
Differential Effects of Losartan and Finerenone on Diabetic Remodeling, Oxidative Stress and ACE Activity in the Gastrointestinal Tract of Streptozotocin-Induced Diabetic Rats
 ,
,  , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
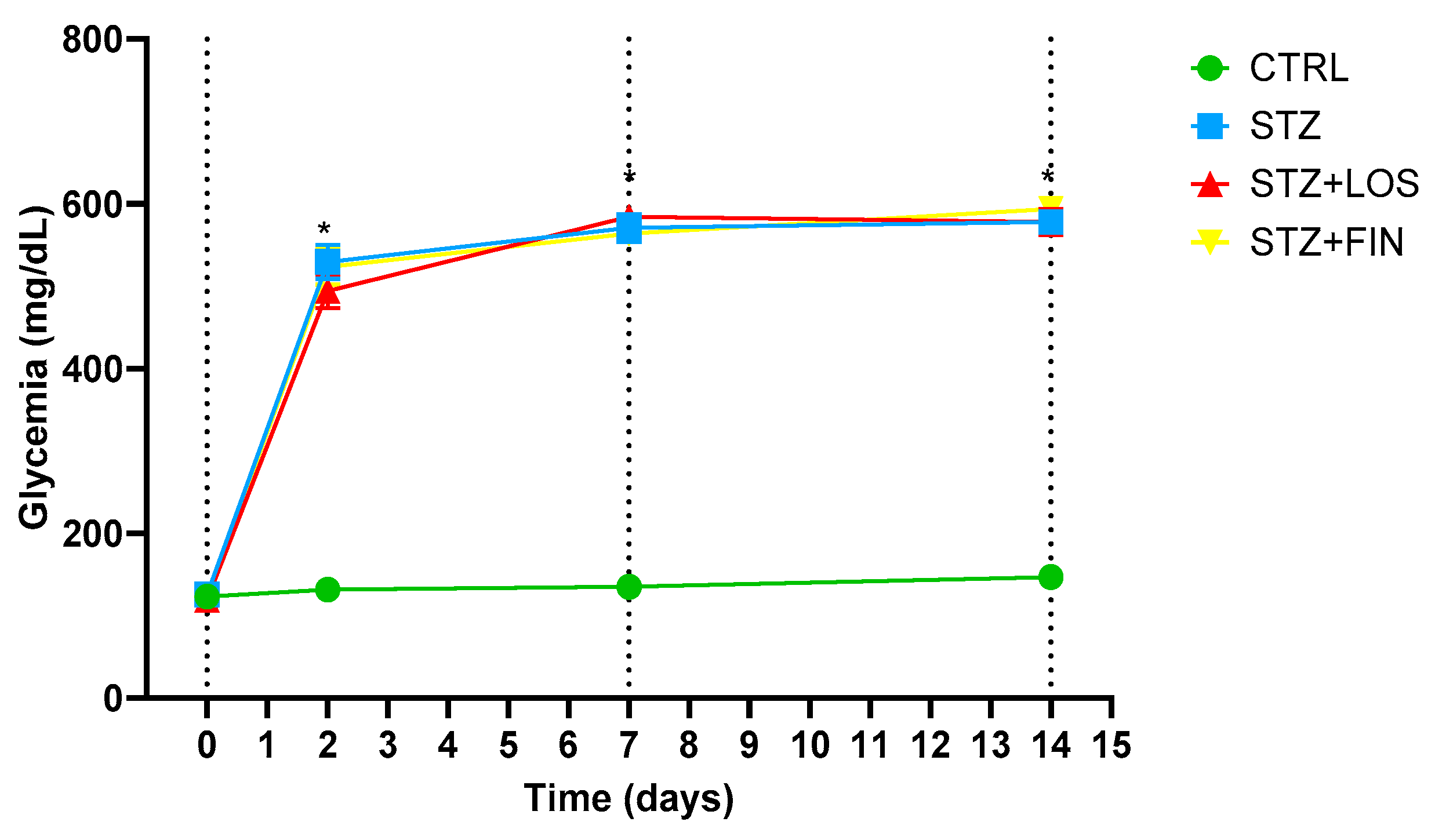
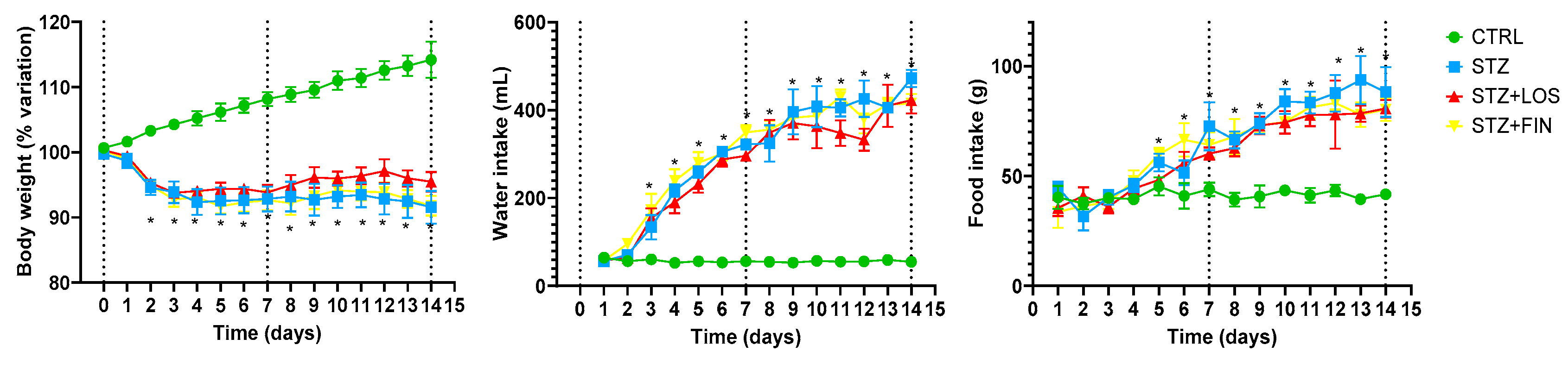
2.1. Animal Monitorization
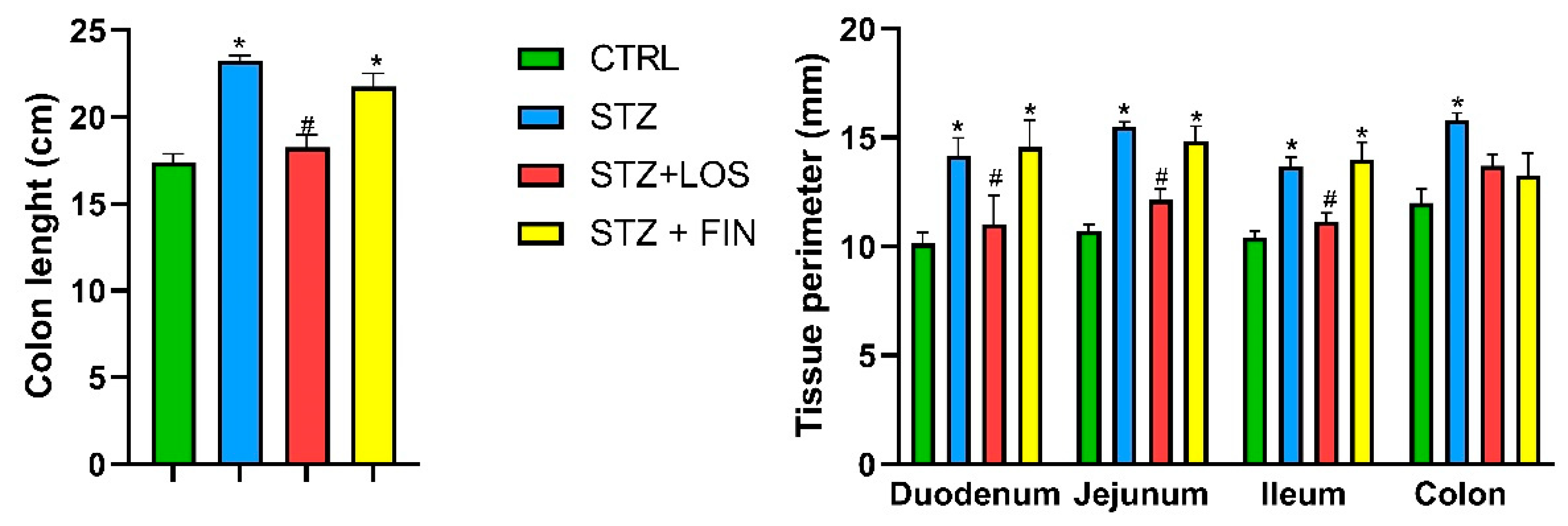
2.2. Macroscopic Evaluation
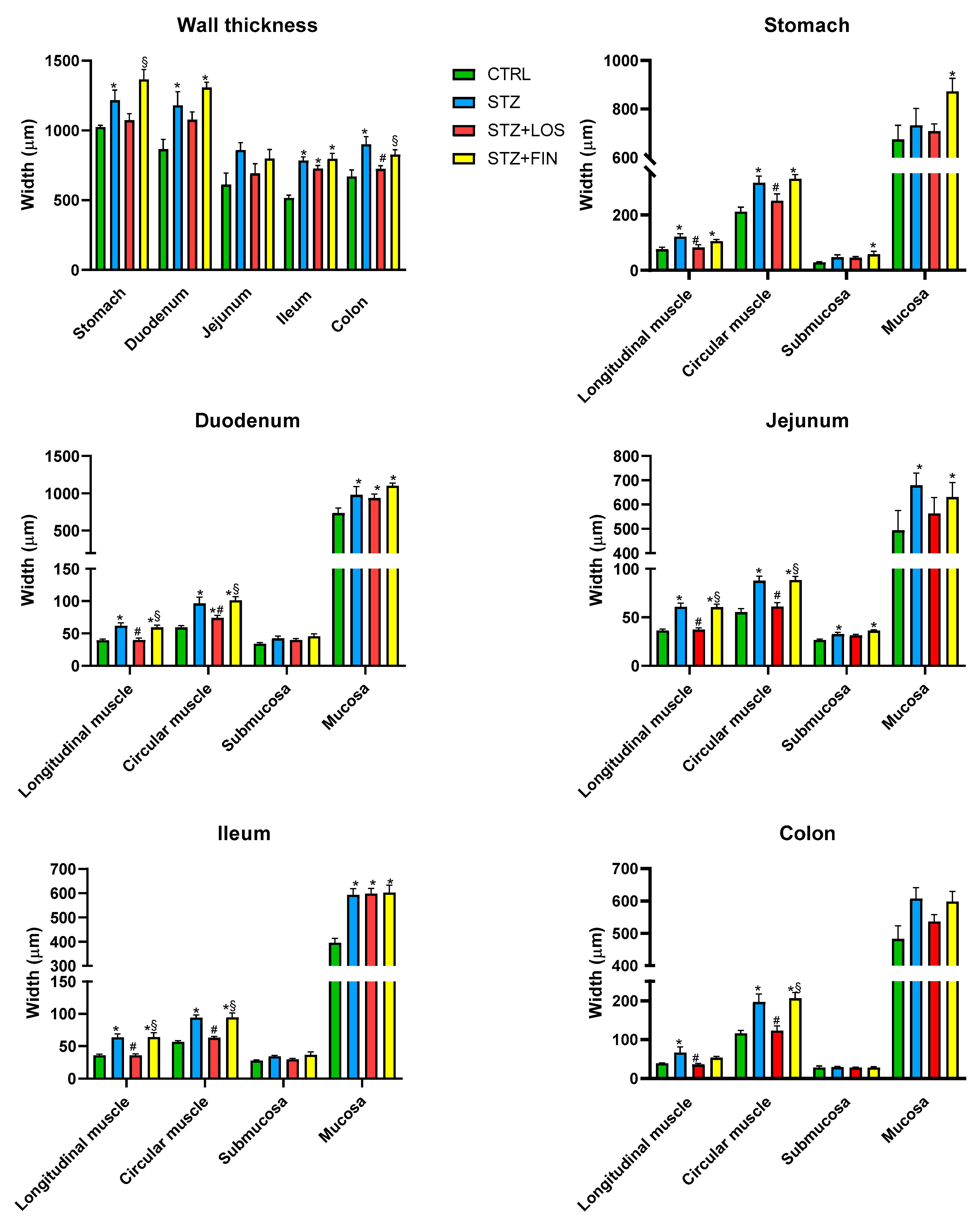
2.3. Microscopic Evaluation
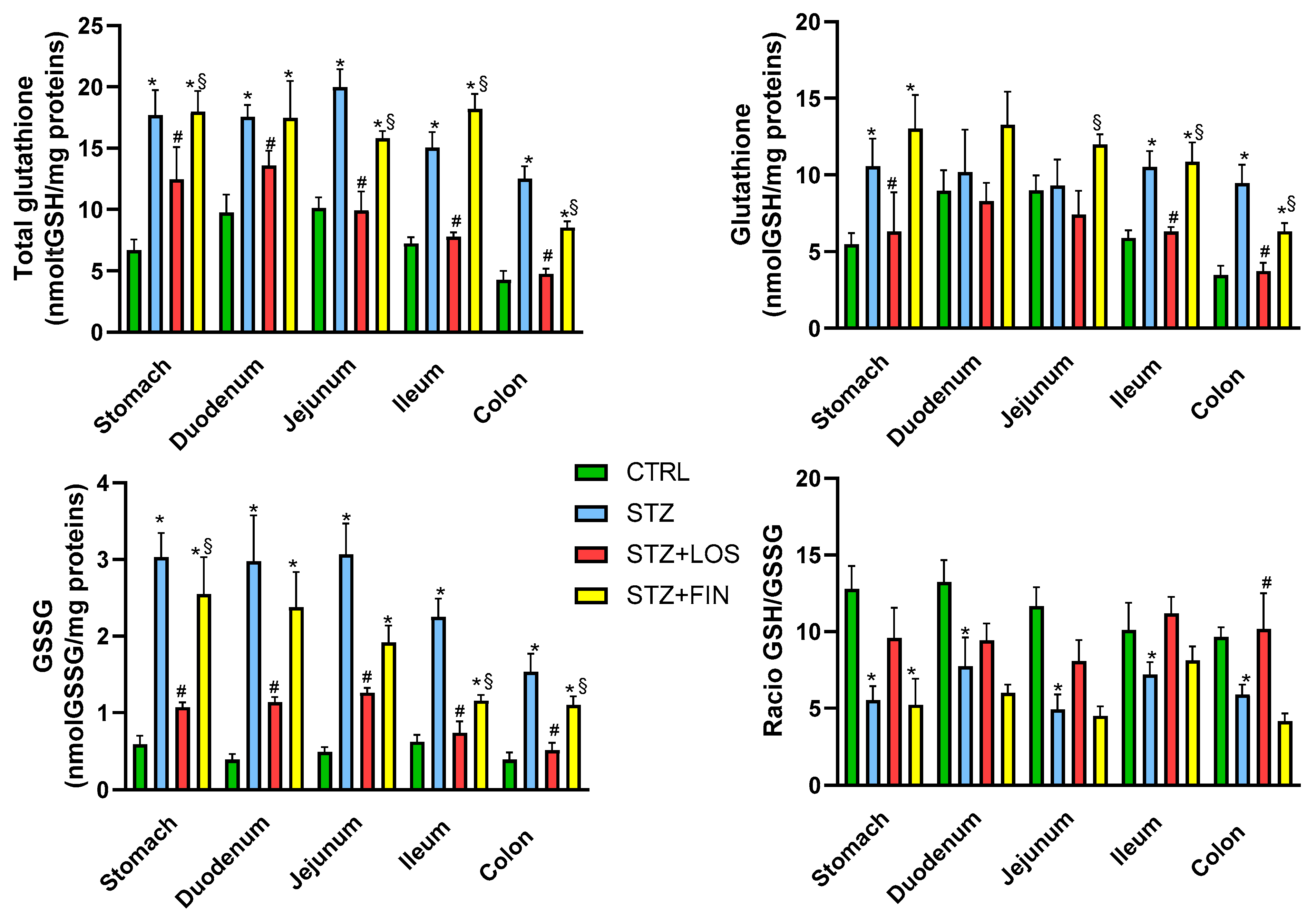
2.4. Total GSH and GSSG Quantification
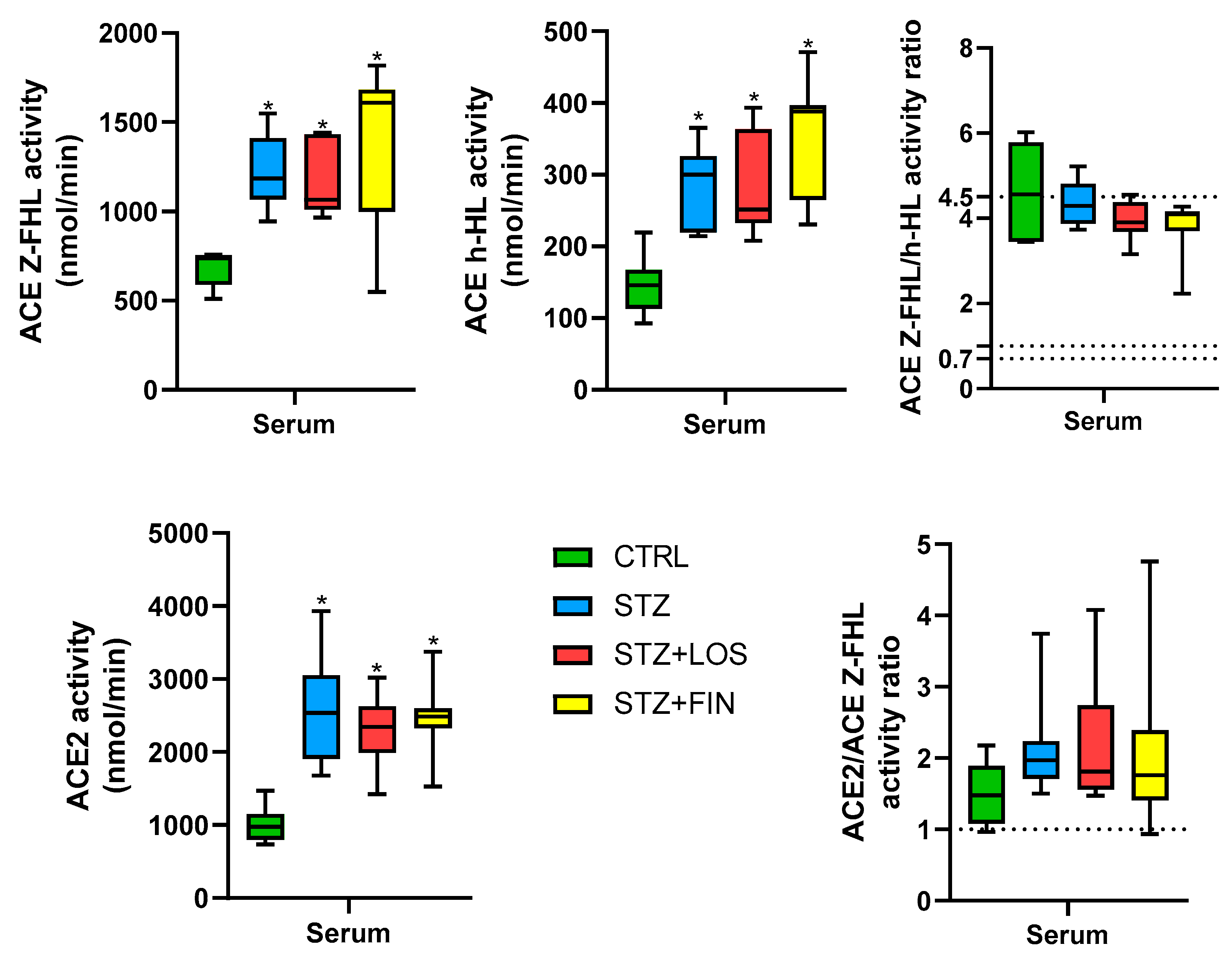
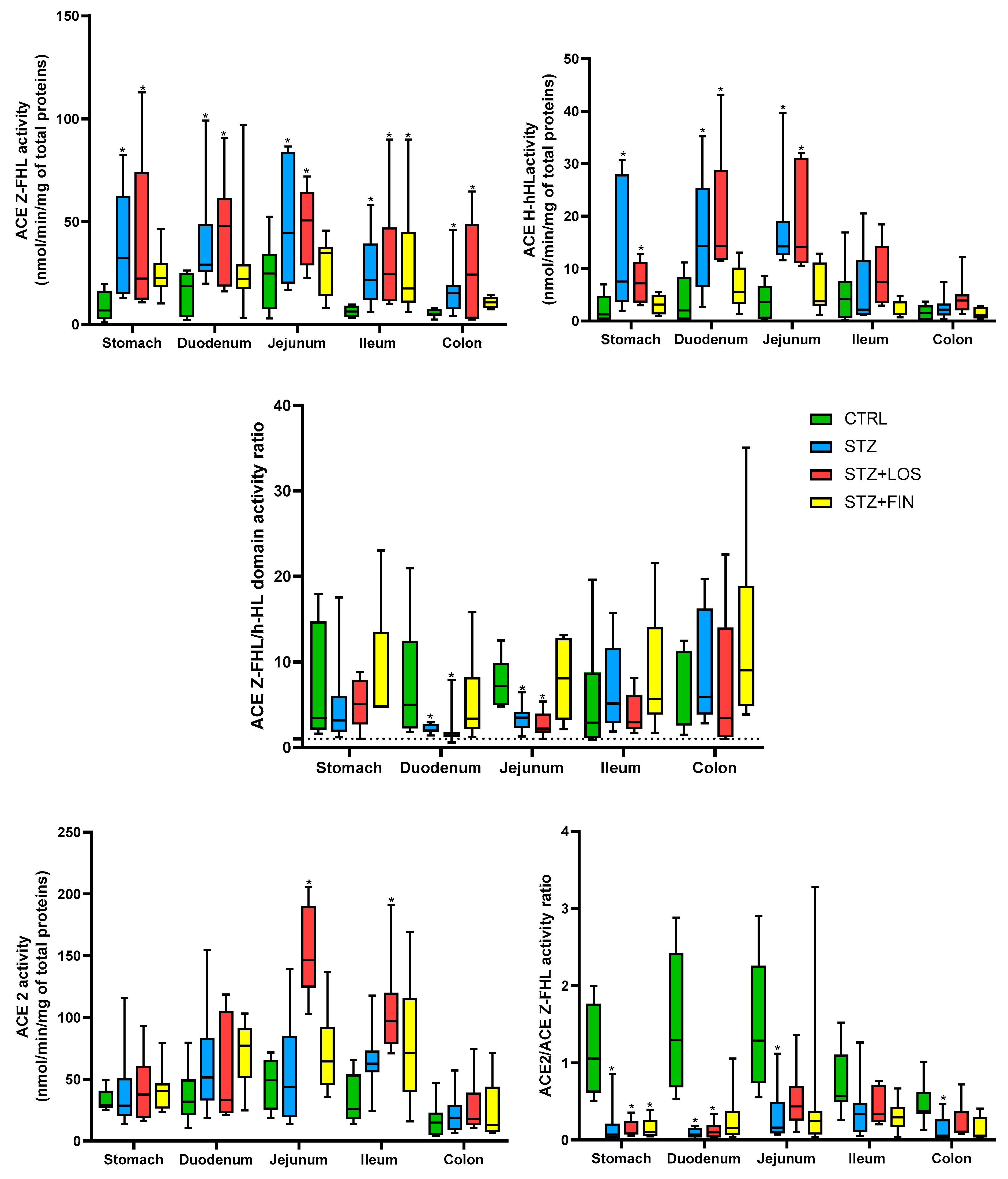
2.5. ACE and ACE 2 Activity
3. Discussion
4. Materials and Methods
4.1. Animals and Housing
4.2. Diabetes Induction
4.3. Animal Monitorization and Welfare Evaluation
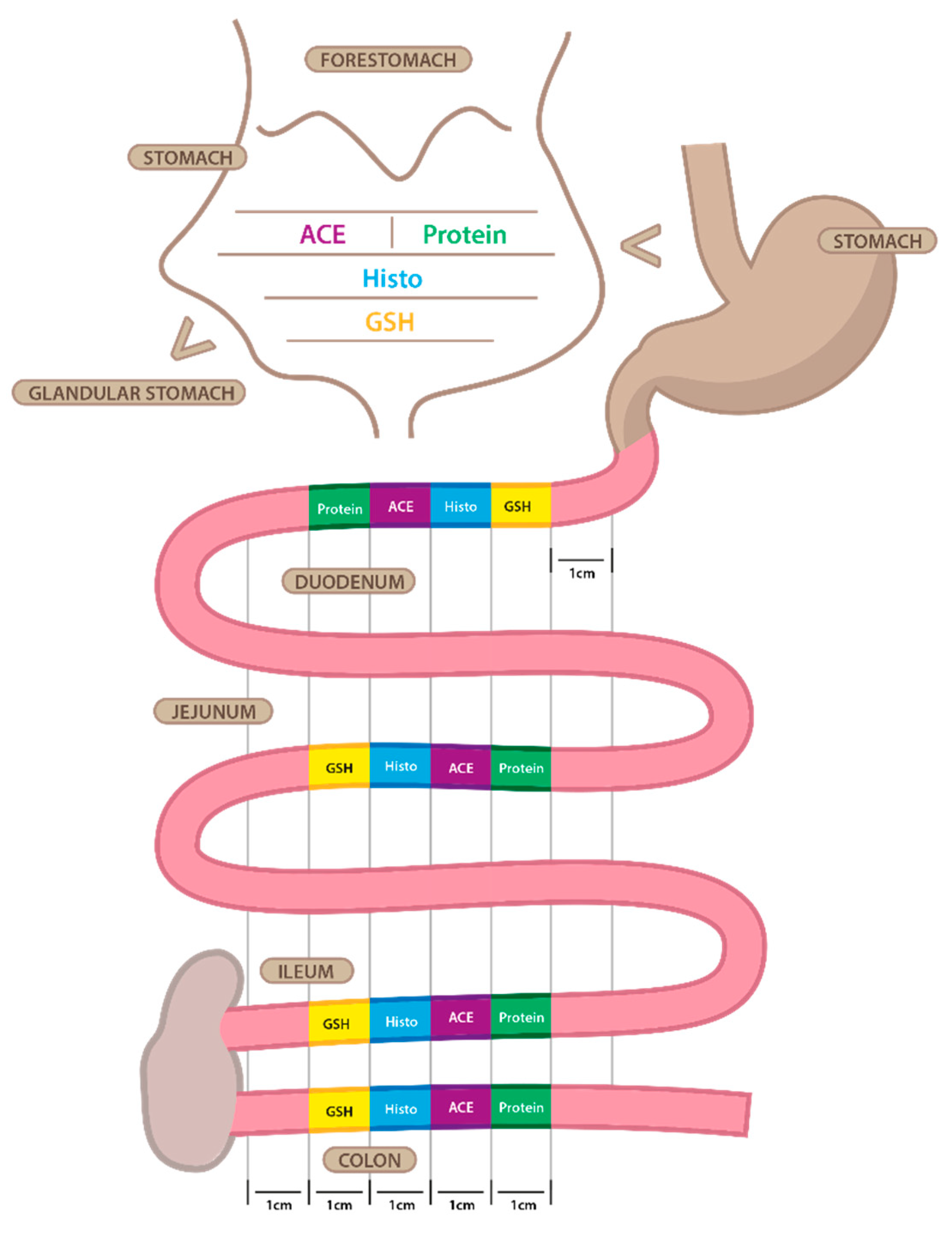
4.4. Tissue Harvesting
4.5. Histology
4.6. tGSH and GSSG Quantification
4.7. ACE and ACE2 Activity
4.8. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Report on Diabetes; WHO Library Cataloguing; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 Diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 Diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.A.; Alcolado, J.C. Animal Models of Diabetes Mellitus. Diabet. Med. 2005, 22, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. 2021, 1, 4. [Google Scholar] [CrossRef]
- Du, Y.T.; Rayner, C.K.; Jones, K.L.; Talley, N.J.; Horowitz, M. Gastrointestinal Symptoms in Diabetes: Prevalence, Assessment, Pathogenesis, and Management. Diabetes Care 2018, 41, 627–637. [Google Scholar] [CrossRef]
- Chandrasekharan, B.; Srinivasan, S. Diabetes and the Enteric Nervous System. Neurogastroenterol. Motil. 2007, 19, 951–960. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Cao, J.; Yu, J.; Tian, J.; Mi, J. Clinical study status of diabetic gastrointestinal diseases. Front. Endocrinol. 2025, 16, 1568552. [Google Scholar] [CrossRef]
- Krishnan, B.; Babu, S.; Walker, J.; Walker, A.B.; Pappachan, J.M. Gastrointestinal Complications of Diabetes Mellitus. World J. Diabetes 2013, 15, 51–63. [Google Scholar] [CrossRef]
- Zhao, M.; Liao, D.; Zhao, J. Diabetes-Induced Mechanophysiological Changes in the Small Intestine and Colon. World J. Diabetes 2017, 8, 249–269. [Google Scholar] [CrossRef]
- Boland, B.S.; Edelman, S.V.; Wolosin, J.D. Gastrointestinal Complications of Diabetes. Endocrinol. Metab. Clin. N. Am. 2013, 42, 809–832. [Google Scholar] [CrossRef]
- Goyal, R.K.; Spiro, H.M. Gastrointestinal Manifestations of Diabetes Mellitus. Med. Clin. N. Am. 1971, 55, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Bulc, M.; Całka, J.; Palus, K. Effect of Streptozotocin-Inducted Diabetes on the Pathophysiology of Enteric Neurons in the Small Intestine Based on the Porcine Diabetes Model. Int. J. Mol. Sci. 2020, 21, 2047. [Google Scholar] [CrossRef] [PubMed]
