
Epigenetic Regulation of Erythropoiesis: From Developmental Programs to Therapeutic Targets

 , and
, and
Abstract
1. Introduction
2. Dynamic Chromatin Remodeling During Development and Lineage Commitment
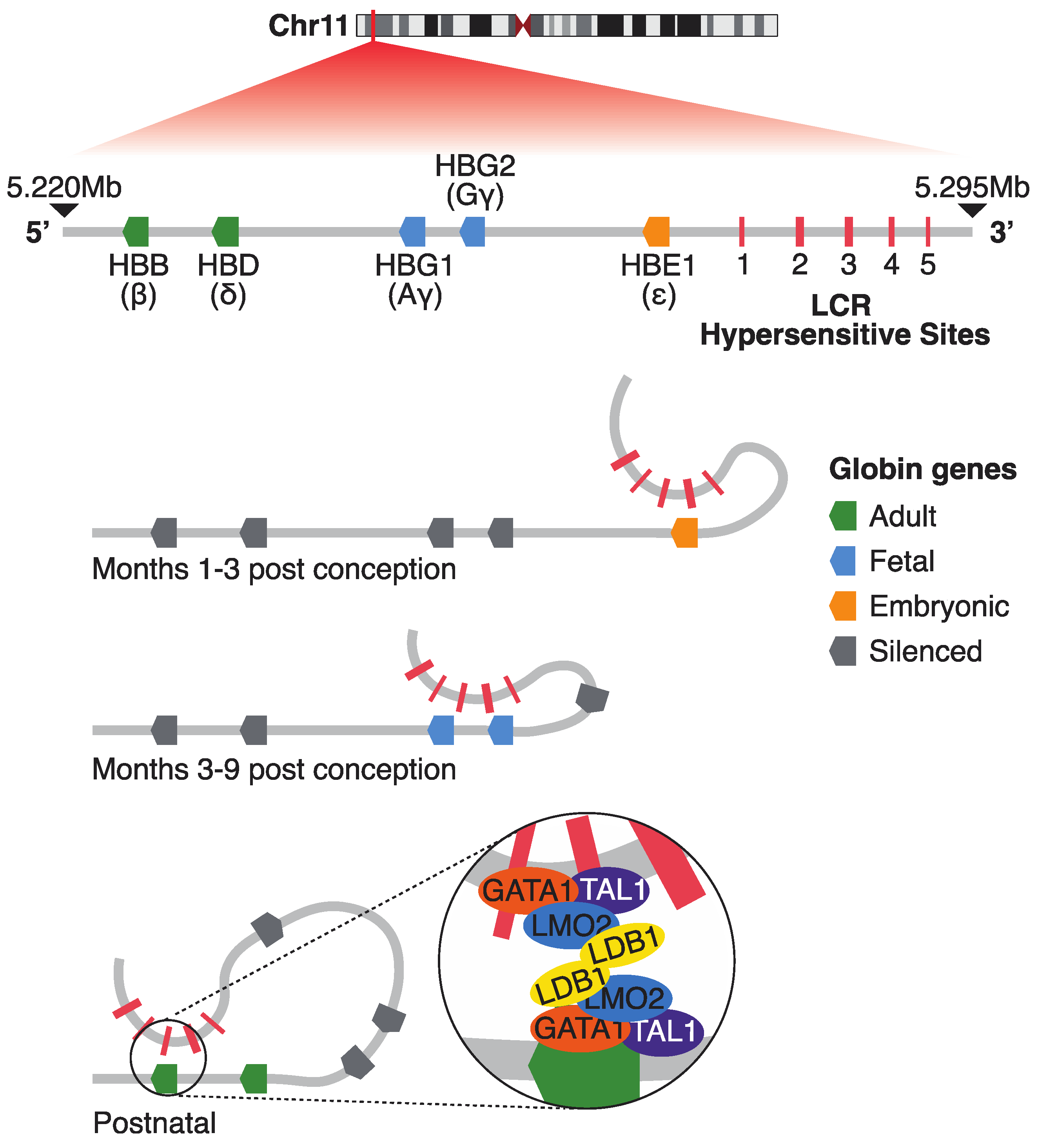
2.1. The β-Globin Locus: A Model of Developmental Gene Regulation
2.2. Chromatin Architecture and the Role of CTCF
2.3. Regulatory Mechanisms at the α-Globin Locus
2.4. Sequence Orientation and Cohesin Flow in Gene Regulation
2.5. Global DNA Methylation Dynamics in Erythropoiesis
3. Role of Erythroid Transcription Factors in Shaping Accessible Chromatin Landscapes
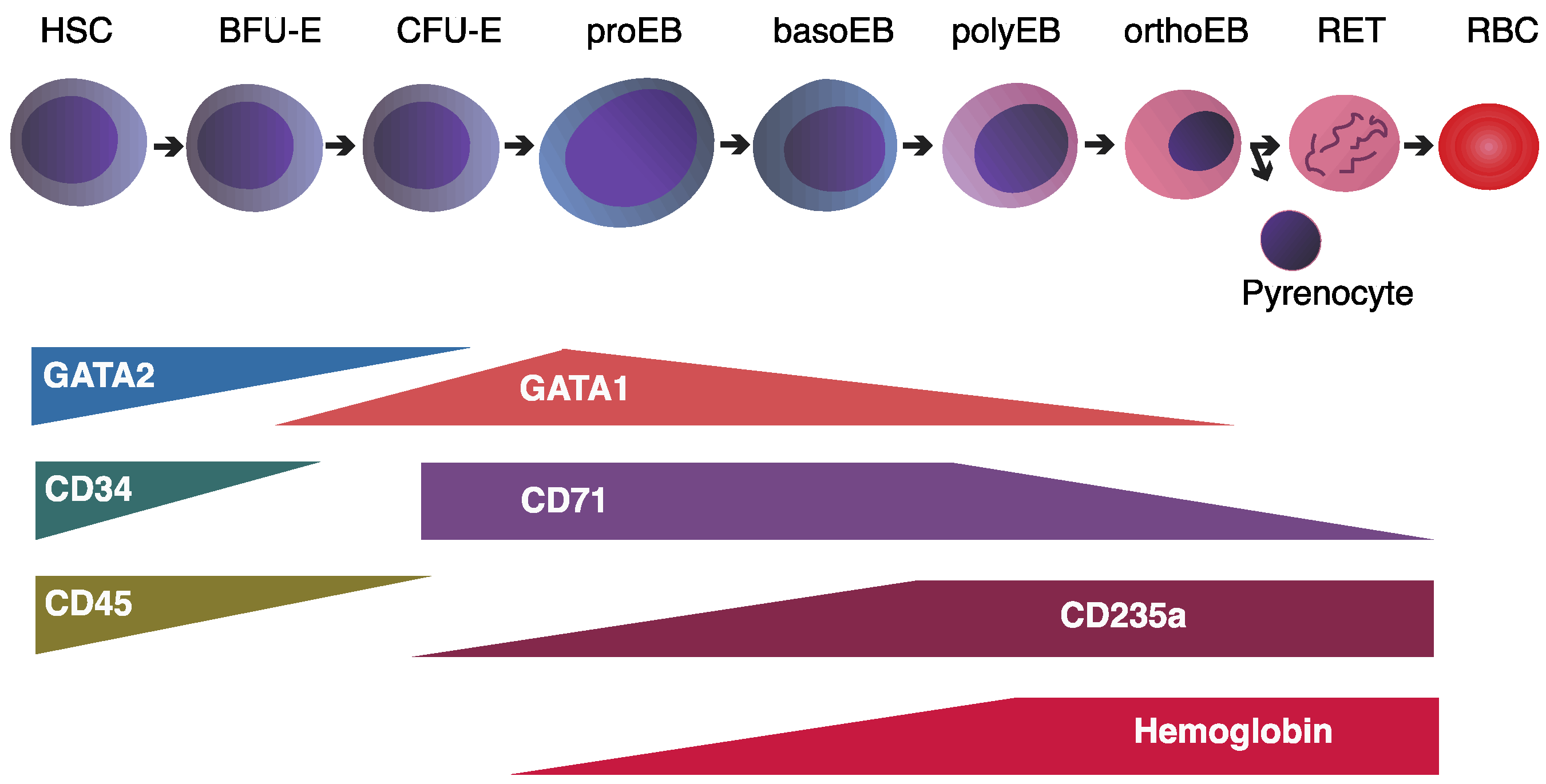
3.1. The GATA2-to-GATA1 Switch: Coordinating Erythroid Lineage Progression
3.2. TAL1: Early Hematopoiesis and β-Globin Regulation
3.3. KLF1: Terminal Erythroid Differentiation and Hemoglobin Switching
4. Non-Coding Variation and Erythroid Phenotypes
4.1. Functional Consequences of Distal Regulatory Variants
4.2. Therapeutic Relevance of Regulatory SNPs
4.3. Non-Coding Variants in Transcription Factor Binding Sites
4.4. Somatic Non-Coding Mutations in Hematologic Malignancies
5. Leveraging Gene Regulation for Translational Applications
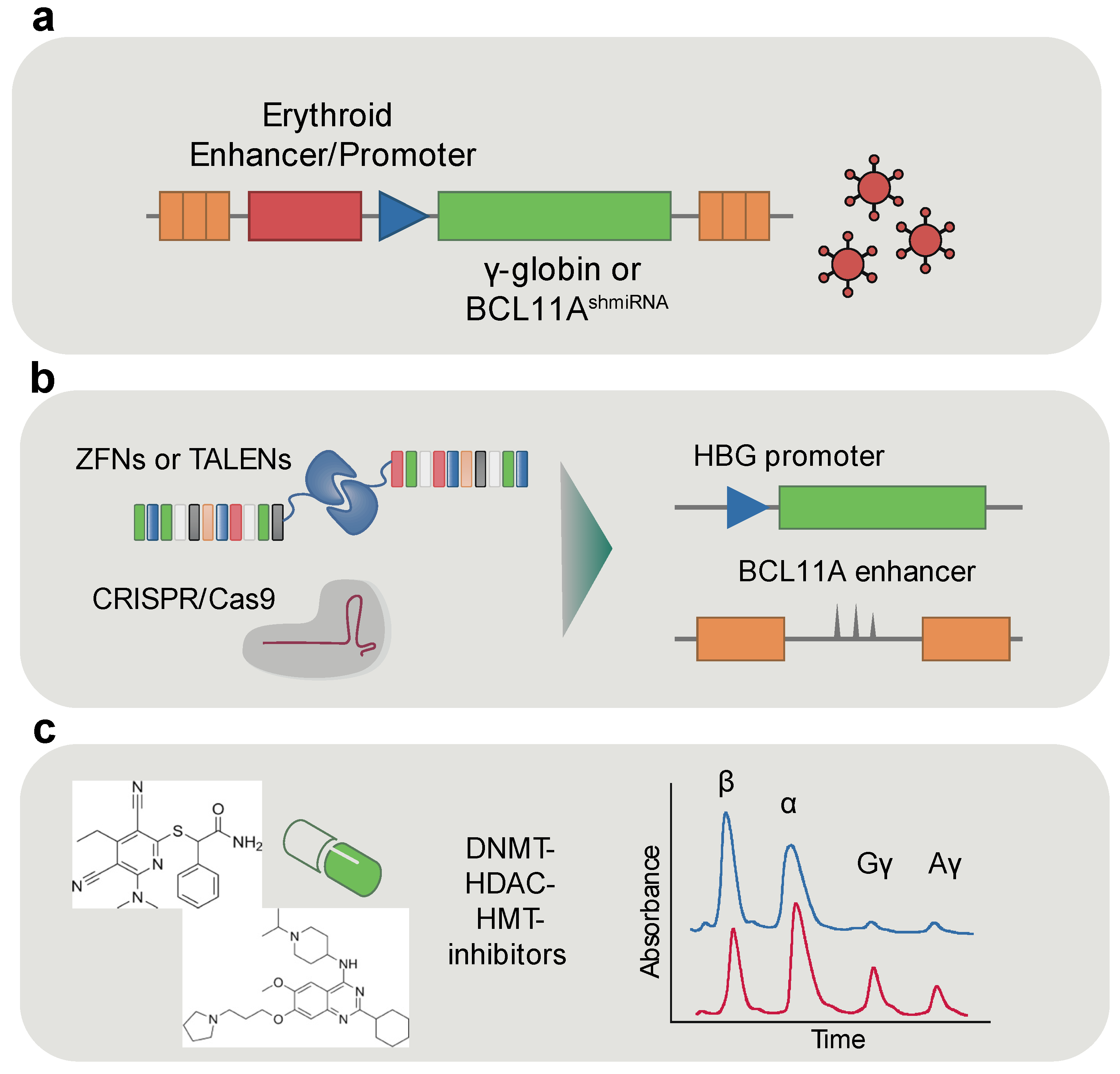
5.1. Erythroid-Specific Enhancers for Gene Therapy
5.2. Fetal Hemoglobin Reactivation
5.3. Epigenetic Therapeutics in Erythroid Disorders
6. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Baron, M.H.; Isern, J.; Fraser, S.T. The Embryonic Origins of Erythropoiesis in Mammals. Blood 2012, 119, 4828–4837. [Google Scholar] [CrossRef] [PubMed]
- Dzierzak, E.; Philipsen, S. Erythropoiesis: Development and Differentiation. Cold Spring Harb. Perspect. Med. 2013, 3, a011601. [Google Scholar] [CrossRef] [PubMed]
- Palis, J. Erythropoiesis in the Mammalian Embryo. Exp. Hematol. 2024, 136, 104283. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Malik, J.; McGrath, K.E.; Kingsley, P.D. Primitive Erythropoiesis in the Mammalian Embryo. Int. J. Dev. Biol. 2010, 54, 1011–1018. [Google Scholar] [CrossRef]
- Palis, J. Primitive and Definitive Erythropoiesis in Mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From Stem Cell to Red Cell: Regulation of Erythropoiesis at Multiple Levels by Multiple Proteins, RNAs, and Chromatin Modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef]
- Schulz, V.P.; Yan, H.; Lezon-Geyda, K.; An, X.; Hale, J.; Hillyer, C.D.; Mohandas, N.; Gallagher, P.G. A Unique Epigenomic Landscape Defines Human Erythropoiesis. Cell Rep. 2019, 28, 2996–3009.e7. [Google Scholar] [CrossRef]
- Georgolopoulos, G.; Psatha, N.; Iwata, M.; Nishida, A.; Som, T.; Yiangou, M.; Stamatoyannopoulos, J.A.; Vierstra, J. Discrete Regulatory Modules Instruct Hematopoietic Lineage Commitment and Differentiation. Nat. Commun. 2021, 12, 6790. [Google Scholar] [CrossRef]
- Martin, E.W.; Rodriguez y Baena, A.; Reggiardo, R.E.; Worthington, A.K.; Mattingly, C.S.; Poscablo, D.M.; Krietsch, J.; McManus, M.T.; Carpenter, S.; Kim, D.H.; et al. Dynamics of Chromatin Accessibility During Hematopoietic Stem Cell Differentiation Into Progressively Lineage-Committed Progeny. Stem Cells 2023, 41, 520–539. [Google Scholar] [CrossRef]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin Architecture Reorganization during Stem Cell Differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef]
- Ludwig, L.S.; Lareau, C.A.; Bao, E.L.; Nandakumar, S.K.; Muus, C.; Ulirsch, J.C.; Chowdhary, K.; Buenrostro, J.D.; Mohandas, N.; An, X.; et al. Transcriptional States and Chromatin Accessibility Underlying Human Erythropoiesis. Cell Rep. 2019, 27, 3228–3240.e7. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, G. Human Hemoglobin Switching. Science 1991, 252, 383. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, G. Control of Globin Gene Expression during Development and Erythroid Differentiation. Exp. Hematol. 2005, 33, 259–271. [Google Scholar] [CrossRef]
- Levings, P.P.; Bungert, J. The Human Β-globin Locus Control Region: A Center of Attraction. Eur. J. Biochem. 2002, 269, 1589–1599. [Google Scholar] [CrossRef]
- Li, Q.; Peterson, K.R.; Fang, X.; Stamatoyannopoulos, G. Locus Control Regions. Blood 2002, 100, 3077–3086. [Google Scholar] [CrossRef]
- Molete, J.M.; Petrykowska, H.; Sigg, M.; Miller, W.; Hardison, R. Functional and Binding Studies of HS3.2 of the Beta-Globin Locus Control Region. Gene 2002, 283, 185–197. [Google Scholar] [CrossRef]
- Bulger, M.; Groudine, M. Functional and Mechanistic Diversity of Distal Transcription Enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef]
- Labbaye, C.; Valtieri, M.; Barberi, T.; Meccia, E.; Masella, B.; Pelosi, E.; Condorelli, G.L.; Testa, U.; Peschle, C. Differential Expression and Functional Role of GATA-2, NF-E2, and GATA-1 in Normal Adult Hematopoiesis. J. Clin. Investig. 1995, 95, 2346–2358. [Google Scholar] [CrossRef]
- Carter, D.; Chakalova, L.; Osborne, C.S.; Dai, Y.; Fraser, P. Long-Range Chromatin Regulatory Interactions in Vivo. Nat. Genet. 2002, 32, 623–626. [Google Scholar] [CrossRef]
- Palstra, R.-J.; Tolhuis, B.; Splinter, E.; Nijmeijer, R.; Grosveld, F.; Laat, W. de The β-Globin Nuclear Compartment in Development and Erythroid Differentiation. Nat. Genet. 2003, 35, 190–194. [Google Scholar] [CrossRef]
- Andrews, N.C.; Erdjument-Bromage, H.; Davidson, M.B.; Tempst, P.; Orkin, S.H. Erythroid Transcription Factor NF-E2 Is a Haematopoietic-Specific Basic–Leucine Zipper Protein. Nature 1993, 362, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Alahouzou, Z.; Miccio, A.; Antoniou, P. Epigenetic Regulation of β-Globin Genes and the Potential to Treat Hemoglobinopathies through Epigenome Editing. Genes 2023, 14, 577. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Kim, S.; Kim, C.G.; Kim, A. The Distinctive Roles of Erythroid Specific Activator GATA-1 and NF-E2 in Transcription of the Human Fetal γ-Globin Genes. Nucleic Acids Res. 2011, 39, 6944–6955. [Google Scholar] [CrossRef]
- Liu, N.; Xu, S.; Yao, Q.; Zhu, Q.; Kai, Y.; Hsu, J.Y.; Sakon, P.; Pinello, L.; Yuan, G.-C.; Bauer, D.E.; et al. Transcription Factor Competition at the γ-Globin Promoters Controls Hemoglobin Switching. Nat. Genet. 2021, 53, 511–520. [Google Scholar] [CrossRef]
- Kim, A.; Dean, A. Chromatin Loop Formation in the β-Globin Locus and Its Role in Globin Gene Transcription. Mol. Cells 2012, 34, 1–6. [Google Scholar] [CrossRef]
- Gurumurthy, A.; Yu, D.T.; Stees, J.R.; Chamales, P.; Gavrilova, E.; Wassel, P.; Li, L.; Stribling, D.; Chen, J.; Brackett, M.; et al. Super-Enhancer Mediated Regulation of Adult β-Globin Gene Expression: The Role of ERNA and Integrator. Nucleic Acids Res. 2021, 49, 1383–1396. [Google Scholar] [CrossRef]
- Kim, Y.W.; Yun, W.J.; Kim, A. Erythroid Activator NF-E2, TAL1 and KLF1 Play Roles in Forming the LCR HSs in the Human Adult β-Globin Locus. Int. J. Biochem. Cell Biol. 2016, 75, 45–52. [Google Scholar] [CrossRef]
- Xu, J.; Sankaran, V.G.; Ni, M.; Menne, T.F.; Puram, R.V.; Kim, W.; Orkin, S.H. Transcriptional Silencing of γ-Globin by BCL11A Involves Long-Range Interactions and Cooperation with SOX6. Genes Dev. 2010, 24, 783–798. [Google Scholar] [CrossRef]
- Jawaid, K.; Wahlberg, K.; Thein, S.L.; Best, S. Binding Patterns of BCL11A in the Globin and GATA1 Loci and Characterization of the BCL11A Fetal Hemoglobin Locus. Blood Cells Mol. Dis. 2010, 45, 140–146. [Google Scholar] [CrossRef]
- Zheng, G.; Orkin, S.H. Transcriptional Repressor BCL11A in Erythroid Cells. Adv. Exp. Med. Biol. 2024, 1459, 199–215. [Google Scholar] [CrossRef]
- Enver, T.; Zhang, J.-W.; Anagnou, N.P.; Stamatoyannopoulos, G.; Papayannopoulou, T. Developmental Programs of Human Erythroleukemia Cells: Globin Gene Expression and Methylation. Mol. Cell. Biol. 1988, 8, 4917–4926. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Li, X.; Azzo, A.; Truong, T.; Dozmorov, M.; Lyons, C.; Manna, A.K.; Williams, D.C.; Ginder, G.D. MBD2a–NuRD Binds to the Methylated γ-Globin Gene Promoter and Uniquely Forms a Complex Required for Silencing of HbF Expression. Proc. Natl. Acad. Sci. USA 2023, 120, e2302254120. [Google Scholar] [CrossRef] [PubMed]
