2.2. Reaction of 1-Hydroxyethyl Radicals with Albumins
Radiolysis of Albumin Solution
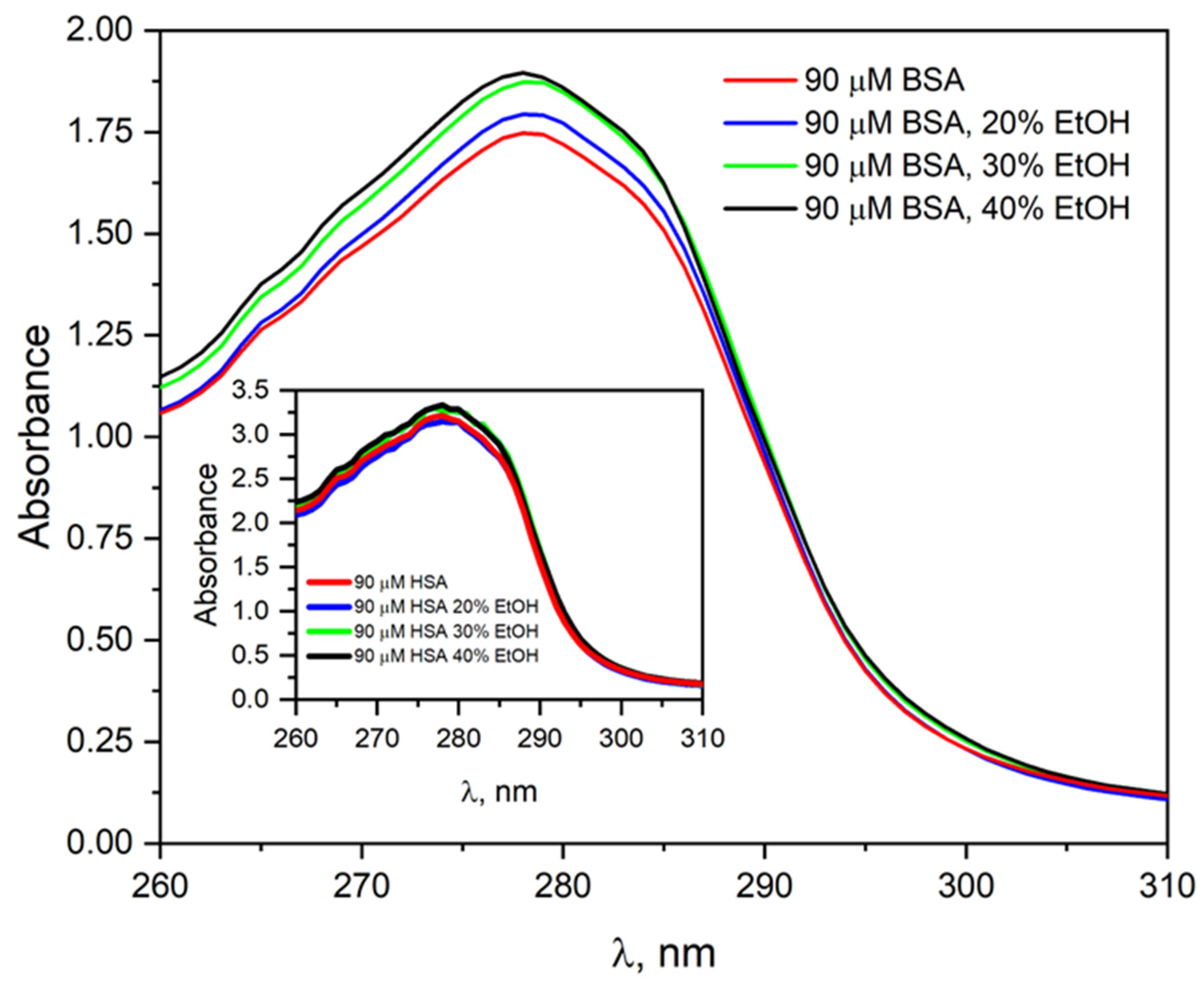
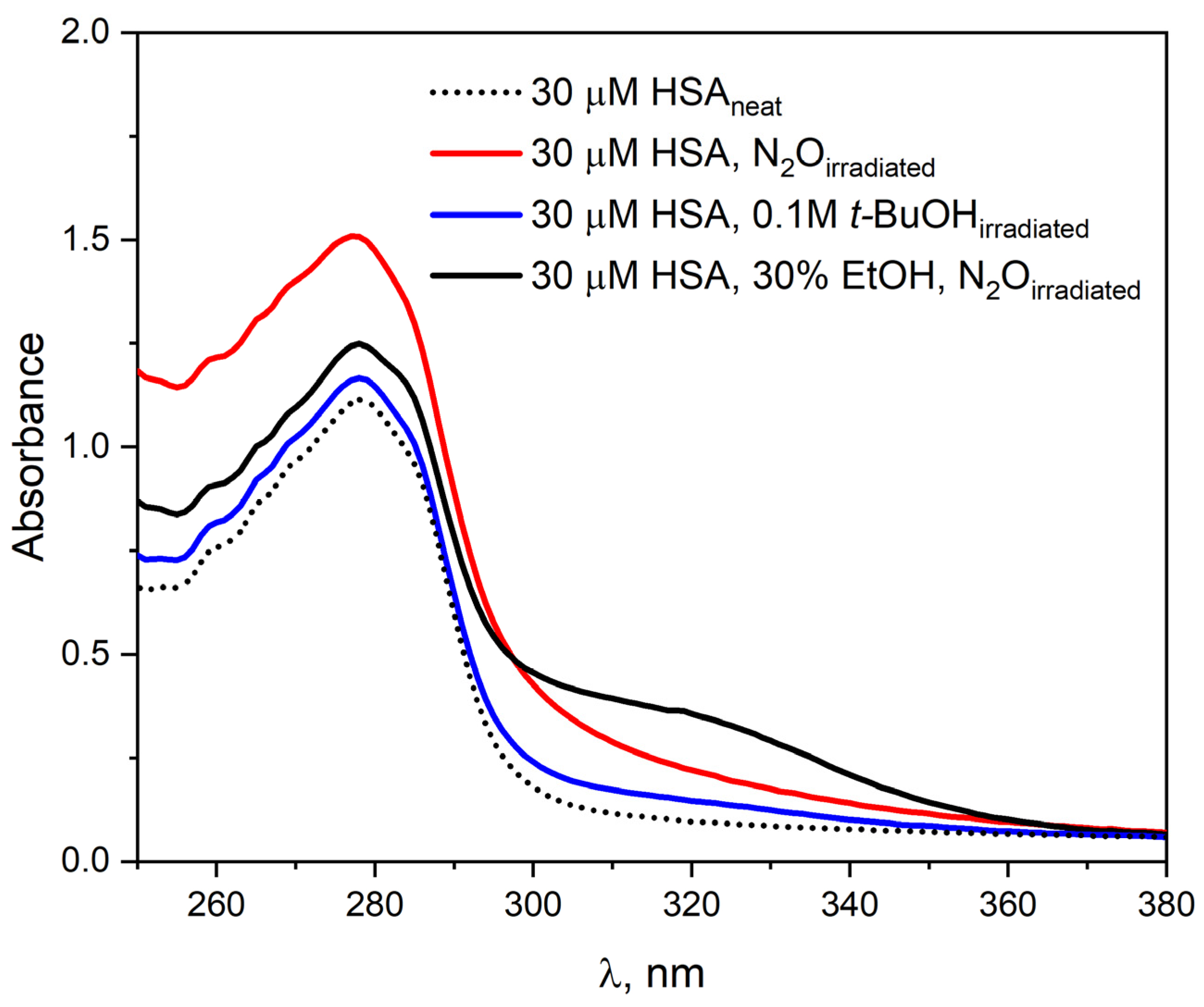
To investigate the effect of ethanol on the absorption properties of HSA, the pulse radiolysis system was used to irradiate a 30 µM HSA solution (4200 Gy) in the presence of EtOH (30% vol.).
