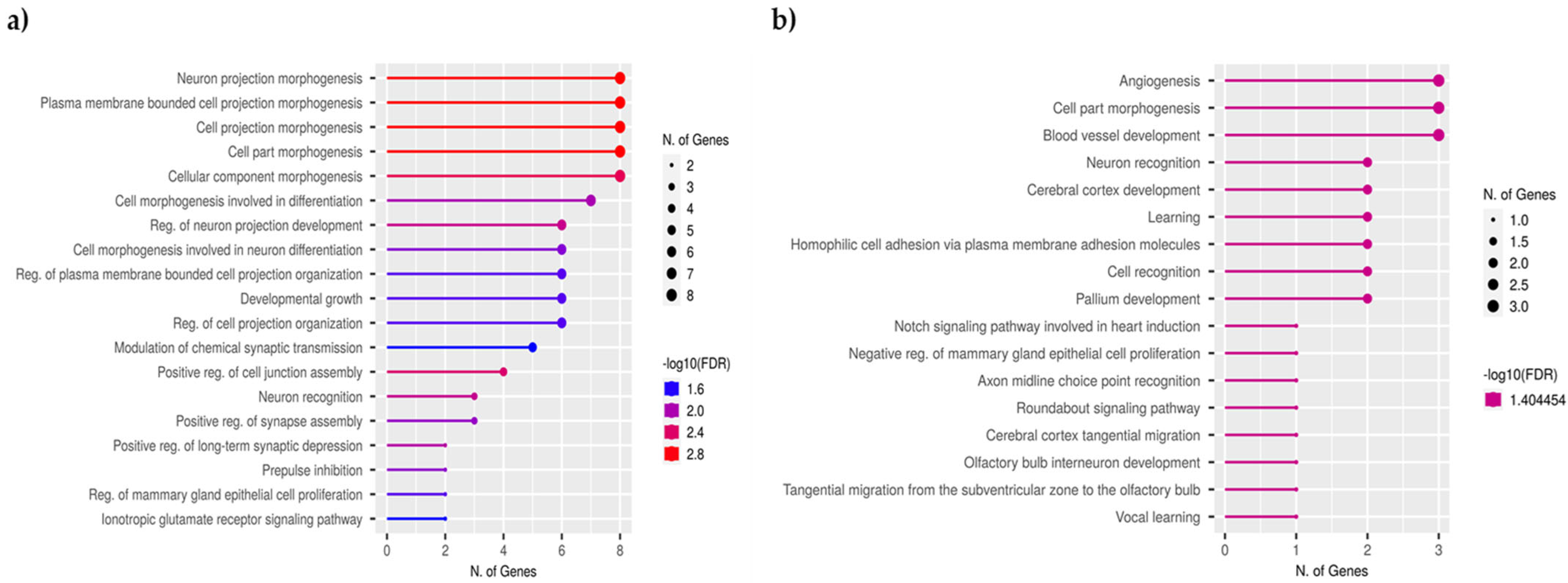
2.5. Functional Annotation of DMGs and TEI-DMGs
3. Discussion
4. Materials and Methods
4.1. Experimental Design, Sample Collection, and Phenotypes
4.2. DNA Extraction and WGBS
4.3. WGBS Alignment
4.4. Differential Methylation Analysis
4.5. Calling Gene Components, DMGs, and TEI-DMGs
4.6. Functional Annotation of the DMGs and TEI-DMGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TEI | Transgenerational epigenetic inheritance |
| DMC | Differentially methylated cytosines |
| DMG | Differentially methylated gene |
| TEI-DMC | Transgenerational epigenetically inherited differentially methylated cytosine |
| TEI-DMG | Transgenerational epigenetically inherited differentially methylated gene |
| BWT | Birth weight |
| WWT | Weaning weight |
| PWT | Post-weaning weight |
| SC | Scrotal circumference |
| ICR | Imprinting control region |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Skinner, M.K. Environmental stress and epigenetic transgenerational inheritance. BMC Med. 2014, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef] [PubMed]
- Khatib, H.; Townsend, J.; Konkel, M.A.; Conidi, G.; Hasselkus, J.A. Calling the question: What is mammalian transgenerational epigenetic inheritance? Epigenetics 2024, 19, 2333586. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch Famine and Its Long-Term Consequences for Adult Health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef]
- Wang, Z.; Li, C.; Yang, Z.; Ma, J.; Zou, Z. Fetal and Infant Exposure to Severe Chinese Famine Increases the Risk of Adult Dyslipidemia: Results from the China Health and Retirement Longitudinal Study. BMC Public. Health 2017, 17, 488. [Google Scholar] [CrossRef]
- Morgan, H.D.; Sutherland, H.G.E.; Martin, D.I.K.; Whitelaw, E. Epigenetic Inheritance at the Agouti Locus in the Mouse. Nat. Genet. 1999, 23, 314. [Google Scholar] [CrossRef]
- Anway, M.D.; Cupp, A.S.; Uzumcu, N.; Skinner, M.K. Toxicology: Epigenetic Transgenerational Actions of Endocrine Disruptors and Male Fertility. Science (1979) 2005, 308, 1466–1469. [Google Scholar] [CrossRef]
- Braz, C.U.; Taylor, T.; Namous, H.; Townsend, J.; Crenshaw, T.; Khatib, H. Paternal Diet Induces Transgenerational Epigenetic Inheritance of DNA Methylation Signatures and Phenotypes in Sheep Model. PNAS Nexus 2022, 1, pgac040. [Google Scholar] [CrossRef]
- Turner, B.M. Epigenetic Responses to Environmental Change and Their Evolutionary Implications. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 3403–3418. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Fitz-James, M.H.; Cavalli, G. Molecular Mechanisms of Transgenerational Epigenetic Inheritance. Nat. Rev. Genet. 2022, 23, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.R.; Veselovska, L.; Kelsey, G. Establishment and Functions of DNA Methylation in the Germline. Epigenomics 2016, 8, 1399–1413. [Google Scholar] [CrossRef]
- Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jerónimo, C. Profiling DNA Methylation Based on Next-Generation Sequencing Approaches: New Insights and Clinical Applications. Genes 2018, 9, 429. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational Epigenetic Inheritance: Myths and Mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef]
- Horsthemke, B. A Critical View on Transgenerational Epigenetic Inheritance in Humans. Nat. Commun. 2018, 9, 2973. [Google Scholar] [CrossRef]
- Horsthemke, B.; Bird, A. Loss of CpG Island Immunity to DNA Methylation Induced by Mutation. Epigenetics Chromatin 2023, 16, 17. [Google Scholar] [CrossRef]
- Díez-Villanueva, A.; Martín, B.; Moratalla-Navarro, F.; Morón-Duran, F.D.; Galván-Femenía, I.; Obón-Santacana, M.; Carreras, A.; de Cid, R.; Peinado, M.A.; Moreno, V. Identification of Intergenerational Epigenetic Inheritance by Whole Genome DNA Methylation Analysis in Trios. Sci. Rep. 2023, 13, 21266. [Google Scholar] [CrossRef]
- Perez, M.F.; Lehner, B. Intergenerational and Transgenerational Epigenetic Inheritance in Animals. Nat. Cell Biol. 2019, 21, 143–151. [Google Scholar] [CrossRef]
- Pastore, A.; Badolati, N.; Manfrevola, F.; Sagliocchi, S.; Laurenzi, V.; Musto, G.; Porreca, V.; Murolo, M.; Chioccarelli, T.; Ciampaglia, R.; et al. Pre-Conceptional Paternal Diet Impacts on Offspring Testosterone Homoeostasis via Epigenetic Modulation of Cyp19a1/Aromatase Activity. npj Metab. Health Dis. 2024, 2, 8. [Google Scholar] [CrossRef]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [CrossRef]
- Daxinger, L.; Whitelaw, E. Understanding Transgenerational Epigenetic Inheritance via the Gametes in Mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Salian, S.; Doshi, T.; Vanage, G. Perinatal Exposure of Rats to Bisphenol a Affects the Fertility of Male Offspring. Life Sci. 2009, 85, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Ponzio, B.F.; Carvalho, M.H.C.; Fortes, Z.B.; Franco, M.D.C. Implications of Maternal Nutrient Restriction in Transgenerational Programming of Hypertension and Endothelial Dysfunction across F1-F3 Offspring. Life Sci. 2012, 90, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K.; Haque, C.G.B.M.; Nilsson, E.; Bhandari, R.; McCarrey, J.R. Environmentally Induced Transgenerational Epigenetic Reprogramming of Primordial Germ Cells and the Subsequent Germ Line. PLoS ONE 2013, 8, e66318. [Google Scholar] [CrossRef]
- Gross, N.; Taylor, T.; Crenshaw, T.; Khatib, H. The Intergenerational Impacts of Paternal Diet on DNA Methylation and Offspring Phenotypes in Sheep. Front. Genet. 2020, 11, e66318. [Google Scholar] [CrossRef]
- Zhang, N. Role of Methionine on Epigenetic Modification of DNA Methylation and Gene Expression in Animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef]