- Enver, T.; Zhang, J.W.; Papayannopoulou, T.; Stamatoyannopoulos, G. DNA Methylation: A Secondary Event in Globin Gene Switching? Genes Dev. 1988, 2, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maurano, M.T.; Qu, H.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sandstrom, R.; et al. Widespread Plasticity in CTCF Occupancy Linked to DNA Methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Handel, B.V.; Mikkola, H.K.A.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human Fetal Hemoglobin Expression Is Regulated by the Developmental Stage-Specific Repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Dean, A.; Blobel, G.A. Controlling Long-Range Genomic Interactions at a Native Locus by Targeted Tethering of a Looping Factor. Cell 2012, 149, 1233–1244. [Google Scholar] [CrossRef]
- Song, S.-H.; Kim, A.; Ragoczy, T.; Bender, M.A.; Groudine, M.; Dean, A. Multiple Functions of Ldb1 Required for β-Globin Activation during Erythroid Differentiation. Blood 2010, 116, 2356–2364. [Google Scholar] [CrossRef]
- Higgs, D.R.; Garrick, D.; Anguita, E.; Gobbi, M.D.; Hughes, J.; Muers, M.; Vernimmen, D.; Lower, K.; Law, M.; Argentaro, A.; et al. Understanding A-Globin Gene Regulation: Aiming to Improve the Management of Thalassemia. Ann. N. York Acad. Sci. 2005, 1054, 92–102. [Google Scholar] [CrossRef]
- Gobbi, M.D.; Anguita, E.; Hughes, J.; Sloane-Stanley, J.A.; Sharpe, J.A.; Koch, C.M.; Dunham, I.; Gibbons, R.J.; Wood, W.G.; Higgs, D.R. Tissue-Specific Histone Modification and Transcription Factor Binding in α Globin Gene Expression. Blood 2007, 110, 4503–4510. [Google Scholar] [CrossRef]
- Higgs, D.R.; Wood, W.G. Long-Range Regulation of α Globin Gene Expression during Erythropoiesis. Curr. Opin. Hematol. 2008, 15, 176–183. [Google Scholar] [CrossRef]
- Vernimmen, D.; Marques-Kranc, F.; Sharpe, J.A.; Sloane-Stanley, J.A.; Wood, W.G.; Wallace, H.A.C.; Smith, A.J.H.; Higgs, D.R. Chromosome Looping at the Human α-Globin Locus Is Mediated via the Major Upstream Regulatory Element (HS −40). Blood 2009, 114, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Garrick, D.; Gobbi, M.D.; Samara, V.; Rugless, M.; Holland, M.; Ayyub, H.; Lower, K.; Sloane-Stanley, J.; Gray, N.; Koch, C.; et al. The Role of the Polycomb Complex in Silencing α-Globin Gene Expression in Nonerythroid Cells. Blood 2008, 112, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Vernimmen, D.; Lynch, M.D.; Gobbi, M.D.; Garrick, D.; Sharpe, J.A.; Sloane-Stanley, J.A.; Smith, A.J.H.; Higgs, D.R. Polycomb Eviction as a New Distant Enhancer Function. Genes Dev. 2011, 25, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Oudelaar, A.M.; Davies, J.O.J.; Hanssen, L.L.P.; Telenius, J.M.; Schwessinger, R.; Liu, Y.; Brown, J.M.; Downes, D.J.; Chiariello, A.M.; Bianco, S.; et al. Single-Allele Chromatin Interactions Identify Regulatory Hubs in Dynamic Compartmentalized Domains. Nat. Genet. 2018, 50, 1744–1751. [Google Scholar] [CrossRef]
- Chiariello, A.M.; Bianco, S.; Oudelaar, A.M.; Esposito, A.; Annunziatella, C.; Fiorillo, L.; Conte, M.; Corrado, A.; Prisco, A.; Larke, M.S.C.; et al. A Dynamic Folded Hairpin Conformation Is Associated with α-Globin Activation in Erythroid Cells. Cell Rep. 2020, 30, 2125–2135.e5. [Google Scholar] [CrossRef]
- Oudelaar, A.M.; Beagrie, R.A.; Gosden, M.; de Ornellas, S.; Georgiades, E.; Kerry, J.; Hidalgo, D.; Carrelha, J.; Shivalingam, A.; El-Sagheer, A.H.; et al. Dynamics of the 4D Genome during in Vivo Lineage Specification and Differentiation. Nat. Commun. 2020, 11, 2722. [Google Scholar] [CrossRef]
- Hanssen, L.L.P.; Kassouf, M.T.; Oudelaar, A.M.; Biggs, D.; Preece, C.; Downes, D.J.; Gosden, M.; Sharpe, J.A.; Sloane-Stanley, J.A.; Hughes, J.R.; et al. Tissue-Specific CTCF–Cohesin-Mediated Chromatin Architecture Delimits Enhancer Interactions and Function in Vivo. Nat. Cell Biol. 2017, 19, 952–961. [Google Scholar] [CrossRef]
- Georgiades, E.; Harrold, C.; Roberts, N.; Kassouf, M.; Riva, S.G.; Sanders, E.; Downes, D.; Francis, H.S.; Blayney, J.; Oudelaar, A.M.; et al. Active Regulatory Elements Recruit Cohesin to Establish Cell Specific Chromatin Domains. Sci. Rep. 2025, 15, 11780. [Google Scholar] [CrossRef]
- Hua, P.; Badat, M.; Hanssen, L.L.P.; Hentges, L.D.; Crump, N.; Downes, D.J.; Jeziorska, D.M.; Oudelaar, A.M.; Schwessinger, R.; Taylor, S.; et al. Defining Genome Architecture at Base-Pair Resolution. Nature 2021, 595, 125–129. [Google Scholar] [CrossRef]
- Sanborn, A.; Rao, S.; Huang, S.; Durand, N.; Huntley, M.; Jewett, A.; Bochkov, I.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin Extrusion Explains Key Features of Loop and Domain Formation in Wild-type and Engineered Genomes. FASEB J. 2016, 30, 588.1. [Google Scholar] [CrossRef]
- Kassouf, M.T.; Francis, H.S.; Gosden, M.; Suciu, M.C.; Downes, D.J.; Harrold, C.; Larke, M.; Oudelaar, M.; Cornell, L.; Blayney, J.; et al. The α-Globin Super-Enhancer Acts in an Orientation-Dependent Manner. Nat. Commun. 2025, 16, 1033. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Liu, Q.; Bungert, J.; Engel, J.D. Effects of Altered Gene Order or Orientation of the Locus Control Region on Human β-Globin Gene Expression in Mice. Nature 1999, 398, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, D.; Branco, M.R.; Splinter, E.; Klous, P.; van IJcken, W.; Swagemakers, S.; Koutsourakis, M.; van der Spek, P.; Pombo, A.; Laat, W. de Transcription and Chromatin Organization of a Housekeeping Gene Cluster Containing an Integrated β-Globin Locus Control Region. PLoS Genet. 2008, 4, e1000016. [Google Scholar] [CrossRef]
- Felder, A.-K.; Tjalsma, S.J.D.; Verhagen, H.J.M.P.; Majied, R.; Verstegen, M.J.A.M.; Verheul, T.C.J.; Mohnani, R.; Gremmen, R.; Krijger, P.H.L.; Philipsen, S.; et al. Reactivation of Developmentally Silenced Globin Genes through Genomic Deletions Reveals That Enhancer Distance Matters. bioRxiv 2025. bioRxiv:2025.01.13.632719. [Google Scholar] [CrossRef]
- Shearstone, J.R.; Pop, R.; Bock, C.; Boyle, P.; Meissner, A.; Socolovsky, M. Global DNA Demethylation During Mouse Erythropoiesis in Vivo. Science 2011, 334, 799–802. [Google Scholar] [CrossRef]
- Bartholdy, B.; Lajugie, J.; Yan, Z.; Zhang, S.; Mukhopadhyay, R.; Greally, J.M.; Suzuki, M.; Bouhassira, E.E. Mechanisms of Establishment and Functional Significance of DNA Demethylation during Erythroid Differentiation. Blood Adv. 2018, 2, 1833–1852. [Google Scholar] [CrossRef]
- Lessard, S.; Beaudoin, M.; Benkirane, K.; Lettre, G. Comparison of DNA Methylation Profiles in Human Fetal and Adult Red Blood Cell Progenitors. Genome Med. 2015, 7, 1. [Google Scholar] [CrossRef]
- Palii, C.G.; Cheng, Q.; Gillespie, M.A.; Shannon, P.; Mazurczyk, M.; Napolitani, G.; Price, N.D.; Ranish, J.A.; Morrissey, E.; Higgs, D.R.; et al. Single-Cell Proteomics Reveal That Quantitative Changes in Co-Expressed Lineage-Specific Transcription Factors Determine Cell Fate. Cell Stem Cell 2019, 24, 812–820.e5. [Google Scholar] [CrossRef]
- Merika, M.; Orkin, S.H. DNA-Binding Specificity of GATA Family Transcription Factors. Mol. Cell. Biol. 1993, 13, 3999–4010. [Google Scholar] [CrossRef]
- Kang, H.; Mesquitta, W.-T.; Jung, H.S.; Moskvin, O.V.; Thomson, J.A.; Slukvin, I.I. GATA2 Is Dispensable for Specification of Hemogenic Endothelium but Promotes Endothelial-to-Hematopoietic Transition. Stem Cell Rep. 2018, 11, 197–211. [Google Scholar] [CrossRef]
- Peters, I.J.A.; de Pater, E.; Zhang, W. The Role of GATA2 in Adult Hematopoiesis and Cell Fate Determination. Front. Cell Dev. Biol. 2023, 11, 1250827. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Ikawa, Y.; Todokoro, K. GATA-1 Transactivates Erythropoietin Receptor Gene, and Erythropoietin Receptor-Mediated Signals Enhance GATA-1 Gene Expression. Nucleic Acids Res. 1991, 19, 3843–3848. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Ito, K.; Merghoub, T.; Poliseno, L.; Hobbs, R.M.; Wang, G.; Dong, L.; Maeda, M.; Dore, L.C.; Zelent, A.; et al. LRF Is an Essential Downstream Target of GATA1 in Erythroid Development and Regulates BIM-Dependent Apoptosis. Dev. Cell 2009, 17, 527–540. [Google Scholar] [CrossRef]
- Vyas, P.; Ault, K.; Jackson, C.W.; Orkin, S.H.; Shivdasani, R.A. Consequences of GATA-1 Deficiency in Megakaryocytes and Platelets. Blood 1999, 93, 2867–2875. [Google Scholar] [CrossRef]