Figure 5 shows the HSA absorbance spectra before and after irradiation.
Before irradiation, the absorption spectrum of HSA is characterized by a single band with a maximum at 278 nm. The addition of EtOH to the HSA solution leads only to a slight increase in absorbance at the maximum of this band. After irradiation of N
2O-saturated albumin solution with ethanol, an increase in the intensity of the HSA band and the appearance of a new absorbance in the spectral range of 295–375 nm were observed. The new absorbance in the albumin spectrum after irradiation of an HSA solution containing ethanol indicates the formation of new species in the sample that specifically absorb light. The apparent increase in absorbance at the maximum of the HSA band after irradiation of the solution is related to light scattering by the sample. The absorption spectra of albumin after irradiation of a solution with added ethanol differ from those recorded after the reaction of HSA with hydrated electron. The differences in the emission spectra may result from the fact that the addition of ethanol already in the initial solution modifies the structure of HSA, and also that the addition of CH
3CH
•OH radicals to HSA molecules was possible.
Figure 5 also shows the HSA spectrum related to steady-state irradiation of N
2O-saturated albumin solution. The experimental results indicate that the reaction of 1-hydroxyethyl radicals does not lead to the formation of a protein oxidation product.
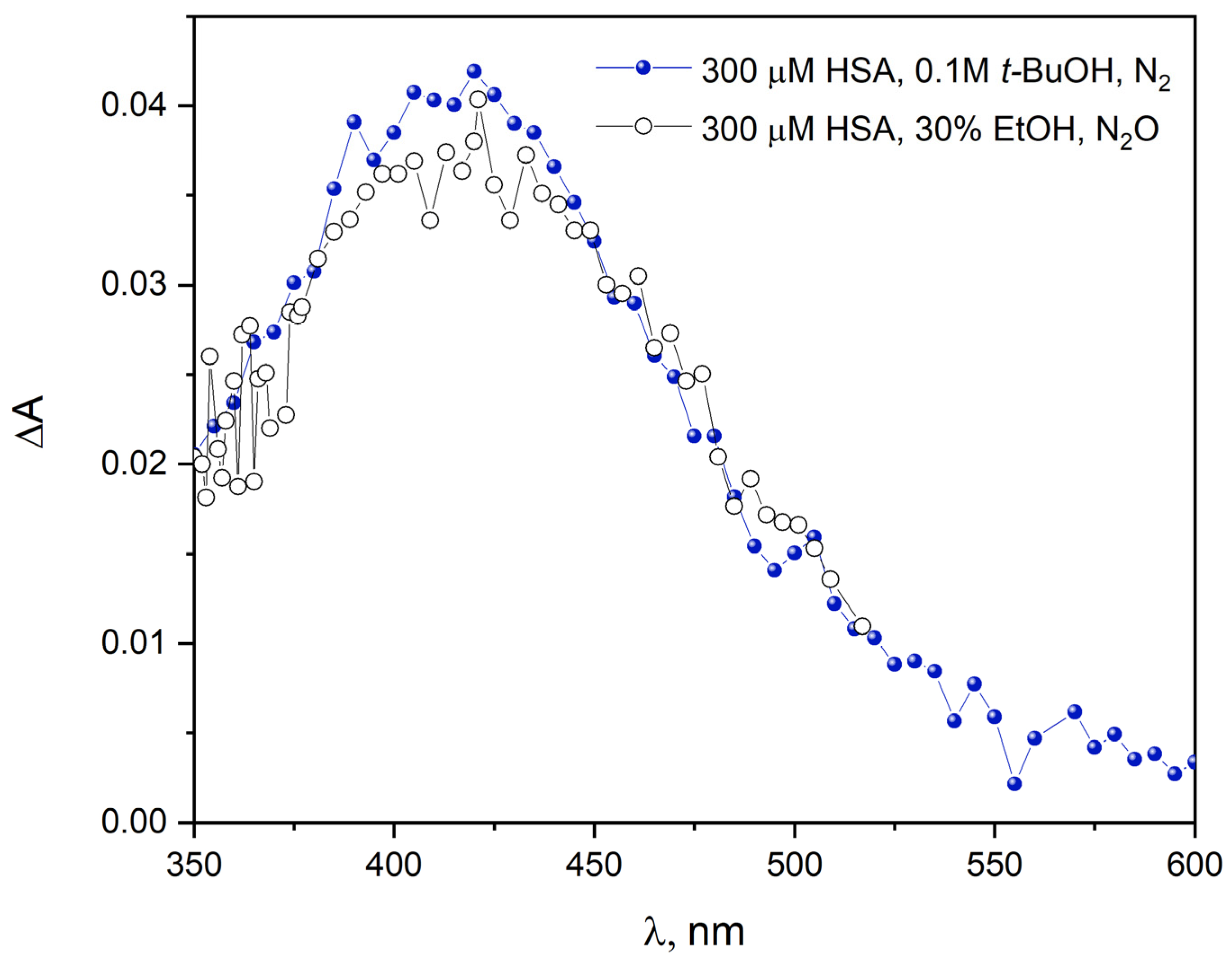
Figure S3 shows the emission spectra recorded before and after pulse radiolysis of an HSA solution (300 µM) containing
t-BuOH (0.1 M) or a solution containing HSA (300 µM) and 20% EtOH (vol.) saturated with N
2O or vacuum-deaerated. In the case of vacuum-deaerated EtOH solution, the emission spectrum of HSA is identical to that of a solution with
t-BuOH. This is obvious because in such deaeration conditions, mainly the hydrated electron reacts with albumin. The intensity of the band with a maximum at around 420 nm in the emission spectrum of HSA after irradiation of a solution containing EtOH and saturated with N
2O is definitely lower compared to the solution containing
t-BuOH. Under such conditions, the reducing species is the 1-hydroxyethyl radical, and the reactivity of those radicals is lower than that of hydrated electrons. However, analyzing the HSA emission spectra in
Figure S3, it can be concluded that the albumin reduction product after reaction with a hydrated electron or CH
3C
•HOH radical is the same in both cases. To confirm the reductive properties of the 1-hydroxyethyl radical, a pulse radiolysis experiment of HSA solution under oxidative conditions (the oxidizing species was the
•OH radical) was performed. The emission spectrum of albumin recorded after reaction with the hydroxyl radical is red-shifted (5 nm) and its intensity is almost two times higher (red line in
Figure S3) compared to the emission spectrum of the HSA solution with
t-BuOH. This excludes the oxidation reaction of proteins with CH
3C
•HOH radicals.