- Dedeli, O.; Pakyuz, S.C.; Daban, U.K.; Kipcak, S.; Sari, D. Prevalence of Gastrointestinal Symptoms and Its Effect on Quality of Life among Patients with Diabetes Mellitus. Am. J. Nurs. Res. 2015, 3, 48–53. [Google Scholar] [CrossRef]
- Garg, M.; Angus, P.W.; Burrell, L.M.; Herath, C.; Gibson, P.R.; Lubel, J.S. Review Article: The Pathophysiological Roles of the Renin—Angiotensin System in the Gastrointestinal Tract. Aliment. Pharmacol. Ther. 2012, 35, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, M.G.; Serio, R. The Renin–Angiotensin System in Gastrointestinal Functions. In Angiotensin; Elsevier: Amsterdam, The Netherlands, 2023; pp. 681–691. [Google Scholar] [CrossRef]
- Atlas, S.A. The Renin-Angiotensin Aldosterone System: Pathophysiological Role and Pharmacologic Inhibition. J. Manag. Care Pharm. 2007, 13 (Suppl. B), 9–20. [Google Scholar] [CrossRef]
- Martyniak, A.; Tomasik, P.J. A New Perspective on the Renin-Angiotensin System. Diagnostics 2022, 13, 16. [Google Scholar] [CrossRef]
- Guang, C.; Phillips, R.D.; Jiang, B.; Milani, F. Three Key Proteases—Angiotensin-I-Converting Enzyme (ACE), ACE2 and Renin—Within and beyond the Renin-Angiotensin System. Arch. Cardiovasc. Dis. 2012, 105, 373–385. [Google Scholar] [CrossRef]
- Lubbe, L.; Cozier, G.E.; Oosthuizen, D.; Acharya, K.R.; Sturrock, E.D. ACE2 and ACE: Structure-Based Insights into Mechanism, Regulation and Receptor Recognition by SARS-CoV. Clin. Sci. 2020, 134, 2851–2871. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a New Regulator of the Renin–Angiotensin System. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Bernardi, S.; Michelli, A.; Zuolo, G.; Candido, R.; Fabris, B. Update on RAAS Modulation for the Treatment of Diabetic Cardiovascular Disease. J. Diabetes Res. 2016, 2016, 8917578. [Google Scholar] [CrossRef]
- Saravi, B.; Li, Z.; Lang, C.N.; Schmid, B.; Lang, F.K.; Grad, S.; Alini, M.; Richards, R.G.; Schmal, H.; Südkamp, N.; et al. The Tissue Renin-Angiotensin System and Its Role in the Pathogenesis of Major Human Diseases: Quo Vadis? Cells 2021, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Esteves-Monteiro, M.; Menezes-Pinto, D.; Ferreira-Duarte, M.; Dias-Pereira, P.; Morato, M.; Duarte-Araújo, M. Histomorphometry Changes and Decreased Reactivity to Angiotensin II in the Ileum and Colon of Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2022, 23, 13233. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, Z.; Moradi, M.; Nasri, H. A Systematic Review of the Role of Renin Angiotensin Aldosterone System Genes in Diabetes Mellitus, Diabetic Retinopathy and Diabetic Neuropathy. J. Res. Med. Sci. 2014, 19, 1090–1098. [Google Scholar] [PubMed]