- Shivdasani, R.A.; Fujiwara, Y.; McDevitt, M.A.; Orkin, S.H. A Lineage-selective Knockout Establishes the Critical Role of Transcription Factor GATA-1 in Megakaryocyte Growth and Platelet Development. EMBO J. 1997, 16, 3965–3973. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Browne, C.P.; Cunniff, K.; Goff, S.C.; Orkin, S.H. Arrested Development of Embryonic Red Cell Precursors in Mouse Embryos Lacking Transcription Factor GATA-1. Proc. Natl. Acad. Sci. USA 1996, 93, 12355–12358. [Google Scholar] [CrossRef]
- Cantor, A.B.; Orkin, S.H. Coregulation of GATA Factors by the Friend of GATA (FOG) Family of Multitype Zinc Finger Proteins. Semin. Cell Dev. Biol. 2005, 16, 117–128. [Google Scholar] [CrossRef]
- Stachura, D.L.; Chou, S.T.; Weiss, M.J. Early Block to Erythromegakaryocytic Development Conferred by Loss of Transcription Factor GATA-1. Blood 2006, 107, 87–97. [Google Scholar] [CrossRef]
- MORCEAU, F.; SCHNEKENBURGER, M.; DICATO, M.; DIEDERICH, M. GATA-1: Friends, Brothers, and Coworkers. Ann. N. Y. Acad. Sci. 2004, 1030, 537–554. [Google Scholar] [CrossRef]
- Lowry, J.A.; Mackay, J.P. GATA-1: One Protein, Many Partners. Int. J. Biochem. Cell Biol. 2006, 38, 6–11. [Google Scholar] [CrossRef]
- Cantor, A.B.; Orkin, S.H. Transcriptional Regulation of Erythropoiesis: An Affair Involving Multiple Partners. Oncogene 2002, 21, 3368–3376. [Google Scholar] [CrossRef] [PubMed]
- Tsang, A.P.; Visvader, J.E.; Turner, C.A.; Fujiwara, Y.; Yu, C.; Weiss, M.J.; Crossley, M.; Orkin, S.H. FOG, a Multitype Zinc Finger Protein, Acts as a Cofactor for Transcription Factor GATA-1 in Erythroid and Megakaryocytic Differentiation. Cell 1997, 90, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Ohneda, K.; Engel, J.D.; Trainor, C.D.; Yamamoto, M. Transgenic Rescue of GATA-1–Deficient Mice with GATA-1 Lacking a FOG-1 Association Site Phenocopies Patients with X-Linked Thrombocytopenia. Blood 2004, 103, 2560–2567. [Google Scholar] [CrossRef] [PubMed]
- Letting, D.L.; Chen, Y.-Y.; Rakowski, C.; Reedy, S.; Blobel, G.A. Context-Dependent Regulation of GATA-1 by Friend of GATA-1. Proc. Natl. Acad. Sci. USA 2004, 101, 476–481. [Google Scholar] [CrossRef]
- Crispino, J.D.; Lodish, M.B.; MacKay, J.P.; Orkin, S.H. Use of Altered Specificity Mutants to Probe a Specific Protein–Protein Interaction in Differentiation the GATA-1:FOG Complex. Mol. Cell 1999, 3, 219–228. [Google Scholar] [CrossRef]
- Chickarmane, V.; Enver, T.; Peterson, C. Computational Modeling of the Hematopoietic Erythroid-Myeloid Switch Reveals Insights into Cooperativity, Priming, and Irreversibility. PLoS Comput. Biol. 2009, 5, e1000268. [Google Scholar] [CrossRef]
- Burda, P.; Curik, N.; Kokavec, J.; Pospisil, V.; Skoultchi, A.I.; Zavadil, J.; Stopka, T. Fog1 and Cebpa Are DNA Targets of GATA-1/PU.1 Antagonism during Leukemia Differentiation. Blood 2007, 110, 4121. [Google Scholar] [CrossRef]
- Fujiwara, T.; Sasaki, K.; Saito, K.; Hatta, S.; Ichikawa, S.; Kobayashi, M.; Okitsu, Y.; Fukuhara, N.; Onishi, Y.; Harigae, H. Forced FOG1 Expression in Erythroleukemia Cells: Induction of Erythroid Genes and Repression of Myelo-Lymphoid Transcription Factor PU.1. Biochem. Biophys. Res. Commun. 2017, 485, 380–387. [Google Scholar] [CrossRef]
- Boyes, J.; Byfield, P.; Nakatani, Y.; Ogryzko, V. Regulation of Activity of the Transcription Factor GATA-1 by Acetylation. Nature 1998, 396, 594–598. [Google Scholar] [CrossRef]
- Hung, H.-L.; Lau, J.; Kim, A.Y.; Weiss, M.J.; Blobel, G.A. CREB-Binding Protein Acetylates Hematopoietic Transcription Factor GATA-1 at Functionally Important Sites. Mol. Cell. Biol. 1999, 19, 3496–3505. [Google Scholar] [CrossRef]
- Papadopoulos, G.L.; Karkoulia, E.; Tsamardinos, I.; Porcher, C.; Ragoussis, J.; Bungert, J.; Strouboulis, J. GATA-1 Genome-Wide Occupancy Associates with Distinct Epigenetic Profiles in Mouse Fetal Liver Erythropoiesis. Nucleic Acids Res. 2013, 41, 4938–4948. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Huang, Z.; Olivey, H.E.; Gurbuxani, S.; Crispino, J.D.; Svensson, E.C. FOG-1-mediated Recruitment of NuRD Is Required for Cell Lineage Re-enforcement during Haematopoiesis. EMBO J. 2010, 29, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.D.; Miccio, A.; Bersenev, A.; Wang, Y.; Hong, W.; Zhang, Z.; Poncz, M.; Tong, W.; Blobel, G.A. FOG1 Requires NuRD to Promote Hematopoiesis and Maintain Lineage Fidelity within the Megakaryocytic-Erythroid Compartment. Blood 2010, 115, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Mancini, E.; Sanjuan-Pla, A.; Luciani, L.; Moore, S.; Grover, A.; Zay, A.; Rasmussen, K.D.; Luc, S.; Bilbao, D.; O’Carroll, D.; et al. FOG-1 and GATA-1 Act Sequentially to Specify Definitive Megakaryocytic and Erythroid Progenitors. EMBO J. 2012, 31, 351–365. [Google Scholar] [CrossRef]
- Ferreira, R.; Wai, A.; Shimizu, R.; Gillemans, N.; Rottier, R.; von Lindern, M.; Ohneda, K.; Grosveld, F.; Yamamoto, M.; Philipsen, S. Dynamic Regulation of Gata Factor Levels Is More Important than Their Identity. Blood 2007, 109, 5481–5490. [Google Scholar] [CrossRef]
- Robb, L.; Elwood, N.J.; Elefanty, A.G.; Köntgen, F.; Li, R.; Barnett, L.D.; Begley, C.G. The Scl Gene Product Is Required for the Generation of All Hematopoietic Lineages in the Adult Mouse. EMBO J. 1996, 15, 4123–4129. [Google Scholar] [CrossRef]
- Porcher, C.; Swat, W.; Rockwell, K.; Fujiwara, Y.; Alt, F.W.; Orkin, S.H. The T Cell Leukemia Oncoprotein SCL/Tal-1 Is Essential for Development of All Hematopoietic Lineages. Cell 1996, 86, 47–57. [Google Scholar] [CrossRef]
- Porcher, C.; Chagraoui, H.; Kristiansen, M.S. SCL/TAL1: A Multifaceted Regulator from Blood Development to Disease. Blood 2017, 129, 2051–2060. [Google Scholar] [CrossRef]
- Palii, C.G.; Perez-Iratxeta, C.; Yao, Z.; Cao, Y.; Dai, F.; Davison, J.; Atkins, H.; Allan, D.; Dilworth, F.J.; Gentleman, R.; et al. Differential Genomic Targeting of the Transcription Factor TAL1 in Alternate Haematopoietic Lineages. EMBO J. 2011, 30, 494–509. [Google Scholar] [CrossRef]
- Finger, L.R.; Kagan, J.; Christopher, G.; Kurtzberg, J.; Hershfield, M.S.; Nowell, P.C.; Croce, C.M. Involvement of the TCL5 Gene on Human Chromosome 1 in T-Cell Leukemia and Melanoma. Proc. Natl. Acad. Sci. USA 1989, 86, 5039–5043. [Google Scholar] [CrossRef]
- Sanda, T.; Lawton, L.N.; Barrasa, M.I.; Fan, Z.P.; Kohlhammer, H.; Gutierrez, A.; Ma, W.; Tatarek, J.; Ahn, Y.; Kelliher, M.A.; et al. Core Transcriptional Regulatory Circuit Controlled by the TAL1 Complex in Human T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, A.; Lin, I.-J.; Barrow, J.; Liang, S.Y.; Masannat, J.; Strouboulis, J.; Huang, S.; Bungert, J. Role of Helix-Loop-Helix Proteins during Differentiation of Erythroid Cells. Mol. Cell. Biol. 2011, 31, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Kassouf, M.T.; Hughes, J.R.; Taylor, S.; McGowan, S.J.; Soneji, S.; Green, A.L.; Vyas, P.; Porcher, C. Genome-Wide Identification of TAL1′s Functional Targets: Insights into Its Mechanisms of Action in Primary Erythroid Cells. Genome Res. 2010, 20, 1064–1083. [Google Scholar] [CrossRef] [PubMed]
- Yun, W.J.; Kim, Y.W.; Kang, Y.; Lee, J.; Dean, A.; Kim, A. The Hematopoietic Regulator TAL1 Is Required for Chromatin Looping between the β-Globin LCR and Human γ-Globin Genes to Activate Transcription. Nucleic Acids Res. 2014, 42, 4283–4293. [Google Scholar] [CrossRef] [PubMed]
- Malyavantham, K.S.; Bhattacharya, S.; Barbeitos, M.; Mukherjee, L.; Xu, J.; Fackelmayer, F.O.; Berezney, R. Identifying Functional Neighborhoods within the Cell Nucleus: Proximity Analysis of Early S-phase Replicating Chromatin Domains to Sites of Transcription, RNA Polymerase II, HP1γ, Matrin 3 and SAF-A. J. Cell. Biochem. 2008, 105, 391–403. [Google Scholar] [CrossRef]
- Porcu, S.; Manchinu, M.F.; Marongiu, M.F.; Sogos, V.; Poddie, D.; Asunis, I.; Porcu, L.; Marini, M.G.; Moi, P.; Cao, A.; et al. Klf1 Affects DNase II-Alpha Expression in the Central Macrophage of a Fetal Liver Erythroblastic Island: A Non-Cell-Autonomous Role in Definitive Erythropoiesis. Mol. Cell. Biol. 2011, 31, 4144–4154. [Google Scholar] [CrossRef]