We studied the reactions of uncharged ethanol radical with human serum albumin (HSA) in pulse radiolysis (
Figure 6).
Figure 6 shows normalized transient absorption spectra of primary products of HSA reduction by 1-hydroxyethyl radical (N
2O-saturated 300 µM HSA solution containing 30% EtOH). These spectra are compared with the spectra obtained after the reduction in HSA by hydrated electron (N
2-saturated 300 µM HSA solution containing 0.1 M
t-BuOH). In the pulse radiolysis measurements of the HSA solution containing ethanol, we used 300 µM HSA because at a concentration of 30 µM HSA, no reaction of CH
3C
•HOH radicals with albumin was observed. Trace amounts of reduced HSA used at a concentration of 30 µM were detected only in the case of very high doses (over 800 Gy) generated by 4 µs accelerator electron pulses. The analysis of the shape of transient absorption spectra of N
2O-saturated HSA solution containing 30% EtOH gave preliminary evidence for the mechanism of the reactions of 1-hydroxyethyl radicals with human serum albumin. The new transient absorption peak with a maximum at 420 nm is formed after pulse irradiation. This spectrum can be attributed to the reduction product of the disulfide bond (CyS–SCy
•–) within albumin. Similar changes have been observed in the case of reduction in the HSA by hydrated electron [
5,
31]. The intensity of the transient absorption spectrum of HSA in the case of 1-hydroxyethyl radicals is 3.2 times lower compared to the spectrum of the protein after the reaction with the hydrated electron (in this work, we show the normalized spectra). This is most likely due to the lower driving force of the CH
3C
•HOH reaction with –S–S– bridges within the HSA compared to the analogous reaction of
with HSA.
The hydrated electron is an extremely effective reducing agent with a reduction potential of –2.9 V. The redox potential of the 1-hydroxyethyl radical is –1.1 V. Another reason for the difference in absorbance at 420 nm generated by the hydrated electron or the CH
3C
•HOH radical could be the fact that some of the 1-hydroxyethyl radicals formed in solution can bind to HSA. A similar mechanism of the reaction of 1-hydroxyethyl radicals with BSA was proposed by Schüssler in 1981 [
17]. It was concluded that initial reactions of the organic radicals result in a conformational change, which makes hidden groups accessible. Comparison of normalized absorption spectra (
Figure 6) recorded during pulse radiolysis of aqueous HSA solutions saturated with N
2 (300 µM) containing
t-BuOH (0.1 M, the reactive species is the hydrated electron) or 30% EtOH vol. (under such conditions, there are two reactive species in the sample: the hydrated electron and the 1-hydroxyethyl radical) indicates that in both cases the reduction in cystine residues of albumin occurs. We did not observe the formation of CH
3C
•HOH radical adduct to the HSA polypeptide chains.
The decay of CyS–SCy
•– in the solution of HSA in the absence and presence of ethanol was also monitored by the pulse radiolysis technique. The absorbance was recorded at 420 nm. The decay of the disulfide radical anion within the HSA structure in both cases is a slow process and takes place on a time scale of seconds (
Figure S4). The kinetics of this band are described by triexponential decay, which is due to initial attachment at different disulfide linkages that have various intrinsic lifetimes. The trace at 420 nm evolves with the same kinetics in both cases. In our opinion, the disappearance of the reduced –S–S– bridges shown in
Figure S4 is related to the formation of hydrogels (under certain conditions, at high albumin concentration, appropriate frequency of ionizing radiation deposition, appropriate solution temperature). To understand the mechanisms of HSA or BSA aggregate formation, including hydrogels, it is necessary to use the scattered laser light detection method combined with the pulse radiolysis technique. We intend to carry out such measurements soon, using our own appropriate equipment. We have shown that under conditions of reductive stress (selectively generated by ionizing radiation, hydrated electrons, or CO
2•– anion radicals) stable protein aggregates can be formed in solutions containing HSA and 0.1 M
t-BuOH. Albumin solution concentrations must be high, close to 300 μM or higher. We reported the possibility of preparing hydrogels at room temperature and at temperatures elevated up to 60–70° C. The key issue for the effective preparation of hydrogels is the method of delivering the electron beam to the neat albumin solutions. In this work, the preparation of albumin aggregates using an electron accelerator for neat solutions containing ethanol was compared with solutions containing 0.1 M
t-BuOH. The general conclusion is that using ethanol concentrations below 40% vol. in albumin stock solutions does not provide any advantage over preparing hydrogels by irradiation using 0.1 M
t-BuOH. Work on the optimization of the hydrogel preparation procedure (pH, solution temperature, albumin concentration, electron beam repetition rate, absorbed dose, etc.) is time-consuming and requires continuation. Certainly, the radiation method of generating albumin aggregates leads to interesting biocompatible materials for medical applications. In some experiments, when the solutions were deaerated by vacuum, clear, transparent gels were obtained. A picture of cuvettes containing albumin hydrogel produced by electron beam is shown in the graphical abstract. Photographs of human serum albumin gels obtained after electron beam irradiation of an aqueous HSA solution containing 0.1 M
t-BuOH (left photo) or an irradiated HSA solution containing a large excess of ethanol (right photo). Air was removed from the solutions by vacuum, and the HSA concentration was 300 µM. In our opinion, only for these conditions (excessive proportion of ethanol in relation to water and HSA or BSA concentration above 300 µM), the use of ionizing radiation gives hope for the commercial production of AN. Certainly, the radiation method, which is effective in protein desolvation, is more advantageous than the method in which chemical reducers are used.