- Yin, T.; König, S. Genetic parameters for body weight from birth to calving and associations between weights with test-day, health, and female fertility traits. J. Dairy Sci. 2018, 101, 2158–2170. [Google Scholar] [CrossRef]
- Patil, V.; Ward, R.L.; Hesson, L.B. The Evidence for Functional Non-CpG Methylation in Mammalian Cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, M.; Wu, H. Mammalian non-CG methylations are conserved and cell-type specific and may have been involved in the evolution of transposon elements. Sci. Rep. 2016, 6, 32207. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Lee, T.H.; Mansoor, S.; Paterson, A.H. Gene Body Methylation Shows Distinct Patterns Associated with Different Gene Origins and Duplication Modes and Has a Heterogeneous Relationship with Gene Expression in Oryza Sativa (Rice). New Phytol. 2013, 198, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Identification and Characterization of a Family of Mammalian Methyl-CpG Binding Proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent Inverse Correlation between DNA Methylation of the First Intron and Gene Expression across Tissues and Species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Shaul, O. How Introns Enhance Gene Expression. Int. J. Biochem. Cell Biol. 2017, 91, 145–155. [Google Scholar] [CrossRef]
- Kim, D.; Shivakumar, M.; Han, S.; Sinclair, M.S.; Lee, Y.J.; Zheng, Y.; Olopade, O.I.; Kim, D.; Lee, Y. Population-Dependent Intron Retention and DNA Methylation in Breast Cancer. Mol. Cancer Res. 2018, 16, 461–469. [Google Scholar] [CrossRef]
- Kubsad, D.; Nilsson, E.E.; King, S.E.; Sadler-Riggleman, I.; Beck, D.; Skinner, M.K. Assessment of Glyphosate Induced Epigenetic Transgenerational Inheritance of Pathologies and Sperm Epimutations: Generational Toxicology. Sci. Rep. 2019, 9, 6372. [Google Scholar] [CrossRef]
- Ben Maamar, M.; Sadler-Riggleman, I.; Beck, D.; McBirney, M.; Nilsson, E.; Klukovich, R.; Xie, Y.; Tang, C.; Yan, W.; Skinner, M.K. Alterations in Sperm DNA Methylation, Non-Coding RNA Expression, and Histone Retention Mediate Vinclozolin-Induced Epigenetic Transgenerational Inheritance of Disease. Environ. Epigenetics 2018, 4, dvy010. [Google Scholar] [CrossRef]
- Blomen, V.A.; Boonstra, J. Stable Transmission of Reversible Modifications: Maintenance of Epigenetic Information through the Cell Cycle. Cell. Mol. Life Sci. 2011, 68, 27–44. [Google Scholar] [CrossRef]
- Lee, G.S.; Conine, C.C. The Transmission of Intergenerational Epigenetic Information by Sperm MicroRNAs. Epigenomes 2022, 6, 12. [Google Scholar] [CrossRef]
- Becker, K.G.; Barnes, K.C.; Bright, T.J.; Wang, S.A. The Genetic Association Database. Nat. Genet. 2004, 36, 431–432. [Google Scholar] [CrossRef]
- Mohamed, F.F.; Ge, C.; Cowling, R.T.; Lucas, D.; Hallett, S.A.; Ono, N.; Binrayes, A.A.; Greenberg, B.; Franceschi, R.T. The Collagen Receptor, Discoidin Domain Receptor 2, Functions in Gli1-Positive Skeletal Progenitors and Chondrocytes to Control Bone Development. Bone Res. 2022, 10, 11. [Google Scholar] [CrossRef]