- Batista, J.P.T.; de Faria, A.O.V.; Ribeiro, T.F.S.; Simões e Silva, A.C. The Role of Renin–Angiotensin System in Diabetic Cardiomyopathy: A Narrative Review. Life 2023, 13, 1598. [Google Scholar] [CrossRef]
- Ustundag, B.; Canatan, H.; Cinkilince, N.; Hali, H. Angiotensin Converting Enzyme (ACE) Activity Levels in Insulin-Independent Diabetes Mellitus and Effect of ACE Levels on Diabetic Patients with Nephropathy. Cell Biochem. Funct. 2000, 28, 23–28. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharmacol. 2014, 18, 1. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Wu, G.; Lupton, J.R.; Turner, N.D.; Fang, Y.-Z.; Yang, S. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The Importance of Glutathione in Human Disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox Status Expressed as GSH:GSSG Ratio as a Marker for Oxidative Stress in Paediatric Tumour Patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef]
- Kuyvenhoven, J.P.; Meinders, A.E. Oxidative Stress and Diabetes Mellitus Pathogenesis of Long-Term Complications. Eur. J. Intern. Med. 1999, 10, 9–19. [Google Scholar] [CrossRef]
- Kashyap, P.; Farrugia, G. Oxidative Stress: Key Player in Gastrointestinal Complications of Diabetes. Neurogastroenterol. Motil. 2011, 23, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Esteves-Monteiro, M.; Ferreira-Duarte, M.; Vitorino-Oliveira, C.; Costa-Pires, J.; Oliveira, S.; Matafome, P.; Morato, M.; Dias-Pereira, P.; Costa, V.M.; Duarte-Araújo, M. Oxidative Stress and Histomorphometric Remodeling: Two Key Intestinal Features of Type 2 Diabetes in Goto–Kakizaki Rats. Int. J. Mol. Sci. 2024, 25, 12115. [Google Scholar] [CrossRef]
- Guthrie, R.A.; Guthrie, D.W. Pathophysiology of Diabetes Mellitus. Crit. Care Nurs. Q. 2004, 27, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, A.; Jeddi, S. Streptozotocin as a Tool for Induction of Rat Models of Diabetes: A Practical Guide. EXCLI J. 2023, 22, 274–294. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.B.; Davis, P.J. Water Metabolism in Diabetes Mellitus. Am. J. Med. 1991, 70, 210–214. [Google Scholar] [CrossRef]
- Havel, P.J.; Sindelar, D.K.; Baskin, D.G.; Dallman, M.F. Effects of Streptozotocin-Induced Diabetes and Insulin Treatment on the Hypothalamic Melanocortin System and Muscle Uncoupling Protein 3 Expression in Rats. Diabetes 2000, 49, 244–252. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, J.; Gregersen, H. Up-Regulated Expression of Advanced Glycation End-Products and Their Receptor in the Small Intestine and Colon of Diabetic Rats. Dig. Dis. Sci. 2012, 57, 48–57. [Google Scholar] [CrossRef]
- Weil, E.J.; Fufaa, G.; Jones, L.I.; Lovato, T.; Lemley, K.V.; Hanson, R.L.; Knowler, W.C.; Bennett, P.H.; Yee, B.; Myers, B.D.; et al. Effect of Losartan on Prevention and Progression of Early Diabetic Nephropathy in American Indians With Type 2 Diabetes. Diabetes 2013, 62, 3224–3231. [Google Scholar] [CrossRef]