The lifetime of hydrated electrons in irradiated aqueous buffer solution and solution containing EtOH is the same and is equal to 3.0 μs. It is well known that the hydrated electron practically does not react with alcohol molecules, including EtOH. Since the beginning of the use of the pulse radiolysis method, the reactions of hydrated electrons with scavengers of various efficiencies have been studied depending on the composition of the solution, which was, among others, a mixture of water and ethanol [
32,
33]. Differences in kinetic behavior reactions of solvated electrons with scavengers in water and aqueous solutions with different ethanol content are caused by differences in the values of reagent diffusion coefficients [
34], solvation energies [
35], and dielectric properties of the binary medium [
33].
The presence of ethanol in the irradiated aqueous solution also affects the structure of the electron solvation shell. In binary solutions, the electron has different properties than those generated during the radiolysis of water and should be called solvated, not hydrated. When comparatively analyzing the decay of electrons in the reaction with HSA in water and in an aqueous solution containing a specific, not too large, amount of ethanol, all the factors mentioned above should be taken into account. The highest proportion of ethanol in our pulse radiolysis measurements was for a solution containing forty percent alcohol by volume. Therefore, this solution contains 17.1 mol (corresponding to 40% vol.) EtOH, and the alcohol concentration is 6.85 mol × dm
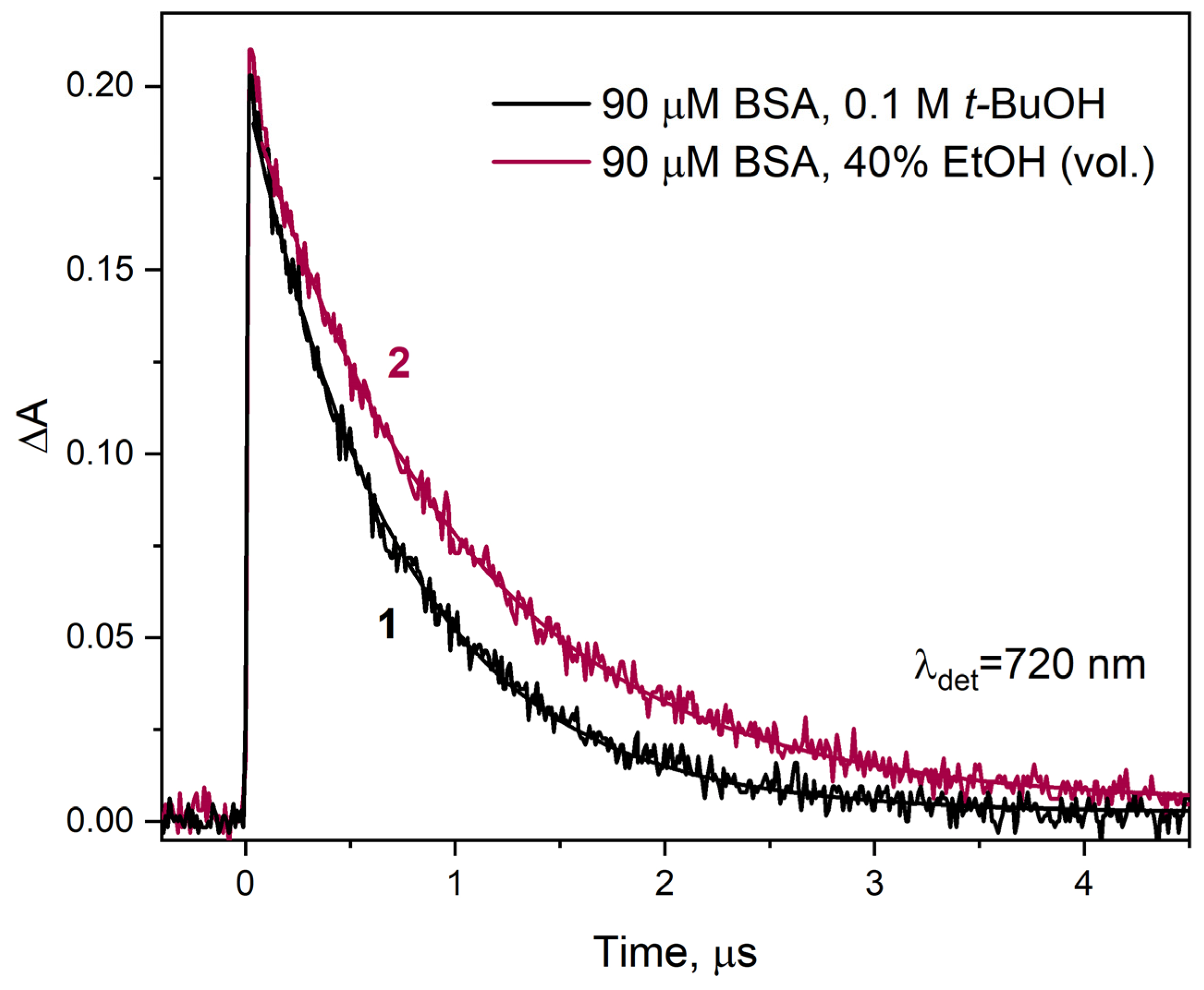
−3. To follow the disappearance of solvated electrons upon reaction with BSA in binary solution (40% EtOH vol.), we recorded the transient electron absorbance at 720 nm as a function of time (
