
Histone Acetylation in Central and Peripheral Nervous System Injuries and Regeneration: Epigenetic Dynamics and Therapeutic Perspectives


Abstract
1. Introduction
2. Post-Translational Histone Modifications
2.1. Histone Acetylation
2.2. Histone Methylation
3. Histone Acetylation and Its Influence on Axonal Regeneration
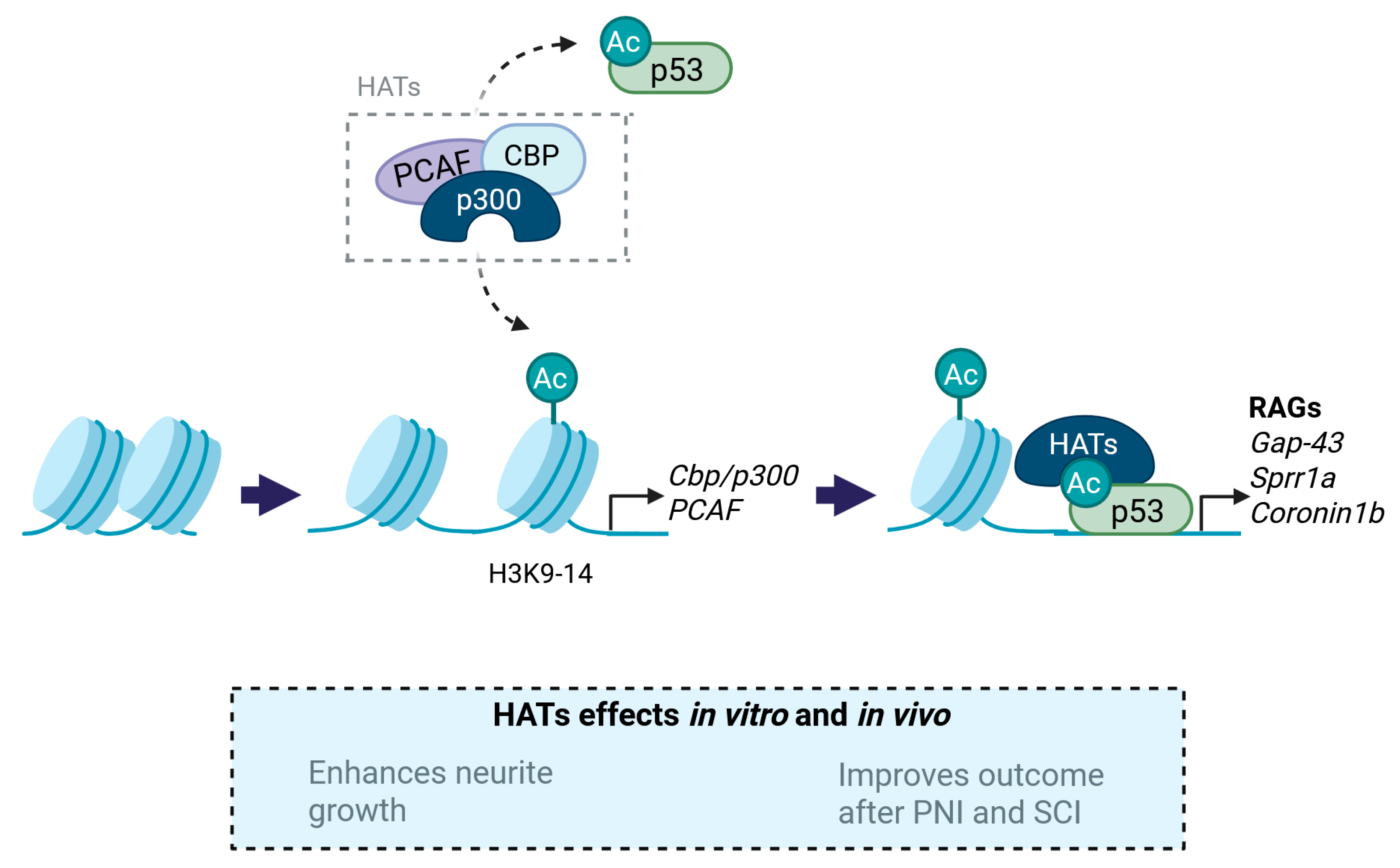
3.1. Histone Acetyltransferases (HATs)
3.1.1. p300/CBP
3.1.2. PCAF
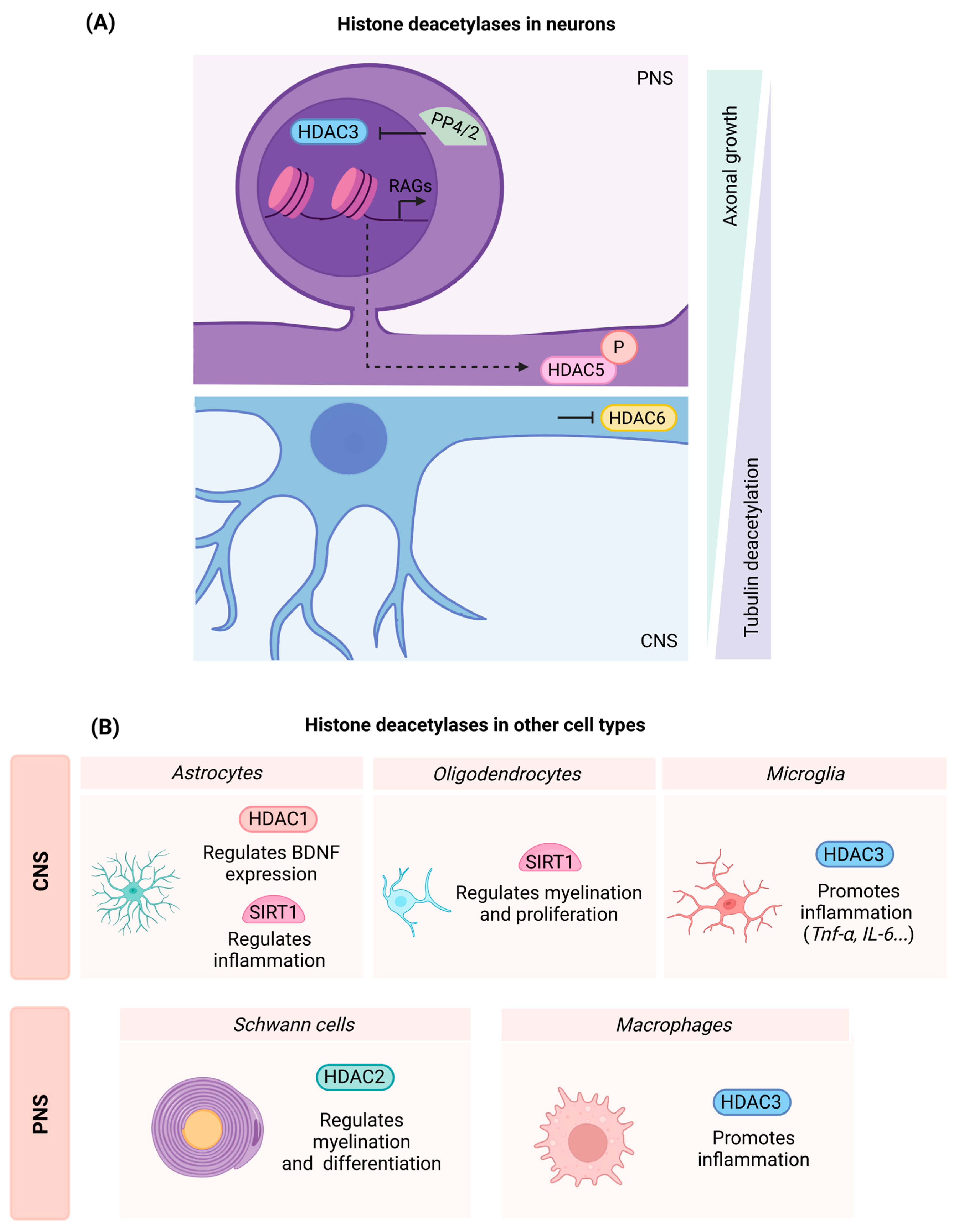
3.2. Histone Deacetylases (HDACs)
3.2.1. Class I HDACs
- HDAC1
- HDAC2
- HDAC3
3.2.2. Class II HDACs
- HDAC5
- HDAC6
3.2.3. Class III HDACs
- Sirtuin 1 (SIRT1)
3.3. Readers of Acetylated Residues
3.3.1. BET Proteins
3.3.2. BRG1
4. Epigenetic Modifications in Non-Neuronal Cells and Consequences for Neural Regeneration
4.1. Myeloid Cells: Microglia and Macrophages
- Histone Deacetylases and the Relevance of HDAC3 in Inflammatory Events
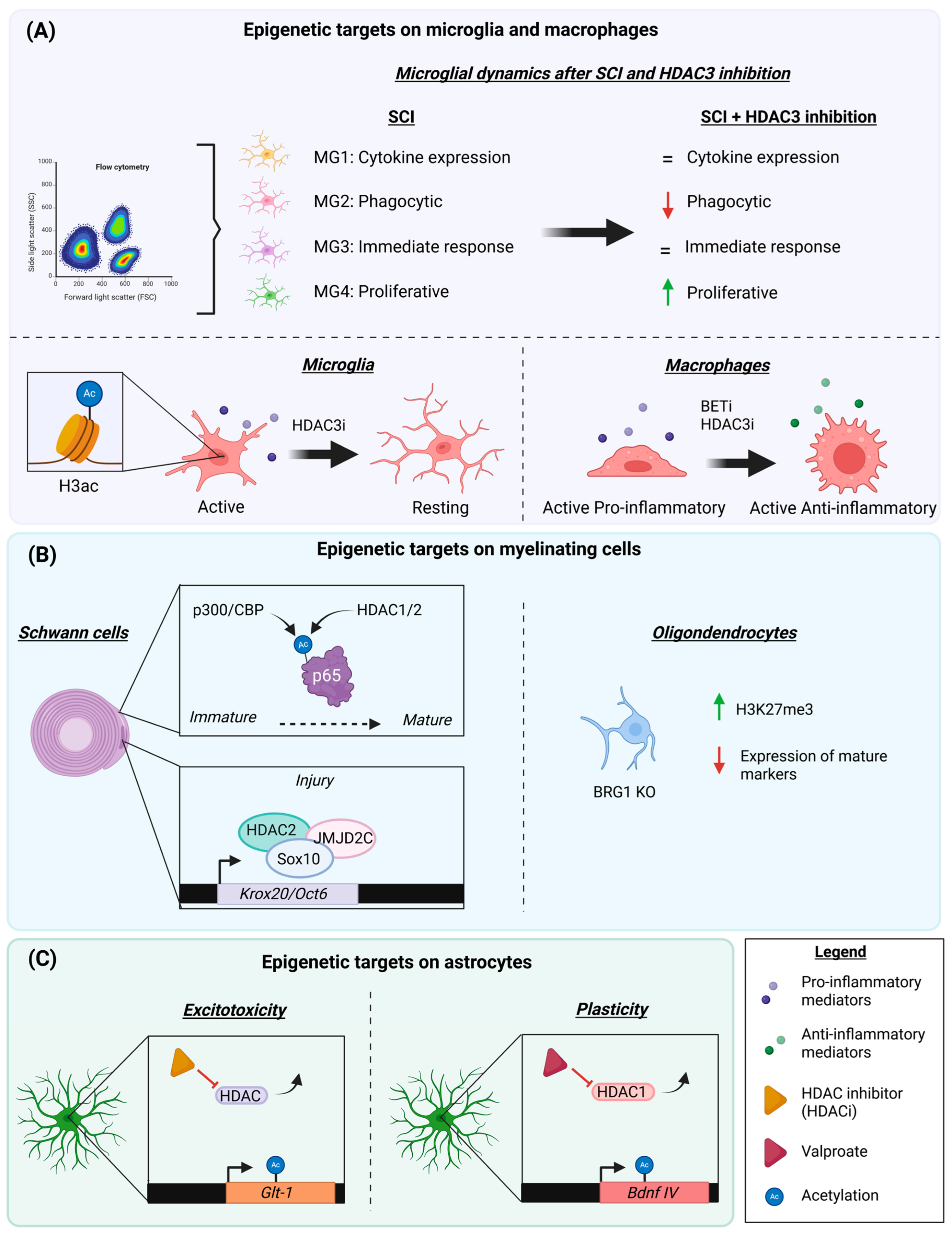
- BET Proteins and Inflammation
4.2. Myelinating Cells: Schwann Cells and Oligodendrocytes
- Influence of Histone Deacetylases
- BRG1
4.3. Astrocytes
- HDACs in Excitotoxicity and Neuroprotection
- Growth and Plasticity: the Importance of BET Proteins and HDACs
- HATs and HDACs in Inflammation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | Basso–Beattie–Bresnahan locomotor test |
| BET | Bromodomain and extra-terminal domain (BET) proteins |
| BDNF | Brain-derived neurotrophic factor |
| BMS | Basso mouse scale |
| BRD4 | Bromodomain-containing protein 4 |
| BRG1 | Brahma-related gene 1 |
| CBP | CREB binding protein |
| CGN | Cerebellum granular neurons |
| CNS | Central nervous system |
| CSPG | Chondroitin sulfate proteoglycan |
| CytC | Cytochrome C |
| DNA | Deoxyribonucleic acid |
| dpi | Days post-injury |
| DRG | Dorsal root ganglion |
| GAP43 | Growth-associated protein 43 |
| GDNF | Glial cell line-derived neurotrophic factor |
| H3 | Histone 3 |
| H4 | Histone 4 |
| HAT | Histone acetylase |
| HDAC | Histone deacetylase |
| hGCN5 | General control non-depressible 5 |