- Zhai, S.; Ma, B.; Chen, W.; Zhao, Q. A Comprehensive Review of Finerenone—A Third-Generation Non-Steroidal Mineralocorticoid Receptor Antagonist. Front. Cardiovasc. Med. 2024, 11, 1476029. [Google Scholar] [CrossRef]
- Laakso, M.; Karjalainen, L.; Lempiäinen-Kuosa, P. Effects of Losartan on Insulin Sensitivity in Hypertensive Subjects. Hypertension 1996, 28, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Veneti, S.; Tziomalos, K. The Role of Finerenone in the Management of Diabetic Nephropathy. Diabetes Ther. 2021, 12, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.; Huizinga, J.D.; Wang, X.Y.; Liu, L.W.; Parsons, M. Increase in Stretch-Induced Rhythmic Motor Activity in the Diabetic Rat Colon Is Associated with Loss of ICC of the Submuscular Plexus. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 315–326. [Google Scholar] [CrossRef]
- Jervis, L.; Levim, R. Anatomic Adaptation of the Alimentary Tract of the Rat to the Hyperphagia of Chronic Alloxan-Diabetes. Nature 1996, 210, 391–393. [Google Scholar] [CrossRef]
- Cripps, A.W.; Williams, V.J. The Effect of Pregnancy and Lactation on Food Intake, Gastrointestinal Anatomy and the Absorptive Capacity of the Small Intestine in the Albino Rat; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar] [CrossRef]
- Brobeck, J.R.; Tepperman, J.; Long, C.N.H. Experimental Hypothalamic Hyperphagia in the Albino Rat. Yale J. Biol. Med. 1943, 15, 831. [Google Scholar] [PubMed] [PubMed Central]
- Zhao, J.; Yang, J.; Gregersen, H. Biomechanical and Morphometric Intestinal Remodelling during Experimental Diabetes in Rats. Diabetologia 2003, 46, 1688–1697. [Google Scholar] [CrossRef]
- Fischer, K.D.; Dhanvantari, S.; Drucker, D.J.; Brubaker, P.L.; Kirk, D.; Dhanvantari, S.; Daniel, J.; Brubaker, P.L. Intestinal Growth Is Associated with Elevated Levels of Glucagon-like Peptide 2 in Diabetic Rats. Am. J. Physiol. 1997, 3, E815–E820. [Google Scholar] [CrossRef]
- Noda, T.; Iwakiri, R.; Fujimoto, K.; Yoshida, T.; Utsumi, H.; Sakata, H.; Hisatomi, A.; Aw, T.Y. Suppression of Apoptosis Is Responsible for Increased Thickness of Intestinal Mucosa in Streptozotocin-Induced Diabetic Rats. Metabolism 2001, 50, 259–264. [Google Scholar] [CrossRef]
- Sha, H.; Tong, X.; Zhao, J. Abnormal Expressions of AGEs, TGF- β1, BDNF and Their Receptors in Diabetic Rat Colon—Associations with Colonic Morphometric and Biomechanical Remodeling. Sci. Rep. 2018, 8, 9437. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, P.; Gregersen, H. Morpho-Mechanical Intestinal Remodeling in Type 2 Diabetic GK Rats-Is It Related to Advanced Glycation End Product Formation? J. Biomech. 2013, 46, 1128–1134. [Google Scholar] [CrossRef]
- Siegman, M.J.; Eto, M.; Butler, T.M. Remodeling of the Rat Distal Colon in Diabetes: Function and Ultrastructure. Am. J. Physiol. Cell Physiol. 2016, 19107, 151–160. [Google Scholar] [CrossRef]