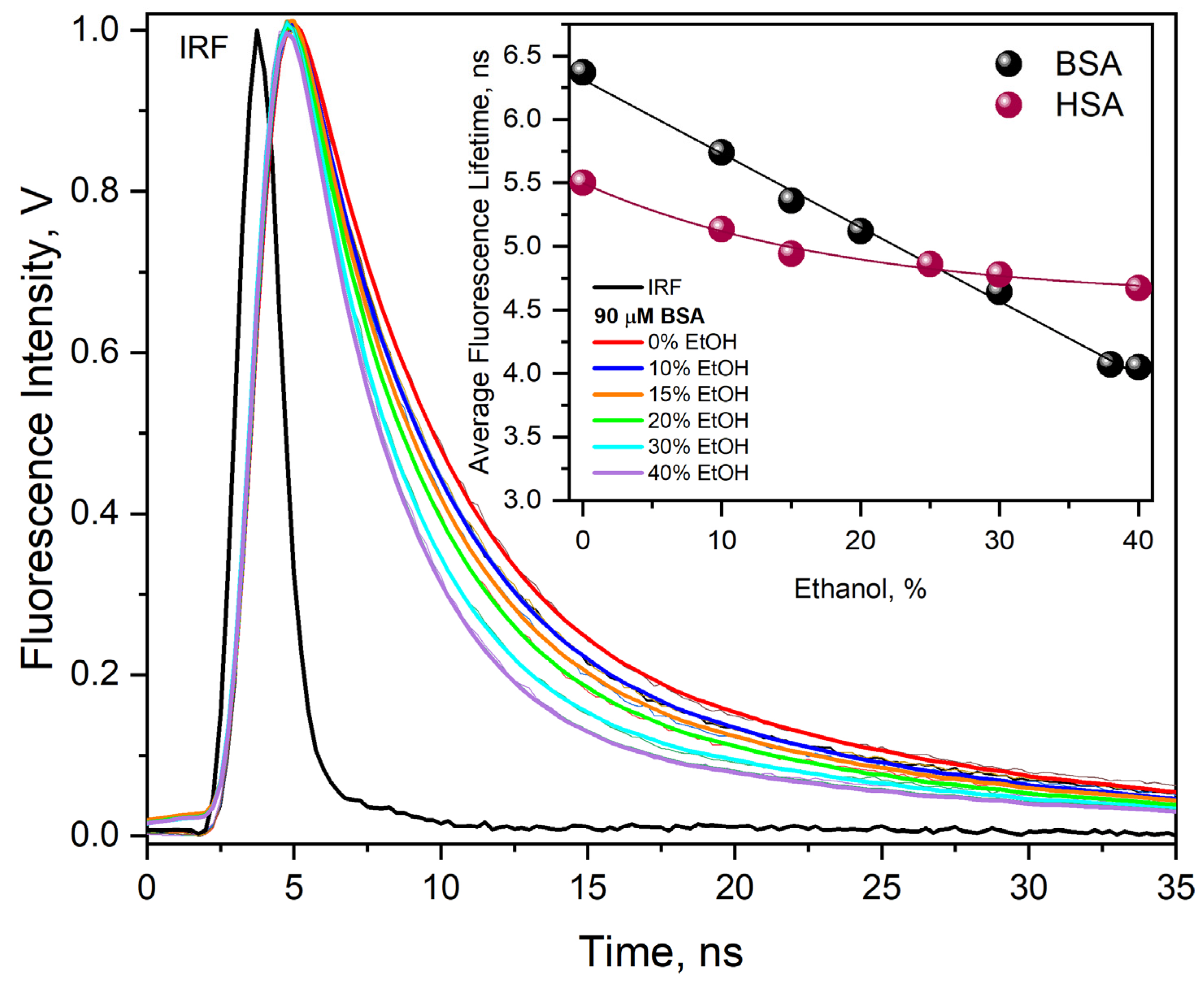
Figure 7). The decay time of the solvated electron absorption band in this case was 1.03 µs (curve 2). In a solution containing BSA without ethanol, the hydrated electron persists for a shorter time; the decay time is 720 ns (curve 1).
Based on the literature data mentioned above, it can be assumed that the difference in electron reactivity in the reaction with BSA in the presence and absence of EtOH is related to the change in the properties of the binary solution in relation to neat water. We believe this is not the only reason. The aggregation of albumin due to desolvation has a greater impact on the efficiency of the one-electron reduction in BSA. This is manifested by a decrease in the reaction rate constant
with BSA with time and aging of the solution. A 40% solution of BSA in ethanol, subjected to pulse radiolysis five hours after preparation, had a solvated electron decay time of over one and a half microseconds (in contrast to 1.03 µs for a freshly prepared sample). We analyzed the decays of the solvated electron absorption band for solutions of 90 µM BSA and containing 5, 10, 20, 30, and 40% EtOH. In the publication, we presented data from pulse radiolysis only for 40% EtOH (
Figure 7). Measurements performed at an interval of 24 h gave identical kinetics of the decay of the absorbance of
and those of the scavenger product recorded at 420 nm for all ethanol contents except for the sample containing 40% EtOH (vol.). The decrease in the efficiency of the –S–S– bridge reduction in the protein interior associated with the formation of albumin aggregates was noted by analyzing the intensity of the absorption band with a maximum of 420 nm. Example kinetic data from pulse radiolysis measurements are presented in