| HIF-1α | Hypoxia-inducible factor 1α |
| HO-1 | Heme oxygenase 1 |
| IL- | Interleukin- |
| IPS | Induced pluripotent stem cell |
| KO | Knockout |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| MBP | Myelin basic protein |
| miR- | MicroRNA- |
| NGF | Nerve growth factor |
| NQO-1 | NAD(P)H quinone dehydrogenase 1 |
| p53 | Tumor protein P53 |
| p300 | Histone acetyltransferase p300 |
| PCAF | P300/CBP associating factor |
| PCR2 | Polycomb repressive complex 2 |
| PNI | Peripheral nerve injury |
| PNS | Peripheral nervous system |
| RAG | Regeneration-associated gene |
| ROS | Reactive oxygen species |
| RNA | Ribonucleic acid |
| SCI | Spinal cord injury |
| SIRT | Sirtuin |
| SMARCA4 | SWI/SNF-related BAF chromatin remodeling complex subunit ATPase 4 |
| SOD | Superoxide dismutase |
| STAT | Signal transducer and activator of transcription |
| SWI-SNF | SWItch/sucrose non-fermentable complex |
| TNF-α | Tumor necrosis factor alpha |
| TSA | Trichostatin A |
| VAMP | Vesicle-associated membrane protein |
| VEGF | Vascular endothelial growth factor |
References
- Navarro, X.; Vivo, M.; Valero-Cabre, A. Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 2007, 82, 163–201. [Google Scholar] [CrossRef]
- Navarro, X.; Geuna, S.; Grothe, C.; Haastert-Talini, K. Introduction: Thematic Papers Issue on Peripheral Nerve Regeneration and Repair. Anat. Rec. 2018, 301, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W. The Struggle to Make CNS Axons Regenerate: Why Has It Been so Difficult? Neurochem. Res. 2020, 45, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Buchli, A.D.; Schwab, M.E. Inhibition of Nogo: A key strategy to increase regeneration, plasticity and functional recovery of the lesioned central nervous system. Ann. Med. 2005, 37, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A. Tuning the orchestra: Transcriptional pathways controlling axon regeneration. Front. Mol. Neurosci. 2011, 4, 60. [Google Scholar] [CrossRef]
- Ma, T.C.; Willis, D.E. What makes a RAG regeneration associated? Front. Mol. Neurosci. 2015, 8, 43. [Google Scholar] [CrossRef]