- Lim, H.S.; MacFadyen, R.J.; Lip, G.Y.H. Diabetes Mellitus, the Renin-Angiotensin-Aldosterone System, and the Heart. Arch. Intern. Med. 2004, 164, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Rein, J.; Bader, M. Renin-Angiotensin System in Diabetes. Protein Pept. Lett. 2017, 24, 833–840. [Google Scholar] [CrossRef]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of Angiotensin II Signaling in the Prevention of Fibrosis. Fibrogenesis Tissue Repair. 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Wyne, K. Renin-Angiotensin-Aldosterone System in Diabetes and Hypertension. J. Clin. Hypertens. 2011, 13, 224–237. [Google Scholar] [CrossRef]
- Jaworska, K.; Kopacz, W.; Koper, M.; Szudzik, M.; Gawryś-Kopczyńska, M.; Konop, M.; Hutsch, T.; Chabowski, D.; Ufnal, M. Enalapril Diminishes the Diabetes-Induced Changes in Intestinal Morphology, Intestinal RAS and Blood SCFA Concentration in Rats. Int. J. Mol. Sci. 2022, 23, 6060. [Google Scholar] [CrossRef]
- Ruiz-ortega, M.; Carvajal, G. Angiotensin II: A Key Factor in the Inflammatory and Fibrotic Response in Kidney Diseases. Nephrol. Dial. Transplant. 2004, 21, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Macconi, D.; Remuzzi, G.; Benigni, A. Key Fibrogenic Mediators: Old Players. Renin–Angiotensin System. Kidney Int. Suppl. 2014, 4, 58–64. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New Roles in Redox Signaling for an Old Antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of Oxidative Stress in Diabetic Complications: A New Perspective on an Old Paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, R.; Mohammadpour, A.; Medoro, A.; Davinelli, S.; Saso, L.; Miroliaei, M. Exploring the Links between Polyphenols, Nrf2, and Diabetes: A Review. Biomed. Pharmacother. 2025, 186, 118020. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE Pathway and Oxidative Stress as a Therapeutic Target in Type II Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Flores, M.; Ibáñez-Hernández, M.A.; Galván, R.E.; Gutiérrez, M.; Durán-Reyes, G.; Medina-Navarro, R.; Pascoe-Lira, D.; Ortega-Camarillo, C.; Vilar-Rojas, C.; Cruz, M.; et al. Glucose-6-Phosphate Dehydrogenase Activity and NADPH/NADP+ Ratio in Liver and Pancreas Are Dependent on the Severity of Hyperglycemia in Rat. Life Sci. 2006, 78, 2601–2607. [Google Scholar] [CrossRef]
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.V.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione Metabolism in Type 2 Diabetes and Its Relationship with Microvascular Complications and Glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef]