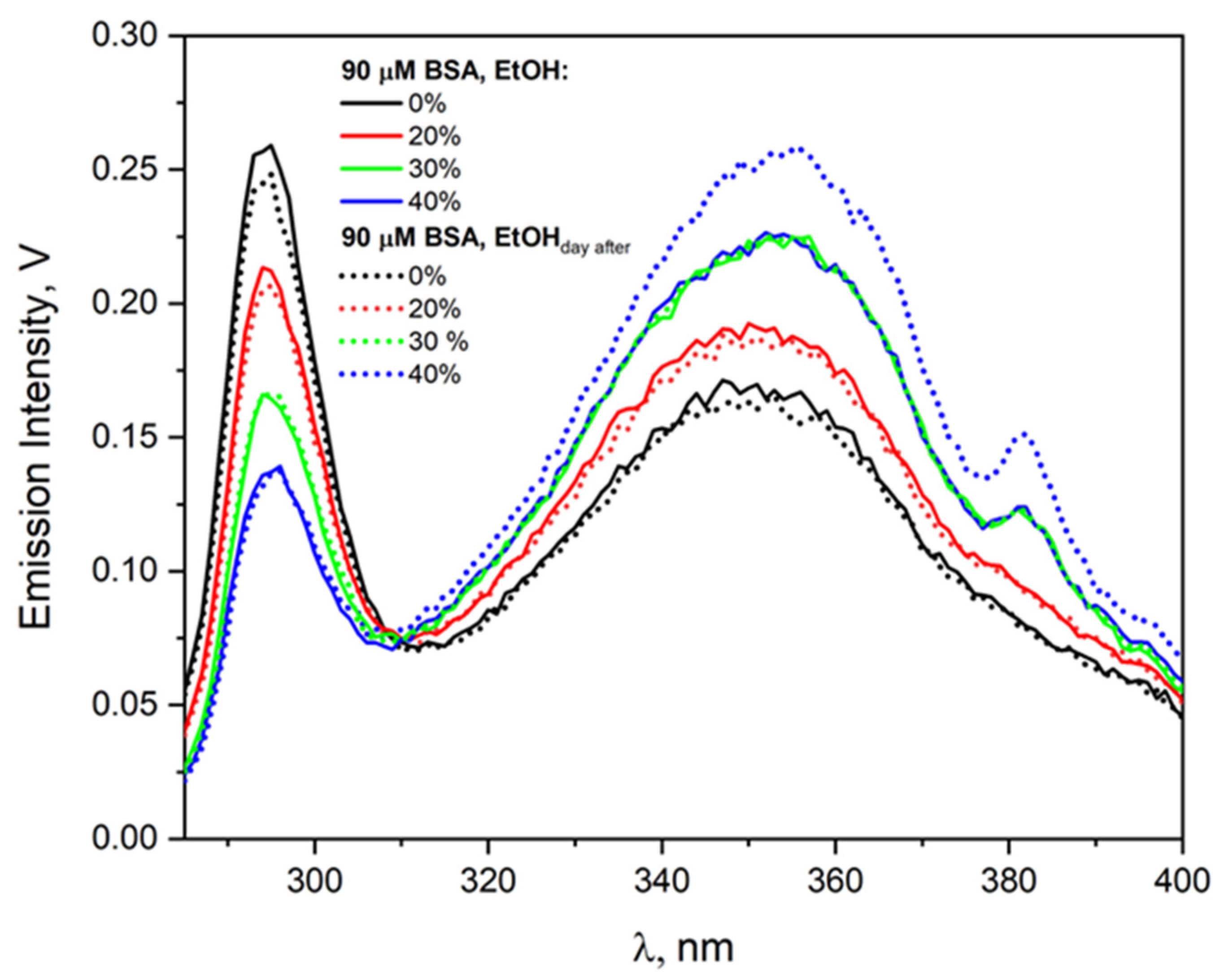
Figure S5 in the Supplementary Materials. Here, as the solution ages (measurement after 5 h and after a day), the intensity of the –S–S– band decreases, but only for the solution with an alcohol concentration of 6.85 M (40% EtOH vol.). Of course, solution parameters such as viscosity and dielectric constant did not change during this time. Aggregation of HSA or BSA induced by the desolvation process of solutions above 40% EtOH (vol.) reduces the number of isolated, single albumin molecules. This causes a decrease in the concentration of the electron scavenger, which prolongs the lifetime of the solvated electron and reduces the efficiency of protein reduction. We demonstrated the instability of albumin solutions containing more than 40% ethanol using emission measurements (
Figure 3 shows increase in the intensity of the emission band of BSA aggregates after one day of solution storage), NIR measurements (see
Figure S1 in Supplementary Materials), and RLS measurements (see
Figure S2 in Supplementary Materials).
For a more detailed analysis of the reaction of the CH
3C
•HOH radical with HSA, we performed pulse radiolysis experiments under various conditions. Samples subjected to electron beam irradiation containing
t-BuOH or EtOH as scavengers of
•OH radicals were deaerated under vacuum. Under such conditions, the reactive species in the solution containing
t-BuOH is the hydrated electron, and in the solution containing EtOH, there are both the hydrated electron and 1-hydroxyethyl radical (
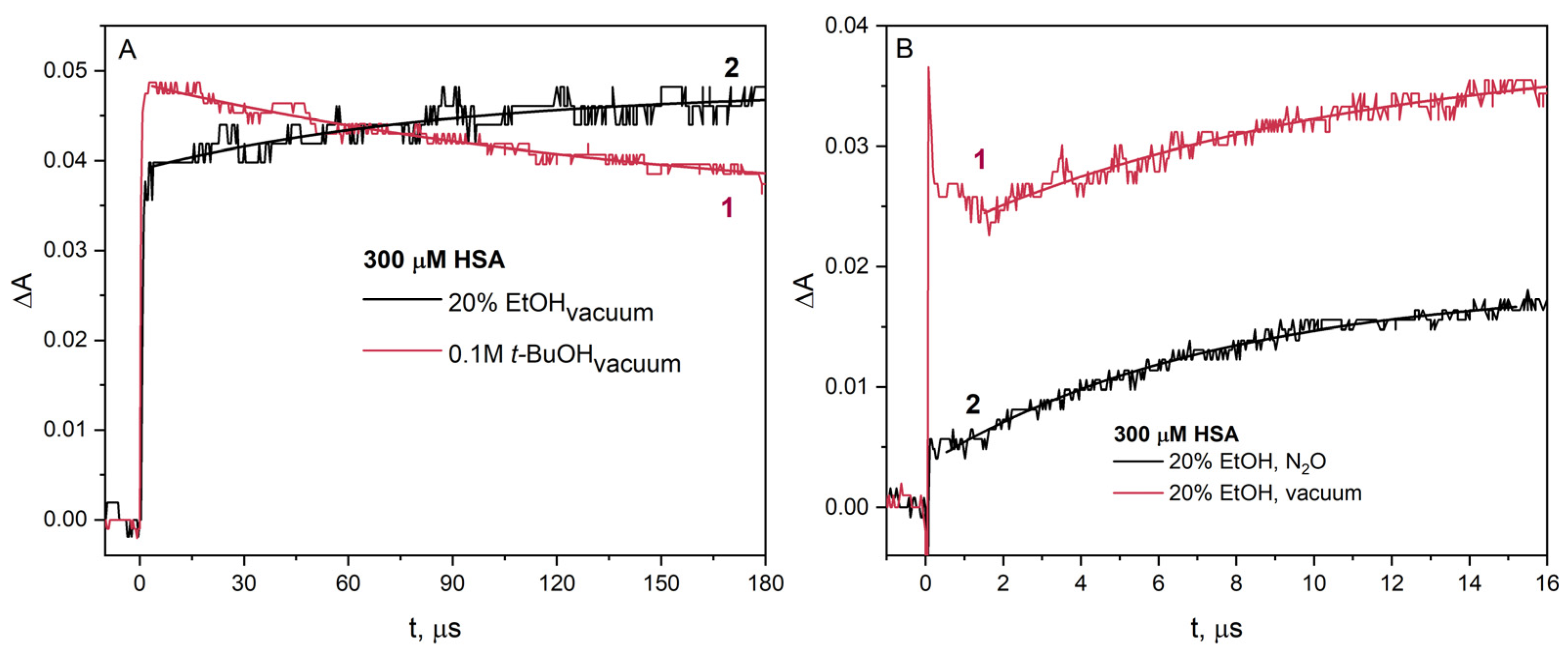
Figure 8A). The difference is in the intensity of absorption of the transient recorded (ΔA
0) immediately after irradiation of the HSA solution containing
t-BuOH (ΔA
0 is about 0.05) or EtOH (ΔA
0 is about 0.04).