- Abe, N.; Cavalli, V. Nerve injury signaling. Curr. Opin. Neurobiol. 2008, 18, 276–283. [Google Scholar] [CrossRef]
- Ma, T.C.; Barco, A.; Ratan, R.R.; Willis, D.E. cAMP-responsive element-binding protein (CREB) and cAMP co-regulate activator protein 1 (AP1)-dependent regeneration-associated gene expression and neurite growth. J. Biol. Chem. 2014, 289, 32914–32925. [Google Scholar] [CrossRef]
- Fagoe, N.D.; Attwell, C.L.; Kouwenhoven, D.; Verhaagen, J.; Mason, M.R. Overexpression of ATF3 or the combination of ATF3, c-Jun, STAT3 and Smad1 promotes regeneration of the central axon branch of sensory neurons but without synergistic effects. Hum. Mol. Genet. 2015, 24, 6788–6800. [Google Scholar] [CrossRef]
- Venkatesh, I.; Mehra, V.; Wang, Z.; Simpson, M.T.; Eastwood, E.; Chakraborty, A.; Beine, Z.; Gross, D.; Cabahug, M.; Olson, G.; et al. Co-occupancy identifies transcription factor co-operation for axon growth. Nat. Commun. 2021, 12, 2555. [Google Scholar] [CrossRef]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Christopher, M.A.; Kyle, S.M.; Katz, D.J. Neuroepigenetic mechanisms in disease. Epigenetics Chromatin 2017, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, I.; Di Giovanni, S. Advances and Limitations of Current Epigenetic Studies Investigating Mammalian Axonal Regeneration. Neurotherapeutics 2018, 15, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Song, H.; Ming, G.L.; Weng, Y.L. Epigenetic and epitranscriptomic regulation of axon regeneration. Mol. Psychiatry 2023, 28, 1440–1450. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Wahane, S.; Halawani, D.; Zhou, X.; Zou, H. Epigenetic Regulation Of Axon Regeneration and Glial Activation in Injury Responses. Front. Genet. 2019, 10, 640. [Google Scholar] [CrossRef]
- Kumar, V.; Rayan, N.A.; Muratani, M.; Lim, S.; Elanggovan, B.; Xin, L.; Lu, T.; Makhija, H.; Poschmann, J.; Lufkin, T.; et al. Comprehensive benchmarking reveals H2BK20 acetylation as a distinctive signature of cell-state-specific enhancers and promoters. Genome Res. 2016, 26, 612–623. [Google Scholar] [CrossRef]