- De Mattia, G.; Bravi, M.C.; Laurenti, O.; Cassone-Faldetta, M.; Armiento, A.; Ferri, C.; Balsano, F. Influence of Reduced Glutathione Infusion on Glucose Metabolism in Patients with Non—Insulin-Dependent Diabetes Mellitus. Metabolism 1998, 47, 993–997. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Leso, V.; Trovato-Salinaro, A.; Ventimiglia, B.; Cavallaro, M.; Scuto, M.; Rizza, S.; Zanoli, L.; Neri, S.; et al. Oxidative Stress, Glutathione Status, Sirtuin and Cellular Stress Response in Type 2 Diabetes. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 729–736. [Google Scholar] [CrossRef]
- Lodovici, M.; Bigagli, E.; Tarantini, F.; Di Serio, C.; Raimondi, L. Losartan Reduces Oxidative Damage to Renal DNA and Conserves Plasma Antioxidant Capacity in Diabetic Rats. Exp. Biol. Med. 2015, 240, 1500–1504. [Google Scholar] [CrossRef]
- Dandona, P.; Kumar, V.; Aljada, A.; Ghanim, H.; Syed, T.; Hofmayer, D.; Mohanty, P.; Tripathy, D.; Garg, R. Angiotensin II Receptor Blocker Valsartan Suppresses Reactive Oxygen Species Generation in Leukocytes, Nuclear Factor-ΚB, in Mononuclear Cells of Normal Subjects: Evidence of an Antiinflammatory Action. J. Clin. Endocrinol. Metab. 2003, 88, 4496–4501. [Google Scholar] [CrossRef]
- Yang, N.; Gonzalez-Vicente, A.; Garvin, J.L. Angiotensin II-Induced Superoxide and Decreased Glutathione in Proximal Tubules: Effect of Dietary Fructose. Am. J. Physiol.-Ren. Physiol. 2020, 318, F183–F192. [Google Scholar] [CrossRef] [PubMed]
- Batlle, D.; Jose Soler, M.; Ye, M. ACE2 and Diabetes: ACE of ACEs? Diabetes 2010, 59, 2994–2996. [Google Scholar] [CrossRef]
- Feman, S.; Mericle, R.A.; Reed, G.W.; May, J.M.; Workman, R.J. Serum angiotensin converting enzyme in diabetic patients. Am. J. Med. Sci. 1993, 305, 280–284. [Google Scholar] [CrossRef]
- Yamaleyeva, L.M.; Gilliam-Davis, S.; Almeida, I.; Brosnihan, K.B.; Lindsey, S.H.; Chappell, M.C. Differential Regulation of Circulating and Renal ACE2 and ACE in Hypertensive MRen2.Lewis Rats with Early-Onset Diabetes. Am. J. Physiol.-Ren. Physiol. 2012, 302, F1374–F1384. [Google Scholar] [CrossRef]
- Enyedi, E.E.; Petukhov, P.A.; Kozuch, A.J.; Dudek, S.M.; Toth, A.; Fagyas, M.; Danilov, S.M. ACE Phenotyping in Human Blood and Tissues: Revelation of ACE Outliers and Sex Differences in ACE Sialylation. Biomedicines 2024, 12, 940. [Google Scholar] [CrossRef]
- Danilov, S.M.; Tikhomirova, V.E.; Metzger, R.; Naperova, I.A.; Bukina, T.M.; Goker-Alpan, O.; Tayebi, N.; Gayfullin, N.M.; Schwartz, D.E.; Samokhodskaya, L.M.; et al. ACE Phenotyping in Gaucher Disease. Mol. Genet. Metab. 2018, 123, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, F.A.; Irigoyen, M.-C.; Casarini, D.E. Association of Somatic and N-Domain Angiotensin-Converting Enzymes from Wistar Rat Tissue with Renal Dysfunction in Diabetes Mellitus. J. Renin-Angiotensin-Aldosterone Syst. 2007, 8, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Anthony, C.S.; Corradi, H.R.; Schwager, S.L.U.; Redelinghuys, P.; Georgiadis, D.; Dive, V.; Acharya, K.R.; Sturrock, E.D. The N Domain of Human Angiotensin-I-Converting Enzyme. J. Biol. Chem. 2010, 285, 35685–35693. [Google Scholar] [CrossRef]