The difference in absorbance value is due to protein aggregation in the presence of ethanol before irradiation. The amount of monomeric albumin decreases due to the formation of spontaneous HSA aggregates and causes a lower initial absorbance value in the pulse radiolysis experiment. In the case of pulse radiolysis of HSA solution containing
t-BuOH, the signal coming (curve 1) from the reduced –S–S– bridge (spectral maximum of 420 nm) disappears in the analyzed time interval. In the radiolysis experiment using EtOH as an
•OH radical scavenger, an increase in the signal of the HSA reduction product recorded at 420 nm was observed (curve 2;
Figure 8A). The increase in absorbance observed at 420 nm (curve 2) is due to the reduction in –S–S– residues of albumin as a result of the reaction of HSA with radicals CH
3C
•HOH. On the long time scale, the decays of the one-electron reduction product of HSA in solutions containing EtOH or
t-BuOH are very similar (see
Figure S4). This experiment confirms that EtOH used at low concentrations behaves as a classical
•OH radical scavenger, namely
t-BuOH.
The kinetic absorption pattern for HSA solution containing ethanol at 420 nm is complex. Recombination of 1-hydroxyethyl radicals also plays a crucial role in the HSA aggregation. 1-hydroxyethyl radicals generated in a water/EtOH solution (70/30% vol.) persist for about 150 µs for a dose of 200 Gy and decay by mutual recombination (see
Figure S6 in Supplementary Materials). 1-hydroxyethyl radicals recombine at a rate constant of 2k = 1.2 × 10
9 M
–1 s
–1, which we calculated using second-order kinetics for the decay in
Figure S6 in Supplementary Materials and the determined value of the molar absorbance coefficient at a wavelength of 260 nm of 800 dm
3 mol
–1 cm
–1 for the CH
3C
•HOH radical. For this reason, the concentration of available –S–S– groups must be rather high, so that the reduction in disulfides becomes measurable. Despite the complex kinetics, we estimated the rate constant of the reaction of the CH
3C
•HOH radical with HSA. The rate constant of this reaction is equal 7 × 10
8 dm
3 mol
–1 s
–1.
The next stage of our research was to study the reaction of the CH
3C
•HOH radical with the –S–S– bridges in the HSA structure. For this purpose, we conducted pulse radiolysis experiments of HSA solutions (300 µM) containing EtOH (up to 30% vol.) and saturated with N
2O or vacuum-deaerated. If an albumin solution containing EtOH was irradiated after N
2O saturation, only CH
3C
•HOH radicals react with the protein molecules. In vacuum-deaerated aqueous solutions, the reducing agents are both
and CH
3C
•HOH radical. As a result of N
2O saturation of albumin solution, the hydrated electrons are converted to
•OH radicals and then to CH
3C
•HOH radicals. For this reason, irradiation of aqueous solutions with the same dose results in the concentration of the CH
3C
•HOH radical being twice as high as the concentration of the hydrated electron. Assuming that all CH
3C
•HOH radicals are scavenged by HSA, the concentration of modified –S–S– bridges in the N
2O-saturated solution should be twice as high as in the vacuum-deaerated solution. However, this was not the case (
Figure 8B). This is due to the low value of the CH
3C
•HOH radical redox potential and other channels of their decay (reactions numbers 3–5). This may indicate that under such measurement conditions, CH
3C
•HOH radicals also attach to the HSA structure. The works of Schüssler [
14,
17] show that 1-hydroxyethyl radicals undergo an addition reaction to albumin during radiolysis. The access of CH
3C
•HOH radicals to disulfide bridges in HSA molecules is difficult, and these radicals may react mainly on the surface of the protein with the primary radicals:
Curve 2 in
Figure 8B, showing the increase in absorbance of the reduction product of selected –S–S– bridges in albumin, is clear evidence of electron transfer from the radical CH
3C
•HOH to HSA. In the case of pulse radiolysis of a solution of identical composition from which air has been removed, a significant increase in transient absorbance is observed. Obviously, this is due to the participation of hydrated electrons in the reduction of albumin. In the vacuum-deaerated solution, in addition to hydrated electrons, 1-hydroxyethyl radicals are also generated, resulting in an increase in the transient absorbance recorded at 425 nm. This increase in absorbance is caused by the reaction of HSA with the 1-hydroxyethyl, and the growth kinetics corresponds to that of the HSA reaction with only CH
3C
•HOH radicals.
Under reducing conditions, the addition of ethanol does not break more disulfide bonds compared to a solution in which –S–S– bonds are reduced only by the hydrated electron or formate radical (regardless of the concentration of HSA) [
11]. Our experimental results showed that the reaction efficiency of hydrated electrons with the same amount of HSA decreases in albumin solution containing EtOH compared to the solution containing 0.1 M
t-BuOH. In the case of HSA solution containing EtOH, the amount of monomeric albumin decreases due to the spontaneous formation of HSA aggregates as a consequence of the addition of alcohol to the solution (
Figure 9).
Albumin molecules are surrounded by layers of water. The addition of alcohol to the protein solution changes the hydration of the albumin, particularly affecting the first layer of water molecules surrounding the HSA. The literature reported the favorable interaction of ethanol with hydrophobic residues (leading to protein denaturation) but the unfavorable interaction with charged residues (causing reduction in protein solubility) [
28]. The presence of EtOH in the protein solution leads to the formation of polypeptide bonds between albumin molecules [
7,
10,
13].
It is important to note that the generation of HSA aggregates in a solution containing ethanol, therefore, consists of two steps: the ethanol-induced spontaneous aggregation of protein molecules and radiation cross-linking within HSA molecules induced by gamma radiation or electron beam. The HSA aggregates are the result of the recombination of –S–S
•– radicals. This leads to the formation of intermolecular –S–S– bridges between HSA molecules, and it is also possible to generate other covalent bridges (including C–C and C–S bonds) [
13].