- Barral, A.; Dejardin, J. The chromatin signatures of enhancers and their dynamic regulation. Nucleus 2023, 14, 2160551. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Taylor, G.C.; Eskeland, R.; Hekimoglu-Balkan, B.; Pradeepa, M.M.; Bickmore, W.A. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013, 23, 2053–2065. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Ninova, M.; Fejes Toth, K.; Aravin, A.A. The control of gene expression and cell identity by H3K9 trimethylation. Development 2019, 146, dev181180. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, Y.; Jia, J.; Fang, Y.; Tang, Y.; Wu, H.; Fang, D. H3K36me3, message from chromatin to DNA damage repair. Cell Biosci. 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Gaub, P.; Tedeschi, A.; Puttagunta, R.; Nguyen, T.; Schmandke, A.; Di Giovanni, S. HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 2010, 17, 1392–1408. [Google Scholar] [CrossRef]
- Gaub, P.; Joshi, Y.; Wuttke, A.; Naumann, U.; Schnichels, S.; Heiduschka, P.; Di Giovanni, S. The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 2011, 134 Pt 7, 2134–2148. [Google Scholar] [CrossRef]
- Choi, M.; Ko, S.Y.; Lee, I.Y.; Wang, S.E.; Lee, S.H.; Oh, D.H.; Kim, Y.-S.; Son, H. Carbamylated erythropoietin promotes neurite outgrowth and neuronal spine formation in association with CBP/p300. Biochem. Biophys. Res. Commun. 2014, 446, 79–84. [Google Scholar] [CrossRef]
- Ding, Z.; Cao, J.; Shen, Y.; Zou, Y.; Yang, X.; Zhou, W.; Guo, Q.; Huang, C. Resveratrol Promotes Nerve Regeneration via Activation of p300 Acetyltransferase-Mediated VEGF Signaling in a Rat Model of Sciatic Nerve Crush Injury. Front. Neurosci. 2018, 12, 341. [Google Scholar] [CrossRef]
- Hutson, T.H.; Kathe, C.; Palmisano, I.; Bartholdi, K.; Hervera, A.; De Virgiliis, F.; McLachlan, E.; Zhou, L.; Kong, G.; Barraud, Q.; et al. Cbp-dependent histone acetylation mediates axon regeneration induced by environmental enrichment in rodent spinal cord injury models. Sci. Transl. Med. 2019, 11, eaaw2064. [Google Scholar] [CrossRef]