- Xue, L.; Galdass, M.; Gnanapragasam, M.N.; Manwani, D.; Bieker, J.J. Extrinsic and Intrinsic Control by EKLF (KLF1) within a Specialized Erythroid Niche. Development 2014, 141, 2245–2254. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and Cross-Reactive SARS-CoV-2 T Cell Epitopes in Unexposed Humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef]
- Frontelo, P.; Manwani, D.; Galdass, M.; Karsunky, H.; Lohmann, F.; Gallagher, P.G.; Bieker, J.J. Novel Role for EKLF in Megakaryocyte Lineage Commitment. Blood 2007, 110, 3871–3880. [Google Scholar] [CrossRef]
- Pilon, A.M.; Arcasoy, M.O.; Dressman, H.K.; Vayda, S.E.; Maksimova, Y.D.; Sangerman, J.I.; Gallagher, P.G.; Bodine, D.M. Failure of Terminal Erythroid Differentiation in EKLF-Deficient Mice Is Associated with Cell Cycle Perturbation and Reduced Expression of E2F2. Mol. Cell. Biol. 2008, 28, 7394–7401. [Google Scholar] [CrossRef]
- Norton, L.J.; Hallal, S.; Stout, E.S.; Funnell, A.P.W.; Pearson, R.C.M.; Crossley, M.; Quinlan, K.G.R. Direct Competition between DNA Binding Factors Highlights the Role of Krüppel-like Factor 1 in the Erythroid/Megakaryocyte Switch. Sci. Rep. 2017, 7, 3137. [Google Scholar] [CrossRef] [PubMed]
- Tallack, M.R.; Whitington, T.; Yuen, W.S.; Wainwright, E.N.; Keys, J.R.; Gardiner, B.B.; Nourbakhsh, E.; Cloonan, N.; Grimmond, S.M.; Bailey, T.L.; et al. A Global Role for KLF1 in Erythropoiesis Revealed by ChIP-Seq in Primary Erythroid Cells. Genome Res. 2010, 20, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.; Coghill, E.; Keys, J.; Maguire, T.; Hartmann, B.; McDowall, A.; Weiss, M.; Grimmond, S.; Perkins, A. A Global Role for EKLF in Definitive and Primitive Erythropoiesis. Blood 2006, 107, 3359–3370. [Google Scholar] [CrossRef]
- Drissen, R.; von Lindern, M.; Kolbus, A.; Driegen, S.; Steinlein, P.; Beug, H.; Grosveld, F.; Philipsen, S. The Erythroid Phenotype of EKLF-Null Mice: Defects in Hemoglobin Metabolism and Membrane Stability. Mol. Cell. Biol. 2005, 25, 5205–5214. [Google Scholar] [CrossRef]
- Nuez, B.; Michalovich, D.; Bygrave, A.; Ploemacher, R.; Grosveld, F. Defective Haematopoiesis in Fetal Liver Resulting from Inactivation of the EKLF Gene. Nature 1995, 375, 316–318. [Google Scholar] [CrossRef]
- Kingsley, P.D.; Malik, J.; Emerson, R.L.; Bushnell, T.P.; McGrath, K.E.; Bloedorn, L.A.; Bulger, M.; Palis, J. “Maturational” Globin Switching in Primary Primitive Erythroid Cells. Blood 2006, 107, 1665–1672. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Xu, J.; Orkin, S.H. Advances in the Understanding of Haemoglobin Switching. Br. J. Haematol. 2010, 149, 181–194. [Google Scholar] [CrossRef]
- Peschle, C.; Mavilio, F.; Carè, A.; Migliaccio, G.; Migliaccio, A.R.; Salvo, G.; Samoggia, P.; Petti, S.; Guerriero, R.; Marinucci, M.; et al. Haemoglobin Switching in Human Embryos: Asynchrony of ζ → α and ε → γ-Globin Switches in Primitive and Definitive Erythropoietic Lineage. Nature 1985, 313, 235–238. [Google Scholar] [CrossRef]
- Parkins, A.C.; Sharpe, A.H.; Orkin, S.H. Lethal β-Thalassaemia in Mice Lacking the Erythroid CACCC-Transcription Factor EKLF. Nature 1995, 375, 318–322. [Google Scholar] [CrossRef]
- Zhang, W.; Kadam, S.; Emerson, B.M.; Bieker, J.J. Site-Specific Acetylation by P300 or CREB Binding Protein Regulates Erythroid Krüppel-Like Factor Transcriptional Activity via Its Interaction with the SWI-SNF Complex. Mol. Cell. Biol. 2001, 21, 2413–2422. [Google Scholar] [CrossRef]
- Waye, J.S.; Eng, B. Krüppel-like Factor 1: Hematologic Phenotypes Associated with KLF1 Gene Mutations. Int. J. Lab. Hematol. 2015, 37, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Tallack, M.R.; Keys, J.R.; Humbert, P.O.; Perkins, A.C. EKLF/KLF1 Controls Cell Cycle Entry via Direct Regulation of E2f2*. J. Biol. Chem. 2009, 284, 20966–20974. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Steiner, L. Epigenetic and Transcriptional Control of Erythropoiesis. Front. Genet. 2022, 13, 805265. [Google Scholar] [CrossRef] [PubMed]
- Jindal, G.A.; Farley, E.K. Enhancer Grammar in Development, Evolution, and Disease: Dependencies and Interplay. Dev. Cell 2021, 56, 575–587. [Google Scholar] [CrossRef]
- Snetkova, V.; Ypsilanti, A.R.; Akiyama, J.A.; Mannion, B.J.; Plajzer-Frick, I.; Novak, C.S.; Harrington, A.N.; Pham, Q.T.; Kato, M.; Zhu, Y.; et al. Ultraconserved Enhancer Function Does Not Require Perfect Sequence Conservation. Nat. Genet. 2021, 53, 521–528. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential Etiologic and Functional Implications of Genome-Wide Association Loci for Human Diseases and Traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- Huang, J.; Liu, X.; Li, D.; Shao, Z.; Cao, H.; Zhang, Y.; Trompouki, E.; Bowman, T.V.; Zon, L.I.; Yuan, G.-C.; et al. Dynamic Control of Enhancer Repertoires Drives Lineage and Stage-Specific Transcription during Hematopoiesis. Dev. Cell 2016, 36, 9–23. [Google Scholar] [CrossRef]
- Wakabayashi, A.; Ulirsch, J.C.; Ludwig, L.S.; Fiorini, C.; Yasuda, M.; Choudhuri, A.; McDonel, P.; Zon, L.I.; Sankaran, V.G. Insight into GATA1 Transcriptional Activity through Interrogation of Cis Elements Disrupted in Human Erythroid Disorders. Proc. Natl. Acad. Sci. USA 2016, 113, 4434–4439. [Google Scholar] [CrossRef]
- Jacob, G.F.; Raper, A.B. Hereditary Persistence of Foetal Haemoglobin Production, and Its Interaction with the Sickle-Cell Trait. Br. J. Haematol. 1958, 4, 138–149. [Google Scholar] [CrossRef]
- Steinberg, M.H. Fetal Hemoglobin in Sickle Hemoglobinopathies: High HbF Genotypes and Phenotypes. J. Clin. Med. 2020, 9, 3782. [Google Scholar] [CrossRef]
- Wonkam, A.; Bitoungui, V.J.N.; Vorster, A.A.; Ramesar, R.; Cooper, R.S.; Tayo, B.; Lettre, G.; Ngogang, J. Association of Variants at BCL11A and HBS1L-MYB with Hemoglobin F and Hospitalization Rates among Sickle Cell Patients in Cameroon. PLoS ONE 2014, 9, e92506. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Boyden, S.E.; Hocutt, C.M.; Lewis, R.G.; Baldwin, E.E.; Vagher, J.; Andrews, A.; Nicholas, T.J.; Chapin, A.; Fan, E.M.; et al. Identification of 2 Novel Noncoding Variants in Patients with Diamond-Blackfan Anemia Syndrome by Whole Genome Sequencing. Blood Adv. 2025, 9, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Hargreaves, V.V.; Zhu, Q.; Kurland, J.V.; Hong, J.; Kim, W.; Sher, F.; Macias-Trevino, C.; Rogers, J.M.; Kurita, R.; et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 2018, 173, 430–442.e17. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Mansour, M.R. The Role of Noncoding Mutations in Blood Cancers. Dis. Model. Mech. 2019, 12, dmm041988. [Google Scholar] [CrossRef]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; Cian, A.D.; Chalumeau, A.; et al. Editing a γ-Globin Repressor Binding Site Restores Fetal Hemoglobin Synthesis and Corrects the Sickle Cell Disease Phenotype. Sci. Adv. 2020, 6, eaay9392. [Google Scholar] [CrossRef]
- Shaikho, E.M.; Farrell, J.J.; Alsultan, A.; Qutub, H.; Al-Ali, A.K.; Figueiredo, M.S.; Chui, D.H.K.; Farrer, L.A.; Murphy, G.J.; Mostoslavsky, G.; et al. A Phased SNP-Based Classification of Sickle Cell Anemia HBB Haplotypes. BMC Genom. 2017, 18, 608. [Google Scholar] [CrossRef]
- Akinbami, A.O.; Campbell, A.D.; Han, Z.J.; Luo, H.-Y.; Chui, D.H.K.; Steinberg, M.H. Hereditary Persistence of Fetal Hemoglobin Caused by Single Nucleotide Promoter Mutations in Sickle Cell Trait and Hb SC Disease. Hemoglobin 2016, 40, 64–65. [Google Scholar] [CrossRef]
- Coleman, M.B.; Adams, J.G.; Steinberg, M.H.; Plonczynski, M.W.; Harrell, A.H.; Castro, O.; Winter, W.P. GγAγ(Β+) Hereditary Persistence of Fetal Hemoglobin: The Gγ–158 C → T Mutation in Cis to the − 175 T → C Mutation of the Aγ-globin Gene Results in Increased Gγ-globin Synthesis. Am. J. Hematol. 1993, 42, 186–190. [Google Scholar] [CrossRef]
- Choudhuri, A.; Trompouki, E.; Abraham, B.J.; Colli, L.M.; Kock, K.H.; Mallard, W.; Yang, M.-L.; Vinjamur, D.S.; Ghamari, A.; Sporrij, A.; et al. Common Variants in Signaling Transcription-Factor-Binding Sites Drive Phenotypic Variability in Red Blood Cell Traits. Nat. Genet. 2020, 52, 1333–1345. [Google Scholar] [CrossRef]