- Kirk, N.A.; Hwang, Y.E.; Lee, S.G.; Kim, K.-B.; Park, K.-S. Robo1 Loss Has Pleiotropic Effects on Postnatal Development and Survival. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lee, S. The Genetic and Epigenetic Association of LDL Receptor Related Protein 1B (LRP1B) Gene with Childhood Obesity. Sci. Rep. 2019, 9, 1815. [Google Scholar] [CrossRef]
- Tarekegn, G.M.; Strandberg, E.; Andonov, S.; Båge, R.; Ask-Gullstrand, P.; Rius-Vilarrasa, E.; Christensen, J.M.; Berglund, B. Single-Step Genome-Wide Association Study Uncovers Known and Novel Candidate Genomic Regions for Endocrine and Classical Fertility Traits in Swedish Red and Holstein Dairy Cows. Livest. Sci. 2021, 253, 104731. [Google Scholar] [CrossRef]
- Guo, J.; Grow, E.J.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Nie, X.; Guo, Y.; Takei, Y.; Yun, J.; Cai, L.; et al. The Adult Human Testis Transcriptional Cell Atlas. Cell Res. 2018, 28, 1141–1157. [Google Scholar] [CrossRef]
- SanMiguel, J.M.; Bartolomei, M.S. DNA Methylation Dynamics of Genomic Imprinting in Mouse Development. Biol. Reprod. 2018, 99, 252–262. [Google Scholar] [CrossRef]
- Schulz, R.; Proudhon, C.; Bestor, T.H.; Woodfine, K.; Lin, C.S.; Lin, S.P.; Prissette, M.; Oakey, R.J.; Bourc’his, D. The Parental Non-Equivalence of Imprinting Control Regions during Mammalian Development and Evolution. PLoS Genet. 2010, 6, e1001214. [Google Scholar] [CrossRef]
- Gigante, S.; Gouil, Q.; Lucattini, A.; Keniry, A.; Beck, T.; Tinning, M.; Gordon, L.; Woodruff, C.; Speed, T.P.; Blewitt, M.E.; et al. Using Long-Read Sequencing to Detect Imprinted DNA Methylation. Nucleic Acids Res. 2019, 47, e46. [Google Scholar] [CrossRef]
- Townsend, J.; Braz, C.U.; Taylor, T.; Khatib, H. Effects of paternal methionine supplementation on sperm DNA methylation and embryo transcriptome in sheep. Environ. Epigenetics 2023, 9, dvac029. [Google Scholar] [CrossRef]
- Matos, B.; Publicover, S.J.; Castro, L.F.C.; Esteves, P.J.; Fardilha, M. Brain and testis: More alike than previously thought? Open Biol. 2021, 11, 200322. [Google Scholar] [CrossRef]
- Guo, J.H.; Huang, Q.; Studholme, D.J.; Wu, C.Q.; Zhao, Z. Transcriptomic analyses support the similarity of gene expression between brain and testis in human as well as mouse. Cytogenet. Genome Res. 2005, 111, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Braz, C.U.; Passamonti, M.M.; Khatib, H. Characterization of genomic regions escaping epigenetic reprogramming in sheep. Environ. Epigenetics 2023, 10, dvad010. [Google Scholar] [CrossRef] [PubMed]
- Mukasa-Mugerwa, E.; Ezaz, Z. Relationship of testicular growth and size to age, body weight and onset of puberty in menz ram lambs. Theriogenology 1992, 38, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Mächler, M.; Bolker, B.M.; Walker, S.C. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 January 2025).