- Müller, F.; De Virgiliis, F.; Kong, G.; Zhou, L.; Serger, E.; Chadwick, J.; Sanchez-Vassopoulos, A.; Singh, A.K.; Eswaramoorthy, M.; Kundu, T.K.; et al. CBP/p300 activation promotes axon growth, sprouting, and synaptic plasticity in chronic experimental spinal cord injury with severe disability. PLoS Biol. 2022, 20, e3001310. [Google Scholar] [CrossRef]
- Puttagunta, R.; Tedeschi, A.; Sória, M.G.; Hervera, A.; Lindner, R.; Rathore, K.I.; Gaub, P.; Joshi, Y.; Nguyen, T.; Schmandke, A.; et al. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat. Commun. 2014, 5, 3527. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell. Sci. 2001, 114 Pt 13, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Zhang, J.; Awasthi, S.; Sharma, A.; Rogers, L.; Matlock, E.F.; Van Lint, C.; Karpova, T.; McNally, J.; Harrod, R. Nerve growth factor receptor signaling induces histone acetyltransferase domain-dependent nuclear translocation of p300/CREB-binding protein-associated factor and hGCN5 acetyltransferases. J. Biol. Chem. 2004, 279, 55667–55674. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Shukla, S.; Shariat-Madar, Z.; Walker, L.A.; Tekwani, B.L. Mechanism for neurotropic action of vorinostat, a pan histone deacetylase inhibitor. Mol. Cell Neurosci. 2016, 77, 11–20. [Google Scholar] [CrossRef]
- Brügger, V.; Duman, M.; Bochud, M.; Münger, E.; Heller, M.; Ruff, S.; Jacob, C. Delaying histone deacetylase response to injury accelerates conversion into repair Schwann cells and nerve regeneration. Nat. Commun. 2017, 8, 14272. [Google Scholar] [CrossRef]
- Finelli, M.J.; Wong, J.K.; Zou, H. Epigenetic regulation of sensory axon regeneration after spinal cord injury. J. Neurosci. 2013, 33, 19664–19676. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef]
- Kuboyama, T.; Wahane, S.; Huang, Y.; Zhou, X.; Wong, J.K.; Koemeter-Cox, A.; Martini, M.; Friedel, R.H.; Zou, H. HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci. Rep. 2017, 7, 8641. [Google Scholar] [CrossRef]