- Menzel, S.; Jiang, J.; Silver, N.; Gallagher, J.; Cunningham, J.; Surdulescu, G.; Lathrop, M.; Farrall, M.; Spector, T.D.; Thein, S.L. The HBS1L-MYB Intergenic Region on Chromosome 6q23.3 Influences Erythrocyte, Platelet, and Monocyte Counts in Humans. Blood 2007, 110, 3624–3626. [Google Scholar] [CrossRef]
- Ropero, P.; Peral, M.; Sánchez-Martínez, L.J.; Rochas, S.; Gómez-Álvarez, M.; Nieto, J.M.; González, F.A.; Villegas, A.; Benavente, C. Phenotype of Sickle Cell Disease. Correlation of Haplotypes and Polymorphisms in Cluster β, BCL11A, and HBS1L−MYB. Pilot Study. Front. Med. 2025, 12, 1347026. [Google Scholar] [CrossRef] [PubMed]
- Munkongdee, T.; Tongsima, S.; Ngamphiw, C.; Wangkumhang, P.; Peerapittayamongkol, C.; Hashim, H.B.; Fucharoen, S.; Svasti, S. Predictive SNPs for Β0-Thalassemia/HbE Disease Severity. Sci. Rep. 2021, 11, 10352. [Google Scholar] [CrossRef] [PubMed]
- Habara, A.H.; Shaikho, E.M.; Steinberg, M.H. Fetal Hemoglobin in Sickle Cell Anemia: The Arab-Indian Haplotype and New Therapeutic Agents. Am. J. Hematol. 2017, 92, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Garrett, M.E.; Telen, M.J.; Ashley-Koch, A.E. Novel Loci Associated with Acute Chest Syndrome in Sickle Cell Disease Patients. Blood 2024, 144, 1110. [Google Scholar] [CrossRef]
- Han, W.; Qi, M.; Ye, K.; He, Q.; Yekefenhazi, D.; Xu, D.; Han, F.; Li, W. Genome-Wide Association Study for Growth Traits with 1066 Individuals in Largemouth Bass (Micropterus salmoides). Front. Mol. Biosci. 2024, 11, 1443522. [Google Scholar] [CrossRef]
- Galarneau, G.; Coady, S.; Garrett, M.E.; Jeffries, N.; Puggal, M.; Paltoo, D.; Soldano, K.; Guasch, A.; Ashley-Koch, A.E.; Telen, M.J.; et al. Gene-Centric Association Study of Acute Chest Syndrome and Painful Crisis in Sickle Cell Disease Patients. Blood 2013, 122, 434–442. [Google Scholar] [CrossRef]
- Papasavva, T.; van IJcken, W.F.J.; Kockx, C.E.M.; van den Hout, M.C.G.N.; Kountouris, P.; Kythreotis, L.; Kalogirou, E.; Grosveld, F.G.; Kleanthous, M. Next Generation Sequencing of SNPs for Non-Invasive Prenatal Diagnosis: Challenges and Feasibility as Illustrated by an Application to β-Thalassaemia. Eur. J. Hum. Genet. 2013, 21, 1403–1410. [Google Scholar] [CrossRef]
- Ulirsch, J.C.; Nandakumar, S.K.; Wang, L.; Giani, F.C.; Zhang, X.; Rogov, P.; Melnikov, A.; McDonel, P.; Do, R.; Mikkelsen, T.S.; et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 2016, 165, 1530–1545. [Google Scholar] [CrossRef]
- Möller, M.; Lee, Y.Q.; Vidovic, K.; Kjellström, S.; Björkman, L.; Storry, J.R.; Olsson, M.L. Disruption of a GATA1-Binding Motif Upstream of XG/PBDX Abolishes Xga Expression and Resolves the Xg Blood Group System. Blood 2018, 132, 334–338. [Google Scholar] [CrossRef]
- Fang, F.; Xu, J.; Kang, Y.; Ren, H.; Muyey, D.M.; Chen, X.; Tan, Y.; Xu, Z.; Wang, H. GATA2 Rs2335052 and GATA2 Rs78245253 Single-nucleotide Polymorphisms in Chinese Patients with Acute Myelocytic Leukemia. Int. J. Lab. Hematol. 2021, 43, 1491–1500. [Google Scholar] [CrossRef]
- Russo, R.; Andolfo, I.; Gambale, A.; Rosa, G.D.; Manna, F.; Arillo, A.; Wandroo, F.; Bisconte, M.G.; Iolascon, A. GATA1 Erythroid-Specific Regulation of SEC23B Expression and Its Implication in the Pathogenesis of Congenital Dyserythropoietic Anemia Type II. Haematologica 2017, 102, e371–e374. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Furuyama, K.; Fujiwara, T.; Kobayashi, R.; Ishida, H.; Harigae, H.; Shibahara, S. Identification of a Novel Erythroid-Specific Enhancer for the ALAS2 Gene and Its Loss-of-Function Mutation Which Is Associated with Congenital Sideroblastic Anemia. Haematologica 2014, 99, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.-A.; Beggs, A.H.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome Sequencing Identifies GATA1 Mutations Resulting in Diamond-Blackfan Anemia. J. Clin. Investig. 2012, 122, 2439–2443. [Google Scholar] [CrossRef] [PubMed]
- Hollanda, L.M.; Lima, C.S.P.; Cunha, A.F.; Albuquerque, D.M.; Vassallo, J.; Ozelo, M.C.; Joazeiro, P.P.; Saad, S.T.O.; Costa, F.F. An Inherited Mutation Leading to Production of Only the Short Isoform of GATA-1 Is Associated with Impaired Erythropoiesis. Nat. Genet. 2006, 38, 807–812. [Google Scholar] [CrossRef]
- Stachorski, L.; Heckl, D.; Thangapandi, V.R.; Maroz, A.; Reinhardt, D.; Klusmann, J.-H. GATA1-Centered Genetic Network on Chromosome 21 Drives Down Syndrome Acute Megakaryoblastic Leukemia. Blood 2014, 124, 4310. [Google Scholar] [CrossRef]
- Kosmidou, A.; Tragiannidis, A.; Gavriilaki, E. Myeloid Leukemia of Down Syndrome. Cancers 2023, 15, 3265. [Google Scholar] [CrossRef]
- Chen, C.-C.; Silberman, R.E.; Ma, D.; Perry, J.A.; Khalid, D.; Pikman, Y.; Amon, A.; Hemann, M.T.; Rowe, R.G. Inherent Genome Instability Underlies Trisomy 21-Associated Myeloid Malignancies. Leukemia 2024, 38, 521–529. [Google Scholar] [CrossRef]
- Baruchel, A.; Bourquin, J.-P.; Crispino, J.; Cuartero, S.; Hasle, H.; Hitzler, J.; Klusmann, J.-H.; Izraeli, S.; Lane, A.A.; Malinge, S.; et al. Down Syndrome and Leukemia: From Basic Mechanisms to Clinical Advances. Haematologica 2023, 108, 2570–2581. [Google Scholar] [CrossRef]
- Malinge, S.; Bliss-Moreau, M.; Kirsammer, G.; Diebold, L.; Chlon, T.; Gurbuxani, S.; Crispino, J.D. Increased Dosage of the Chromosome 21 Ortholog Dyrk1a Promotes Megakaryoblastic Leukemia in a Murine Model of Down Syndrome. J. Clin. Investig. 2012, 122, 948–962. [Google Scholar] [CrossRef]
- Dore, L.C.; Amigo, J.D.; dos Santos, C.O.; Zhang, Z.; Gai, X.; Tobias, J.W.; Yu, D.; Klein, A.M.; Dorman, C.; Wu, W.; et al. A GATA-1-Regulated MicroRNA Locus Essential for Erythropoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 3333–3338. [Google Scholar] [CrossRef]
- Pase, L.; Layton, J.E.; Kloosterman, W.P.; Carradice, D.; Waterhouse, P.M.; Lieschke, G.J. miR-451 Regulates Zebrafish Erythroid Maturation in Vivo via Its Target Gata2. Blood 2009, 113, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wu, F.; Yang, L.; Wang, F.; Zhang, T.; Deng, X.; Zhang, X.; Yuan, X.; Yan, Y.; Li, Y.; et al. miR-144/451 Inhibits C-Myc to Promote Erythroid Differentiation. FASEB J. 2020, 34, 13194–13210. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of Non-Coding Sequence Variants in Cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Zhou, H.; Yang, J.; Li, M.; Niu, T. Epigenetic Regulation in Hematopoiesis and Its Implications in the Targeted Therapy of Hematologic Malignancies. Signal Transduct. Target. Ther. 2023, 8, 71. [Google Scholar] [CrossRef]
- Sharma, V.; Wright, K.L.; Epling-Burnette, P.K.; Reuther, G.W. Metabolic Vulnerabilities and Epigenetic Dysregulation in Myeloproliferative Neoplasms. Front. Immunol. 2020, 11, 604142. [Google Scholar] [CrossRef]
- Mejía-Ochoa, M.; Toro, P.A.A.; Cardona-Arias, J.A. Systematization of Analytical Studies of Polycythemia Vera, Essential Thrombocythemia and Primary Myelofibrosis, and a Meta-Analysis of the Frequency of JAK2, CALR and MPL Mutations: 2000–2018. BMC Cancer 2019, 19, 590. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Green, A.R. Molecular Determinants of Pathogenesis and Clinical Phenotype in Myeloproliferative Neoplasms. Haematologica 2017, 102, 7–17. [Google Scholar] [CrossRef]
- Oddsson, A.; Kristinsson, S.Y.; Helgason, H.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Steingrimsdottir, H.; Vidarsson, B.; et al. The Germline Sequence Variant Rs2736100_C in TERT Associates with Myeloproliferative Neoplasms. Leukemia 2014, 28, 1371–1374. [Google Scholar] [CrossRef]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Group, U.S.S.; Crawley, C.; Craig, J.; Scott, M.A.; et al. Leukemia-Associated Somatic Mutations Drive Distinct Patterns of Age-Related Clonal Hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Khan, S.N.; Jankowska, A.M.; Mahfouz, R.; Dunbar, A.J.; Sugimoto, Y.; Hosono, N.; Hu, Z.; Cheriyath, V.; Vatolin, S.; Przychodzen, B.; et al. Multiple Mechanisms Deregulate EZH2 and Histone H3 Lysine 27 Epigenetic Changes in Myeloid Malignancies. Leukemia 2013, 27, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Papayannopoulou, T.; Schweiger, S.G.; Stamatoyannopoulos, G.; Nienhuis, A.W. Retroviral-Mediated Transfer of Genomic Globin Genes Leads to Regulated Production of RNA and Protein. Proc. Natl. Acad. Sci. USA 1987, 84, 2411–2415. [Google Scholar] [CrossRef]