- Krueger, F. Trim Galore!: A Wrapper Around Cutadapt and FastQC to Consistently Apply Adapter and Quality Trimming to FastQ Files, with Extra Functionality for RRBS Data; Babraham Institute: Cambridge, UK, 2015. [Google Scholar]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Feng, H.; Conneely, K.N.; Wu, H. A Bayesian Hierarchical Model to Detect Differentially Methylated Loci from Single Nucleotide Resolution Sequencing Data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef]
- Dolzhenko, E.; Smith, A.D. Using Beta-Binomial Regression for High-Precision Differential Methylation Analysis in Multifactor Whole-Genome Bisulfite Sequencing Experiments. BMC Bioinform. 2014, 15, 215. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Akalin, A.; Franke, V.; Vlahoviček, K.; Mason, C.E.; Schübeler, D. Genomation: A Toolkit to Summarize, Annotate and Visualize Genomic Intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F3 Generation | F4 Generation | |||||
|---|---|---|---|---|---|---|
| Trait | N | Effect Size | p-Value | N | Effect Size | p-Value |
| BWT (kg) | 178 | −0.22 | 0.026 | 224 | 0.23 | 0.033 |
| WWT (kg) | 178 | 1.02 | 0.052 | 215 | 2.30 | 0.001 |
| PWT (kg) | 162 | 2.72 | 0.081 | 202 | 2.94 | 0.001 |
| LMD (mm) | 81 | −1.56 | 0.015 | 95 | NS | NS |
| SC (cm) | 81 | −0.76 | 0.079 | 95 | NS | NS |
| Generation | F0 | F1 | F2 | F3 | F4 |
|---|---|---|---|---|---|
| F0 | 7286 | ||||
| F1 | 1007 | 3483 | |||
| F2 | 347 | 272 | 4630 | ||
| F3 | 1 | 1 | 8 | 2981 | |
| F4 | 0 | 0 | 1 | 16 | 1726 |
| Generation | DMGs |
|---|---|
| F0 | 1388 |
| F1 | 817 |
| F2 | 1069 |
| F3 | 798 |
| F4 | 553 |
| F0-F1 | 414 |
| F0-F1-F2 | 181 |
| F0-F1-F2-F3 | 41 |
| F0-F1-F2-F3-F4 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kizilaslan, M.; Braz, C.U.; Townsend, J.; Taylor, T.; Crenshaw, T.D.; Khatib, H. Transgenerational Epigenetic and Phenotypic Inheritance Across Five Generations in Sheep. Int. J. Mol. Sci. 2025, 26, 6412. https://doi.org/10.3390/ijms26136412
Kizilaslan M, Braz CU, Townsend J, Taylor T, Crenshaw TD, Khatib H. Transgenerational Epigenetic and Phenotypic Inheritance Across Five Generations in Sheep. International Journal of Molecular Sciences. 2025; 26(13):6412. https://doi.org/10.3390/ijms26136412
Chicago/Turabian StyleKizilaslan, Mehmet, Camila U. Braz, Jessica Townsend, Todd Taylor, Thomas D. Crenshaw, and Hasan Khatib. 2025. "Transgenerational Epigenetic and Phenotypic Inheritance Across Five Generations in Sheep" International Journal of Molecular Sciences 26, no. 13: 6412. https://doi.org/10.3390/ijms26136412
APA StyleKizilaslan, M., Braz, C. U., Townsend, J., Taylor, T., Crenshaw, T. D., & Khatib, H. (2025). Transgenerational Epigenetic and Phenotypic Inheritance Across Five Generations in Sheep. International Journal of Molecular Sciences, 26(13), 6412. https://doi.org/10.3390/ijms26136412