- Chen, J.; Laramore, C.; Shifman, M.I. Differential expression of HDACs and KATs in high and low regeneration capacity neurons during spinal cord regeneration. Exp. Neurol. 2016, 280, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shifman, M.I. The expression of histone deacetylases and the regenerative abilities of spinal-projecting neurons after injury. Neural Regen. Res. 2016, 11, 1577–1578. [Google Scholar] [PubMed]
- Sada, N.; Fujita, Y.; Mizuta, N.; Ueno, M.; Furukawa, T.; Yamashita, T. Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell Death Dis. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Garraway, S.M.; Huie, J.R. Spinal Plasticity and Behavior: BDNF-Induced Neuromodulation in Uninjured and Injured Spinal Cord. Neural Plast. 2016, 2016, 9857201. [Google Scholar] [CrossRef]
- Hervera, A.; Zhou, L.; Palmisano, I.; McLachlan, E.; Kong, G.; Hutson, T.H.; Danzi, M.C.; Lemmon, V.P.; Bixby, J.L.; Matamoros-Angles, A.; et al. PP4-dependent HDAC3 dephosphorylation discriminates between axonal regeneration and regenerative failure. EMBO J. 2019, 38, e101032. [Google Scholar] [CrossRef]
- Chen, S.; Ye, J.; Wu, G.; Shi, J.; Li, X.; Chen, X.; Wu, W. Histone Deacetylase 3 Inhibition Ameliorates Microglia-Mediated Neuro-Inflammation Via the SIRT1/Nrf2 Pathway After Traumatic Spinal Cord Injury. Neurorehabil. Neural Repair 2023, 37, 503–518. [Google Scholar] [CrossRef]
- Sanchez, S.; Lemmens, S.; Baeten, P.; Sommer, D.; Dooley, D.; Hendrix, S.; Fabregas, M.G. HDAC3 Inhibition Promotes Alternative Activation of Macrophages but Does Not Affect Functional Recovery after Spinal Cord Injury. Exp. Neurobiol. 2018, 27, 437–452. [Google Scholar] [CrossRef]
- Kamal, S.R.; Potukutchi, S.; Gelovani, D.J.; Bonomi, R.E.; Kallakuri, S.; Cavanaugh, J.M.; Mangner, T.; Conti, A.; Liu, R.S.; Pasqualini, R.; et al. Spatial and temporal dynamics of HDACs class IIa following mild traumatic brain injury in adult rats. Mol. Psychiatry 2022, 27, 1683–1693. [Google Scholar] [CrossRef]