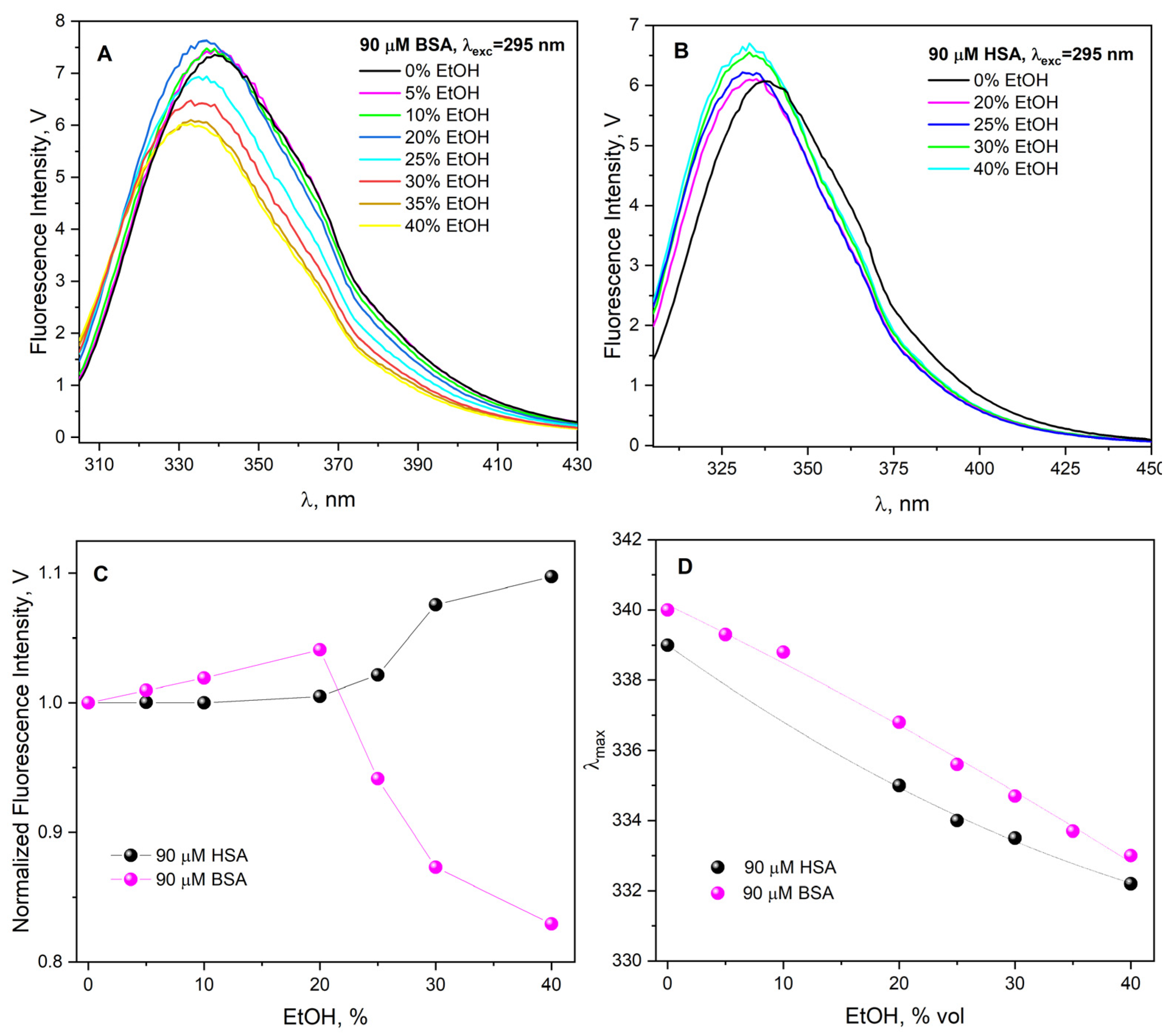
The reaction of reactive species with albumins can influence the structural integrity of proteins and lead to changes in their fluorescence lifetime. In order to more precisely analyze the process of HSA molecule aggregation in the presence of alcohol, time-resolved measurements of the emission decay of the studied solutions were performed (
Figure S7). The experiment for non-irradiated HSA solution (20 μM) after the addition of ethanol (30% vol.) showed a decrease in the fluorescence lifetime of Trp214, τ
avg = 4.31 ns (A
1 = 14%, τ
1 = 1.44 ns, A
2 = 86%, τ
2 = 4.78 ns) in comparison to the neat albumin solution (before the addition of EtOH). The average fluorescence lifetime of Trp214 in HSA solution (native form) is 5.49 ns (A
1 = 15%, τ
1 = 1.65 ns, A
2 = 85%, τ
2 = 6.17 ns). Due to the fact that tryptophan in the ground state occurs in two isomeric forms, L-tryptophan and D-tryptophan (conformational isomers), different fluorescence lifetimes of this amino acid are observed in time-resolved measurements [
36,
37]. The lifetime of Trp fluorescence in proteins is approximately 2 and 6 ns, depending on the microenvironment in which it is located. The decrease in the Trp214 fluorescence lifetime and the shift of the maximum band in the HSA emission spectrum confirmed the structural changes in albumin in the presence of ethanol. However, analogous measurements were performed for HSA solutions (20 μM) containing 30% EtOH (vol.) and irradiated with an electron beam with different doses (0–7200 Gy) (
Figure S7). Irradiation of these solutions with increasingly higher doses leads to a shorter fluorescence lifetime of Trp214 compared to the neat HSA solution. Our previous measurements show that the reaction of the hydrated electron with Trp induces a decrease in the fluorescence of Trp but does not cause any change in the fluorescence lifetime [
31].
Valuable information was also obtained from the analysis of emission time measurements recorded at 420 nm (after excitation with a laser pulse of wavelength 337 nm) of BSA solutions (300 µM) saturated with N
2O in the absence (blue line) and in the presence of 20% EtOH vol. (pink line). Both solutions were irradiated with a dose of 9000 Gy (
Figure S8). Excitation of BSA solutions with 337 nm laser light selectively enables the detection of emission from excited protein aggregates, as this wavelength is not absorbed by monomeric albumin. BSA aggregates formed as a result of the irradiation of a solution containing ethanol are characterized by different emission decay kinetics recorded on an oscilloscope than the emission pattern of a BSA solution in which the aggregates were generated in the reaction with
•OH radicals (
Figure S8). The emission decay at 420 nm is characterized by a two-exponential function with an average time of 3.37 ns in the case of ethanolic solution of BSA and 2.69 ns for the irradiated solution without EtOH (only the hydroxyl radicals are involved in the formation of aggregates). These measurements provide further evidence that the reaction of 1-hydroxyethyl radicals with protein causes the reduction in disulfide bridges and does not lead to the formation of dityrosine bridges (in the oxidation process).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}