- Dzierzak, E.A.; Papayannopoulou, T.; Mulligan, R.C. Lineage-Specific Expression of a Human β-Globin Gene in Murine Bone Marrow Transplant Recipients Reconstituted with Retrovirus-Transduced Stem Cells. Nature 1988, 331, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, M.; deBoer, E.; Habets, G.; Grosveld, F. The Human Beta-globin Gene Contains Multiple Regulatory Regions: Identification of One Promoter and Two Downstream Enhancers. EMBO J. 1988, 7, 377–384. [Google Scholar] [CrossRef]
- Collis, P.; Antoniou, M.; Grosveld, F. Definition of the Minimal Requirements within the Human Beta-globin Gene and the Dominant Control Region for High Level Expression. EMBO J. 1990, 9, 233–240. [Google Scholar] [CrossRef]
- Forrester, W.C.; Takegawa, S.; Papayannopoulou, T.; Stamatoyannopoulos, G.; Groudine, M. Evidence for a Locus Activation Region: The Formation of Developmentally Stable Hypersensitive Sites in Globin-Expressing Hybrids. Nucleic Acids Res. 1987, 15, 10159–10177. [Google Scholar] [CrossRef]
- Chow, C.-M.; Athanassiadou, A.; Raguz, S.; Psiouri, L.; Harland, L.; Malik, M.; Aitken, M.; Grosveld, F.; Antoniou, M. LCR-Mediated, Long-Term Tissue-Specific Gene Expression within Replicating Episomal Plasmid and Cosmid Vectors. Gene Ther. 2002, 9, 327–336. [Google Scholar] [CrossRef]
- Sadelain, M.; Wang, C.H.; Antoniou, M.; Grosveld, F.; Mulligan, R.C. Generation of a High-Titer Retroviral Vector Capable of Expressing High Levels of the Human Beta-Globin Gene. Proc. Natl. Acad. Sci. USA 1995, 92, 6728–6732. [Google Scholar] [CrossRef]
- May, C.; Rivella, S.; Callegari, J.; Heller, G.; Gaensler, K.M.L.; Luzzatto, L.; Sadelain, M. Therapeutic Haemoglobin Synthesis in β-Thalassaemic Mice Expressing Lentivirus-Encoded Human β-Globin. Nature 2000, 406, 82–86. [Google Scholar] [CrossRef]
- Pawliuk, R.; Westerman, K.A.; Fabry, M.E.; Payen, E.; Tighe, R.; Bouhassira, E.E.; Acharya, S.A.; Ellis, J.; London, I.M.; Eaves, C.J.; et al. Correction of Sickle Cell Disease in Transgenic Mouse Models by Gene Therapy. Science 2001, 294, 2368–2371. [Google Scholar] [CrossRef]
- Imren, S.; Payen, E.; Westerman, K.A.; Pawliuk, R.; Fabry, M.E.; Eaves, C.J.; Cavilla, B.; Wadsworth, L.D.; Beuzard, Y.; Bouhassira, E.E.; et al. Permanent and Panerythroid Correction of Murine β Thalassemia by Multiple Lentiviral Integration in Hematopoietic Stem Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14380–14385. [Google Scholar] [CrossRef] [PubMed]
- Negre, O.; Bartholomae, C.; Beuzard, Y.; Cavazzana, M.; Christiansen, L.; Courne, C.; Deichmann, A.; Denaro, M.; de Dreuzy, E.; Finer, M.; et al. Preclinical Evaluation of Efficacy and Safety of an Improved Lentiviral Vector for the Treatment of β-Thalassemia and Sickle Cell Disease. Curr. Gene Ther. 2014, 15, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Miccio, A.; Cesari, R.; Lotti, F.; Rossi, C.; Sanvito, F.; Ponzoni, M.; Routledge, S.J.E.; Chow, C.-M.; Antoniou, M.N.; Ferrari, G. In Vivo Selection of Genetically Modified Erythroblastic Progenitors Leads to Long-Term Correction of β-Thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 10547–10552. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Guda, S.; Renella, R.; Bauer, D.E.; Canver, M.C.; Kim, Y.-J.; Heeney, M.M.; Klatt, D.; Fogel, J.; Milsom, M.D.; et al. Lineage-Specific BCL11A Knockdown Circumvents Toxicities and Reverses Sickle Phenotype. J. Clin. Investig. 2016, 126, 3868–3878. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion Independence and HMGA2 Activation after Gene Therapy of Human β-Thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Lee, Y.T.; de Vasconcellos, J.F.; Yuan, J.; Byrnes, C.; Noh, S.-J.; Meier, E.R.; Kim, K.S.; Rabel, A.; Kaushal, M.; Muljo, S.A.; et al. LIN28B-Mediated Expression of Fetal Hemoglobin and Production of Fetal-like Erythrocytes from Adult Human Erythroblasts Ex Vivo. Blood 2013, 122, 1034–1041. [Google Scholar] [CrossRef]
- Liu, M.; Maurano, M.T.; Wang, H.; Qi, H.; Song, C.-Z.; Navas, P.A.; Emery, D.W.; Stamatoyannopoulos, J.A.; Stamatoyannopoulos, G. Genomic Discovery of Potent Chromatin Insulators for Human Gene Therapy. Nat. Biotechnol. 2015, 33, 198–203. [Google Scholar] [CrossRef]
- Papayanni, P.-G.; Psatha, N.; Christofi, P.; Li, X.-G.; Melo, P.; Volpin, M.; Montini, E.; Liu, M.; Kaltsounis, G.; Yiangou, M.; et al. Investigating the Barrier Activity of Novel, Human Enhancer-Blocking Chromatin Insulators for Hematopoietic Stem Cell Gene Therapy. Hum. Gene Ther. 2021, 32, 1186–1199. [Google Scholar] [CrossRef]
- Roselli, E.A.; Mezzadra, R.; Frittoli, M.C.; Maruggi, G.; Biral, E.; Mavilio, F.; Mastropietro, F.; Amato, A.; Tonon, G.; Refaldi, C.; et al. Correction of Β-thalassemia Major by Gene Transfer in Haematopoietic Progenitors of Pediatric Patients. EMBO Mol. Med. 2010, 2, 315–328. [Google Scholar] [CrossRef]
- Morgan, R.A.; Ma, F.; Unti, M.J.; Brown, D.; Ayoub, P.G.; Tam, C.; Lathrop, L.; Aleshe, B.; Kurita, R.; Nakamura, Y.; et al. Creating New β-Globin-Expressing Lentiviral Vectors by High-Resolution Mapping of Locus Control Region Enhancer Sequences. Mol. Ther. Methods Clin. Dev. 2020, 17, 999–1013. [Google Scholar] [CrossRef]
- Nemeth, M.J.; Bodine, D.M.; Garrett, L.J.; Lowrey, C.H. An Erythroid-Specific Chromatin Opening Element Reorganizes β-Globin Promoter Chromatin Structure and Augments Gene Expression. Blood Cells Mol. Dis. 2001, 27, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Peslak, S.A.; Demirci, S.; Chandra, V.; Ryu, B.; Bhardwaj, S.K.; Jiang, J.; Rupon, J.W.; Throm, R.E.; Uchida, N.; Leonard, A.; et al. Forced Enhancer-Promoter Rewiring to Alter Gene Expression in Animal Models. Mol. Ther. Nucleic Acids 2023, 31, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Reitman, M.; Lee, E.; Westphal, H.; Felsenfeld, G. An Enhancer/Locus Control Region Is Not Sufficient To Open Chromatin. Mol. Cell. Biol. 1993, 13, 3990–3998. [Google Scholar] [CrossRef] [PubMed]
- Buzina, A.; Lo, M.Y.M.; Moffett, A.; Hotta, A.; Fussner, E.; Bharadwaj, R.R.; Pasceri, P.; Garcia-Martinez, J.V.; Bazett-Jones, D.P.; Ellis, J. β-Globin LCR and Intron Elements Cooperate and Direct Spatial Reorganization for Gene Therapy. PLoS Genet. 2008, 4, e1000051. [Google Scholar] [CrossRef]
- Gazouli, M.; Katsantoni, E.; Kosteas, T.; Anagnou, N.P. Persistent Fetal γ-Globin Expression in Adult Transgenic Mice Following Deletion of Two Silencer Elements Located 3′ to the Human Aγ-Globin Gene. Mol. Med. 2009, 15, 415–424. [Google Scholar] [CrossRef]
- Katsantoni, E.Z.; de Krom, M.; Kong-a-San, J.; Imam, A.M.A.; Grosveld, F.; Anagnou, N.P.; Strouboulis, J. An Embryonic-Specific Repressor Element Located 3′ to the Aγ-Globin Gene Influences Transcription of the Human β-Globin Locus in Transgenic Mice. Exp. Hematol. 2004, 32, 224–233. [Google Scholar] [CrossRef]
- Katsantoni, E.Z.; Langeveld, A.; Wai, A.W.K.; Drabek, D.; Grosveld, F.; Anagnou, N.P.; Strouboulis, J. Persistent γ-Globin Expression in Adult Transgenic Mice Is Mediated by HPFH-2, HPFH-3, and HPFH-6 Breakpoint Sequences. Blood 2003, 102, 3412–3419. [Google Scholar] [CrossRef]
- Drakopoulou, E.; Georgomanoli, M.; Lederer, C.W.; Panetsos, F.; Kleanthous, M.; Voskaridou, E.; Valakos, D.; Papanikolaou, E.; Anagnou, N.P. The Optimized γ-Globin Lentiviral Vector GGHI-MB-3D Leads to Nearly Therapeutic HbF Levels In Vitro in CD34+ Cells from Sickle Cell Disease Patients. Viruses 2022, 14, 2716. [Google Scholar] [CrossRef]
- Papanikolaou, E.; Georgomanoli, M.; Stamateris, E.; Panetsos, F.; Karagiorga, M.; Tsaftaridis, P.; Graphakos, S.; Anagnou, N.P. The New Self-Inactivating Lentiviral Vector for Thalassemia Gene Therapy Combining Two HPFH Activating Elements Corrects Human Thalassemic Hematopoietic Stem Cells. Hum. Gene Ther. 2012, 23, 15–31. [Google Scholar] [CrossRef]
- Morgan, R.A.; Unti, M.J.; Aleshe, B.; Brown, D.; Osborne, K.S.; Koziol, C.; Ayoub, P.G.; Smith, O.B.; O’Brien, R.; Tam, C.; et al. Improved Titer and Gene Transfer by Lentiviral Vectors Using Novel, Small β-Globin Locus Control Region Elements. Mol. Ther. 2020, 28, 328–340. [Google Scholar] [CrossRef]