- Janke, C.; Kneussel, M. Tubulin post-translational modifications: Encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010, 33, 362–372. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012, 31, 3063–3078. [Google Scholar] [CrossRef]
- Cho, Y.; Sloutsky, R.; Naegle, K.M.; Cavalli, V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 2013, 155, 894–908. [Google Scholar] [CrossRef] [PubMed]
- Rivieccio, M.A.; Brochier, C.; Willis, D.E.; Walker, B.A.; D’ANnibale, M.A.; McLaughlin, K.; Siddiq, A.; Kozikowski, A.P.; Jaffrey, S.R.; Twiss, J.L.; et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 19599–19604. [Google Scholar] [CrossRef] [PubMed]
- Erturk, A.; Hellal, F.; Enes, J.; Bradke, F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 2007, 27, 9169–9180. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhou, Y.; Ye, L.; Lu, Q.; Zhang, K.; Zhang, J.; Xie, L.; Wu, Y.; Xu, K.; Zhang, H.; et al. Histone deacetylase 6 inhibition restores autophagic flux to promote functional recovery after spinal cord injury. Exp. Neurol. 2020, 324, 113138. [Google Scholar] [CrossRef]
- Sakamoto, K.; Ozaki, T.; Ko, Y.-C.; Tsai, C.-F.; Gong, Y.; Morozumi, M.; Ishikawa, Y.; Uchimura, K.; Nadanaka, S.; Kitagawa, H.; et al. Glycan sulfation patterns define autophagy flux at axon tip via PTPRsigma-cortactin axis. Nat. Chem. Biol. 2019, 15, 699–709. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Forés, J.; Herrando-Grabulosa, M.; Valls, R.; Leiva-Rodríguez, T.; Galea, E.; González-Pérez, F.; Navarro, X.; Petegnief, V.; Bosch, A.; et al. Neuroprotective Drug for Nerve Trauma Revealed Using Artificial Intelligence. Sci. Rep. 2018, 8, 1879. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Leiva-Rodriguez, T.; Fores, J.; Casas, C. Improved Motor Nerve Regeneration by SIRT1/Hif1a-Mediated Autophagy. Cells 2019, 8, 1354. [Google Scholar] [CrossRef]
- Liu, C.M.; Wang, R.Y.; Saijilafu Jiao, Z.X.; Zhang, B.Y.; Zhou, F.Q. MicroRNA-138 and SIRT1 form a mutual negative feedback loop to regulate mammalian axon regeneration. Genes. Dev. 2013, 27, 1473–1483. [Google Scholar] [CrossRef]