- Sharma, R.K.; Douglas, R.G.; Louw, S.; Chibale, K.; Sturrock, E.D. New Ketomethylene Inhibitor Analogues: Synthesis and Assessment of Structural Determinants for N-Domain Selective Inhibition of Angiotensin-Converting Enzyme. Biol. Chem. 2012, 393, 485–493. [Google Scholar] [CrossRef]
- Mizuiri, S.; Hemmi, H.; Arita, M.; Ohashi, Y.; Tanaka, Y.; Miyagi, M.; Sakai, K.; Ishikawa, Y.; Shibuya, K.; Hase, H.; et al. Expression of ACE and ACE2 in Individuals With Diabetic Kidney Disease and Healthy Controls. Am. J. Kidney Dis. 2008, 51, 613–623. [Google Scholar] [CrossRef]
- Bezerra, E.M.; de Alvarenga, É.C.; dos Santos, R.P.; de Sousa, J.S.; Fulco, U.L.; Freire, V.N.; Albuquerque, E.L.; da Costa, R.F. Losartan as an ACE Inhibitor: A Description of the Mechanism of Action through Quantum Biochemistry. RSC Adv. 2022, 12, 28395–28404. [Google Scholar] [CrossRef] [PubMed]
- Eleazu, C.O.; Eleazu, K.C.; Chukwuma, S.; Essien, U.N. Review of the Mechanism of Cell Death Resulting from Streptozotocin Challenge in Experimental Animals, Its Practical Use and Potential Risk to Humans. J. Diabetes Metab. Disord. 2013, 12, 60. [Google Scholar] [CrossRef]
- Cavusoglu, T.; Karadeniz, T.; Cagiltay, E.; Karadeniz, M.; Yigitturk, G.; Acikgoz, E.; Uyanikgil, Y.; Ates, U.; Tuglu, M.; Erbas, O. The Protective Effect of Losartan on Diabetic Neuropathy in a Diabetic Rat Model. Exp. Clin. Endocrinol. Diabetes 2015, 123, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Kamper, M.; Tsimpoukidi, O.; Chatzigeorgiou, A.; Lymberi, M.; Kamper, E.F. The Antioxidant Effect of Angiotensin II Receptor Blocker, Losartan, in Streptozotocin-Induced Diabetic Rats. Transl. Res. 2010, 156, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Diogo, L.N.; Faustino, I.V.; Afonso, R.A.; Pereira, S.A.; Monteiro, E.C.; Santos, A.I. Voluntary Oral Administration of Losartan in Rats. J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 549–556. [Google Scholar] [PubMed]
- Sanz-Gómez, M.; Manzano-Lista, F.J.; Vega-Martín, E.; González-Moreno, D.; Alcalá, M.; Gil-Ortega, M.; Somoza, B.; Pizzamiglio, C.; Ruilope, L.M.; Aránguez, I.; et al. Finerenone Protects against Progression of Kidney and Cardiovascular Damage in a Model of Type 1 Diabetes through Modulation of Proinflammatory and Osteogenic Factors. Biomed. Pharmacother. 2023, 168, 115661. [Google Scholar] [CrossRef]
- Lima Posada, I.; Soulié, M.; Stephan, Y.; Palacios Ramirez, R.; Bonnard, B.; Nicol, L.; Pitt, B.; Kolkhof, P.; Mulder, P.; Jaisser, F. Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone Improves Diastolic Dysfunction in Preclinical Nondiabetic Chronic Kidney Disease. J. Am. Heart Assoc. 2024, 13, e032971. [Google Scholar] [CrossRef]