- Uchida, N.; Ferrara, F.; Drysdale, C.M.; Yapundich, M.; Gamer, J.; Nassehi, T.; DiNicola, J.; Shibata, Y.; Wielgosz, M.; Kim, Y.-S.; et al. Sustained Fetal Hemoglobin Induction in Vivo Is Achieved by BCL11A Interference and Coexpressed Truncated Erythropoietin Receptor. Sci. Transl. Med. 2021, 13, eabb0411. [Google Scholar] [CrossRef] [PubMed]
- Tallack, M.R.; Magor, G.W.; Dartigues, B.; Sun, L.; Huang, S.; Fittock, J.M.; Fry, S.V.; Glazov, E.A.; Bailey, T.L.; Perkins, A.C. Novel Roles for KLF1 in Erythropoiesis Revealed by mRNA-Seq. Genome Res. 2012, 22, 2385–2398. [Google Scholar] [CrossRef] [PubMed]
- Siatecka, M.; Bieker, J.J. The Multifunctional Role of EKLF/KLF1 during Erythropoiesis. Blood 2011, 118, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Borg, J.; Papadopoulos, P.; Georgitsi, M.; Gutiérrez, L.; Grech, G.; Fanis, P.; Phylactides, M.; Verkerk, A.J.M.H.; van der Spek, P.J.; Scerri, C.A.; et al. Haploinsufficiency for the Erythroid Transcription Factor KLF1 Causes Hereditary Persistence of Fetal Hemoglobin. Nat. Genet. 2010, 42, 801–805. [Google Scholar] [CrossRef]
- Voit, R.A.; Liao, X.; Caulier, A.; Antoszewski, M.; Cohen, B.; Armant, M.; Lu, H.Y.; Fleming, T.J.; Kamal, E.; Wahlster, L.; et al. Regulated GATA1 Expression as a Universal Gene Therapy for Diamond-Blackfan Anemia. Cell Stem Cell 2025, 32, 38–52.e6. [Google Scholar] [CrossRef]
- Psatha, N.; Sova, P.; Georgolopoulos, G.; Paschoudi, K.; Iwata, M.; Bloom, J.; Ulyanova, T.; Wang, H.; Kirtsou, A.; Vasiloudis, N.-I.; et al. Large-Scale Discovery of Potent, Compact and Erythroid Specific Enhancers for Gene Therapy Vectors. Nat. Commun. 2025, 16, 4325. [Google Scholar] [CrossRef]
- Lodish, H. PPARα Agonists and TGFβ Inhibitors Stimulate Red Blood Cell Production by Enhancing Self-Renewal of BFU-E Erythroid Progenitors. Blood 2016, 128, SCI-48. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Gao, X.; Barrasa, M.I.; Li, H.; Elmes, R.R.; Peters, L.L.; Lodish, H.F. PPAR-α and Glucocorticoid Receptor Synergize to Promote Erythroid Progenitor Self-Renewal. Nature 2015, 522, 474–477. [Google Scholar] [CrossRef]
- Jane, S.M.; Cunningham, J.M. Understanding Fetal Globin Gene Expression: A Step Towards Effective Hbf Reactivation in Haemoglobinopathies. Br. J. Haematol. 1998, 102, 415–423. [Google Scholar] [CrossRef]
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef]
- Psatha, N.; Papayanni, P.-G.; Yannaki, E. A New Era for Hemoglobinopathies: More Than One Curative Option. Curr. Gene Ther. 2018, 17, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Paschoudi, K.; Yannaki, E.; Psatha, N. Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies. Int. J. Mol. Sci. 2023, 24, 9527. [Google Scholar] [CrossRef] [PubMed]
- Psatha, N.; Paschoudi, K.; Papadopoulou, A.; Yannaki, E. In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations. Genes 2022, 13, 2222. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Keller, C.A.; Giardine, B.; Grevet, J.D.; Davies, J.O.J.; Hughes, J.R.; Kurita, R.; Nakamura, Y.; Hardison, R.C.; Blobel, G.A. Comparative Analysis of Three-Dimensional Chromosomal Architecture Identifies a Novel Fetal Hemoglobin Regulatory Element. Genes Dev. 2017, 31, 1704–1713. [Google Scholar] [CrossRef]
- Kiefer, C.M.; Lee, J.; Hou, C.; Dale, R.K.; Lee, Y.T.; Meier, E.R.; Miller, J.L.; Dean, A. Distinct Ldb1/NLI Complexes Orchestrate γ-Globin Repression and Reactivation through ETO2 in Human Adult Erythroid Cells. Blood 2011, 118, 6200–6208. [Google Scholar] [CrossRef]
- FORGET, B.G. Molecular Basis of Hereditary Persistence of Fetal Hemoglobin. Ann. N. York Acad. Sci. 1998, 850, 38–44. [Google Scholar] [CrossRef]
- Chakalova, L.; Osborne, C.S.; Dai, Y.-F.; Goyenechea, B.; Metaxotou-Mavromati, A.; Kattamis, A.; Kattamis, C.; Fraser, P. The Corfu Δβ Thalassemia Deletion Disrupts γ-Globin Gene Silencing and Reveals Post-Transcriptional Regulation of HbF Expression. Blood 2005, 105, 2154–2160. [Google Scholar] [CrossRef]
- Lux, C.T.; Pattabhi, S.; Berger, M.; Nourigat, C.; Flowers, D.A.; Negre, O.; Humbert, O.; Yang, J.G.; Lee, C.; Jacoby, K.; et al. TALEN-Mediated Gene Editing of HBG in Human Hematopoietic Stem Cells Leads to Therapeutic Fetal Hemoglobin Induction. Mol. Ther. Methods Clin. Dev. 2019, 12, 175–183. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome Editing Using CRISPR-Cas9 to Create the HPFH Genotype in HSPCs: An Approach for Treating Sickle Cell Disease and β-Thalassemia. Proc. Natl. Acad. Sci. USA 2016, 113, 10661–10665. [Google Scholar] [CrossRef]
- Traxler, E.A.; Yao, Y.; Wang, Y.-D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A Genome-Editing Strategy to Treat β-Hemoglobinopathies That Recapitulates a Mutation Associated with a Benign Genetic Condition. Nat. Med. 2016, 22, 987–990. [Google Scholar] [CrossRef]
- Li, C.; Psatha, N.; Sova, P.; Gil, S.; Wang, H.; Kim, J.; Kulkarni, C.; Valensisi, C.; Hawkins, R.D.; Stamatoyannopoulos, G.; et al. Reactivation of γ-Globin in Adult β-YAC Mice after Ex Vivo and in Vivo Hematopoietic Stem Cell Genome Editing. Blood 2018, 131, 2915–2928. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A Enhancer Dissection by Cas9-Mediated in Situ Saturating Mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Humbert, O.; Peterson, C.W.; Norgaard, Z.K.; Radtke, S.; Kiem, H.-P. A Nonhuman Primate Transplantation Model to Evaluate Hematopoietic Stem Cell Gene Editing Strategies for β-Hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018, 8, 75–86. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly Efficient Therapeutic Gene Editing of Human Hematopoietic Stem Cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Psatha, N.; Georgakopoulou, A.; Li, C.; Nandakumar, V.; Georgolopoulos, G.; Acosta, R.; Paschoudi, K.; Nelson, J.; Chee, D.; Athanasiadou, A.; et al. Enhanced HbF Reactivation by Multiplex Mutagenesis of Thalassemic CD34+ Cells in Vitro and in Vivo. Blood 2021, 138, 1540–1553. [Google Scholar] [CrossRef]
- Locatelli, F.; Lang, P.; Wall, D.; Meisel, R.; Corbacioglu, S.; Li, A.M.; de la Fuente, J.; Shah, A.J.; Carpenter, B.; Kwiatkowski, J.L.; et al. Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2024, 390, 1663–1676. [Google Scholar] [CrossRef]
- Janoudi, T.; Jagdale, M.; Wu, M.; Gorlla, S.; Zhang, P.; Shao, Y.; Li, L.; Bowley, S.R.; Marco, E.; Chang, K.-H. Nonclinical Evaluation of HBG1/2 and BCL11A as Genome-Editing Targets for the Treatment of β-Hemoglobinopathies. Blood Adv. 2025, 9, 808–813. [Google Scholar] [CrossRef]
- Frangoul, H.; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Molinari, L.; Wall, D.; Liem, R.I.; Telfer, P.; Shah, A.J.; et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N. Engl. J. Med. 2024, 390, 1649–1662. [Google Scholar] [CrossRef]
- Singh, A.; Irfan, H.; Fatima, E.; Nazir, Z.; Verma, A.; Akilimali, A. Revolutionary Breakthrough: FDA Approves CASGEVY, the First CRISPR/Cas9 Gene Therapy for Sickle Cell Disease. Ann. Med. Surg. 2024, 86, 4555. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base Editing: Advances and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.K.; Song, B.; Bae, S. Current Status and Challenges of DNA Base Editing Tools. Mol. Ther. 2020, 28, 1938–1952. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Ma, Y.; Hu, H.; Li, Q.; Yang, Y.; Liu, W.; Yin, S.; Li, W.; Fu, B.; et al. Reactivation of γ-Globin Expression through Cas9 or Base Editor to Treat β-Hemoglobinopathies. Cell Res. 2020, 30, 276–278. [Google Scholar] [CrossRef]
- Li, C.; Georgakopoulou, A.; Mishra, A.; Gil, S.; Hawkins, R.D.; Yannaki, E.; Lieber, A. In Vivo HSPC Gene Therapy with Base Editors Allows for Efficient Reactivation of Fetal γ-Globin in β-YAC Mice. Blood Adv. 2021, 5, 1122–1135. [Google Scholar] [CrossRef]
- Li, C.; Georgakopoulou, A.; Newby, G.A.; Everette, K.A.; Nizamis, E.; Paschoudi, K.; Vlachaki, E.; Gil, S.; Anderson, A.K.; Koob, T.; et al. In Vivo Base Editing by a Single Intravenous Vector Injection for Treatment of Hemoglobinopathies. JCI Insight 2022, 7, e162939. [Google Scholar] [CrossRef]