- Leung, V.; Zhang, E.; Pang, D.S. Real-Time Application of the Rat Grimace Scale as a Welfare Refinement in Laboratory Rats. Sci. Rep. 2016, 6, 31667. [Google Scholar] [CrossRef]
- Bastos, M.L.; Carvalho, M.; Milhazes, N.; Remiro, F.; Borges, F.; Fernandes, E.; Amado, F.; Monks, T.J.; Carvalho, F. Hepatotoxicity of 3, 4-Methylenedioxyamphetamine and Methyldopamine in Isolated Rat Hepatocytes: Formation of Glutathione Conjugates. Arch. Toxicol. 2004, 78, 16–24. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Duarte, M.; Oliveira, L.C.G.; Quintas, C.; Esteves-Monteiro, M.; Duarte-Araújo, M.; Sousa, T.; Casarini, D.E.; Morato, M. ACE and ACE2 Catalytic Activity in the Fecal Content along the Gut. Neurogastroenterol. Motil. 2023, 35, e14598. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.A.; Danilov, S.; Alhenc-Gelas, F.; Soubrier, F. A Study of Chimeras Constructed with the Two Domains of Angiotensin I-Converting Enzyme. Biochem. Pharmacol. 1996, 51, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteves-Monteiro, M.; Vitorino-Oliveira, C.; Castanheira-Moreira, J.; Ferreira-Duarte, M.; Dias-Pereira, P.; Costa, V.M.; Morato, M.; Duarte-Araújo, M. Differential Effects of Losartan and Finerenone on Diabetic Remodeling, Oxidative Stress and ACE Activity in the Gastrointestinal Tract of Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2025, 26, 6294. https://doi.org/10.3390/ijms26136294
Esteves-Monteiro M, Vitorino-Oliveira C, Castanheira-Moreira J, Ferreira-Duarte M, Dias-Pereira P, Costa VM, Morato M, Duarte-Araújo M. Differential Effects of Losartan and Finerenone on Diabetic Remodeling, Oxidative Stress and ACE Activity in the Gastrointestinal Tract of Streptozotocin-Induced Diabetic Rats. International Journal of Molecular Sciences. 2025; 26(13):6294. https://doi.org/10.3390/ijms26136294
Chicago/Turabian StyleEsteves-Monteiro, Marisa, Cláudia Vitorino-Oliveira, Joana Castanheira-Moreira, Mariana Ferreira-Duarte, Patrícia Dias-Pereira, Vera Marisa Costa, Manuela Morato, and Margarida Duarte-Araújo. 2025. "Differential Effects of Losartan and Finerenone on Diabetic Remodeling, Oxidative Stress and ACE Activity in the Gastrointestinal Tract of Streptozotocin-Induced Diabetic Rats" International Journal of Molecular Sciences 26, no. 13: 6294. https://doi.org/10.3390/ijms26136294
APA StyleEsteves-Monteiro, M., Vitorino-Oliveira, C., Castanheira-Moreira, J., Ferreira-Duarte, M., Dias-Pereira, P., Costa, V. M., Morato, M., & Duarte-Araújo, M. (2025). Differential Effects of Losartan and Finerenone on Diabetic Remodeling, Oxidative Stress and ACE Activity in the Gastrointestinal Tract of Streptozotocin-Induced Diabetic Rats. International Journal of Molecular Sciences, 26(13), 6294. https://doi.org/10.3390/ijms26136294