- Antoniou, P.; Hardouin, G.; Martinucci, P.; Frati, G.; Felix, T.; Chalumeau, A.; Fontana, L.; Martin, J.; Masson, C.; Brusson, M.; et al. Base-Editing-Mediated Dissection of a γ-Globin Cis-Regulatory Element for the Therapeutic Reactivation of Fetal Hemoglobin Expression. Nat. Commun. 2022, 13, 6618. [Google Scholar] [CrossRef]
- Mayuranathan, T.; Newby, G.A.; Feng, R.; Yao, Y.; Mayberry, K.D.; Lazzarotto, C.R.; Li, Y.; Levine, R.M.; Nimmagadda, N.; Dempsey, E.; et al. Potent and Uniform Fetal Hemoglobin Induction via Base Editing. Nat. Genet. 2023, 55, 1210–1220. [Google Scholar] [CrossRef]
- Butterfield, G.L.; Rohm, D.; Roberts, A.; Nethery, M.A.; Rizzo, A.J.; Morone, D.J.; Garnier, L.; Iglesias, N.; Barrangou, R.; Gersbach, C.A. Characterization of Diverse Cas9 Orthologs for Genome and Epigenome Editing. Proc. Natl. Acad. Sci. USA 2025, 122, e2417674122. [Google Scholar] [CrossRef]
- Bou-Fakhredin, R.; Franceschi, L.D.; Motta, I.; Cappellini, M.D.; Taher, A.T. Pharmacological Induction of Fetal Hemoglobin in β-Thalassemia and Sickle Cell Disease: An Updated Perspective. Pharmaceuticals 2022, 15, 753. [Google Scholar] [CrossRef]
- Chin, J.; Singh, M.; Banzon, V.; Vaitkus, K.; Ibanez, V.; Kouznetsova, T.; Mahmud, N.; DeSimone, J.; Lavelle, D. Transcriptional Activation of the γ-Globin Gene in Baboons Treated with Decitabine and in Cultured Erythroid Progenitor Cells Involves Different Mechanisms. Exp. Hematol. 2009, 37, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; DeSimone, J.; Noguchi, C.T.; Turner, P.H.; Schechter, A.N.; Heller, P.; Nienhuis, A.W. 5-Azacytidine Increases γ-Globin Synthesis and Reduces the Proportion of Dense Cells in Patients With Sickle Cell Anemia. Blood 1983, 62, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Dorn, L.; Bressler, L.; Molokie, R.; Lavelle, D.; Talischy, N.; Hoffman, R.; van Overveld, W.; DeSimone, J. 2-Deoxy 5-Azacytidine and Fetal Hemoglobin Induction in Sickle Cell Anemia. Blood 2000, 96, 2379–2384. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Groy, A.; Gore, E.R.; Atkins, C.; Long, E.R.; Montoute, M.N.; Wu, Z.; Halsey, W.; McNulty, D.E.; Ennulat, D.; et al. In Vitro and in Vivo Induction of Fetal Hemoglobin with a Reversible and Selective DNMT1 Inhibitor. Haematologica 2020, 106, 1979–1987. [Google Scholar] [CrossRef]
- Shearstone, J.R.; Golonzhka, O.; Chonkar, A.; Tamang, D.; van Duzer, J.H.; Jones, S.S.; Jarpe, M.B. Chemical Inhibition of Histone Deacetylases 1 and 2 Induces Fetal Hemoglobin through Activation of GATA2. PLoS ONE 2016, 11, e0153767. [Google Scholar] [CrossRef]
- Nualkaew, T.; Khamphikham, P.; Pongpaksupasin, P.; Kaewsakulthong, W.; Songdej, D.; Paiboonsukwong, K.; Sripichai, O.; Engel, J.D.; Hongeng, S.; Fucharoen, S.; et al. UNC0638 Induces High Levels of Fetal Hemoglobin Expression in β-Thalassemia/HbE Erythroid Progenitor Cells. Ann. Hematol. 2020, 99, 2027–2036. [Google Scholar] [CrossRef]
- Chen, X.; Skutt-Kakaria, K.; Davison, J.; Ou, Y.-L.; Choi, E.; Malik, P.; Loeb, K.; Wood, B.; Georges, G.; Torok-Storb, B.; et al. G9a/GLP-Dependent Histone H3K9me2 Patterning during Human Hematopoietic Stem Cell Lineage Commitment. Genes Dev. 2012, 26, 2499–2511. [Google Scholar] [CrossRef]
- Renneville, A.; Galen, P.V.; Canver, M.C.; McConkey, M.; Krill-Burger, J.M.; Dorfman, D.M.; Holson, E.B.; Bernstein, B.E.; Orkin, S.H.; Bauer, D.E.; et al. EHMT1 and EHMT2 Inhibition Induces Fetal Hemoglobin Expression. Blood 2015, 126, 1930–1939. [Google Scholar] [CrossRef]
- Krivega, I.; Byrnes, C.; de Vasconcellos, J.F.; Lee, Y.T.; Kaushal, M.; Dean, A.; Miller, J.L. Inhibition of G9a Methyltransferase Stimulates Fetal Hemoglobin Production by Facilitating LCR/γ-Globin Looping. Blood 2015, 126, 665–672. [Google Scholar] [CrossRef]
- Takase, S.; Hiroyama, T.; Shirai, F.; Maemoto, Y.; Nakata, A.; Arata, M.; Matsuoka, S.; Sonoda, T.; Niwa, H.; Sato, S.; et al. A Specific G9a Inhibitor Unveils BGLT3 LncRNA as a Universal Mediator of Chemically Induced Fetal Globin Gene Expression. Nat. Commun. 2023, 14, 23. [Google Scholar] [CrossRef]
- Holshouser, S.; Cafiero, R.; Robinson, M.; Kirkpatrick, J.; Casero, R.A.; Hyacinth, H.I.; Woster, P.M. Epigenetic Reexpression of Hemoglobin F Using Reversible LSD1 Inhibitors: Potential Therapies for Sickle Cell Disease. ACS Omega 2020, 5, 14750–14758. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, V.; Vaitkus, K.; Ruiz, M.A.; Lei, Z.; Maienschein-Cline, M.; Arbieva, Z.; Lavelle, D. Effect of the LSD1 Inhibitor RN-1 on γ-Globin and Global Gene Expression during Erythroid Differentiation in Baboons (Papio Anubis). PLoS ONE 2023, 18, e0289860. [Google Scholar] [CrossRef] [PubMed]
- Rivers, A.; Vaitkus, K.; Jagadeeswaran, R.; Ruiz, M.A.; Ibanez, V.; Ciceri, F.; Cavalcanti, F.; Molokie, R.E.; Saunthararajah, Y.; Engel, J.D.; et al. Oral Administration of the LSD1 Inhibitor ORY-3001 Increases Fetal Hemoglobin in Sickle Cell Mice and Baboons. Exp. Hematol. 2018, 67, 60–64.e2. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Habara, A.; Le, C.Q.; Nguyen, N.; Chen, R.; Murphy, G.J.; Chui, D.H.K.; Steinberg, M.H.; Cui, S. Pharmacologic Induction of PGC-1α Stimulates Fetal Haemoglobin Gene Expression. Br. J. Haematol. 2022, 197, 97–109. [Google Scholar] [CrossRef]
- Sun, Y.; Benmhammed, H.; Abdullatif, S.A.; Habara, A.; Fu, E.; Brady, J.; Williams, C.; Ilinski, A.; Sharma, A.; Mahdaviani, K.; et al. PGC-1α Agonism Induces Fetal Hemoglobin and Exerts Antisickling Effects in Sickle Cell Disease. Sci. Adv. 2024, 10, eadn8750. [Google Scholar] [CrossRef]
- Fernandes, P.; Waldron, N.; Chatzilygeroudi, T.; Naji, N.S.; Karantanos, T. Acute Erythroid Leukemia: From Molecular Biology to Clinical Outcomes. Int. J. Mol. Sci. 2024, 25, 6256. [Google Scholar] [CrossRef]
- Fagnan, A.; Piqué-Borràs, M.; Tauchmann, S.; Mercher, T.; Schwaller, J. Molecular Landscapes and Models of Acute Erythroleukemia. HemaSphere 2021, 5, e558. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epigenetic Agent Class | Example Compounds | Mechanism or Target | Therapeutic Effect |
|---|---|---|---|
| DNMT Inhibitors | Azacytidine, Decitabine, GSK3482364 | Hypomethylation of γ-globin promoters; DNMT1 selective inhibition | Reactivation of γ-globin in β-thalassemia and SCD |
| HDAC Inhibitors | Butyrate, Trichostatin A, ACY-957 | Affect p38 MAPK and STAT5 signaling; histone acetylation; GATA2 activation | Reactivation of γ-globin in SCD |
| HMT Inhibitors | UNC0638, RK-701 | Decrease H3K9me2; promote chromatin looping; increase BGLT3 expression | Enhanced γ-globin reactivation |
| BET Inhibitors | Apabetalone (RVX-208), JQ1, I-BET762 (Molibresib) | Target bromodomain and extra-terminal domains | Increased γ-globin expression and HbF levels in SCD and β-thalassemia models |
| LSD1 Inhibitors | TCP, RN-1, ORY-3001 | Target LSD1 | Reactivation of γ-globin in SCD, β-YAC mice and baboons |
| PGC-1a modulators | SR-18292, ZLN005 | Increase PGC1a expression | Increased γ-globin expression in human primary cells, SCD, and β-thalassemia mouse models |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasiloudis, N.I.; Paschoudi, K.; Beta, C.; Georgolopoulos, G.; Psatha, N. Epigenetic Regulation of Erythropoiesis: From Developmental Programs to Therapeutic Targets. Int. J. Mol. Sci. 2025, 26, 6342. https://doi.org/10.3390/ijms26136342
Vasiloudis NI, Paschoudi K, Beta C, Georgolopoulos G, Psatha N. Epigenetic Regulation of Erythropoiesis: From Developmental Programs to Therapeutic Targets. International Journal of Molecular Sciences. 2025; 26(13):6342. https://doi.org/10.3390/ijms26136342
Chicago/Turabian StyleVasiloudis, Ninos Ioannis, Kiriaki Paschoudi, Christina Beta, Grigorios Georgolopoulos, and Nikoletta Psatha. 2025. "Epigenetic Regulation of Erythropoiesis: From Developmental Programs to Therapeutic Targets" International Journal of Molecular Sciences 26, no. 13: 6342. https://doi.org/10.3390/ijms26136342
APA StyleVasiloudis, N. I., Paschoudi, K., Beta, C., Georgolopoulos, G., & Psatha, N. (2025). Epigenetic Regulation of Erythropoiesis: From Developmental Programs to Therapeutic Targets. International Journal of Molecular Sciences, 26(13), 6342. https://doi.org/10.3390/ijms26136342