- Jiang, T.; Qin, T.; Gao, P.; Tao, Z.; Wang, X.; Wu, M.; Gu, J.; Chu, B.; Zheng, Z.; Yi, J.; et al. SIRT1 attenuates blood-spinal cord barrier disruption after spinal cord injury by deacetylating p66Shc. Redox Biol. 2023, 60, 102615. [Google Scholar] [CrossRef]
- Chen, H.; Ji, H.; Zhang, M.; Liu, Z.; Lao, L.; Deng, C.; Chen, J.; Zhong, G. An Agonist of the Protective Factor SIRT1 Improves Functional Recovery and Promotes Neuronal Survival by Attenuating Inflammation after Spinal Cord Injury. J. Neurosci. 2017, 37, 2916–2930. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, J.; Zhang, J.; Lin, J.; Wu, Y.; Wang, X. Inhibition of Brd4 by JQ1 Promotes Functional Recovery From Spinal Cord Injury by Activating Autophagy. Front. Cell Neurosci. 2020, 14, 555591. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Jin, H.; Lin, D.; Chen, Y.; Chen, X.; Wang, B.; Hu, S.; Wu, Y.; Wu, Y.; et al. BRD4 inhibition attenuates inflammatory response in microglia and facilitates recovery after spinal cord injury in rats. J. Cell Mol. Med. 2019, 23, 3214–3223. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ventura, J.; Amo-Aparicio, J.; Navarro, X.; Penas, C. BET protein inhibition regulates cytokine production and promotes neuroprotection after spinal cord injury. J. Neuroinflammation 2019, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- Palomes-Borrajo, G.; Navarro, X.; Penas, C. BET protein inhibition in macrophages enhances dorsal root ganglion neurite outgrowth in female mice. J. Neurosci. Res. 2022, 100, 1331–1346. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, M.; Chang, C.W.; Wang, C.; Shi, X.; Zhan, X.; Birnbaum, S.G.; Bezprozvanny, I.; Huber, K.M.; Wu, J.I. Autism-Associated Chromatin Regulator Brg1/SmarcA4 Is Required for Synapse Development and Myocyte Enhancer Factor 2-Mediated Synapse Remodeling. Mol. Cell Biol. 2016, 36, 70–83. [Google Scholar] [CrossRef]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFkappaB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. [Google Scholar] [CrossRef]
- Berryer, M.H.; Rizki, G.; Nathanson, A.; Klein, J.A.; Trendafilova, D.; Susco, S.G.; Lam, D.; Messana, A.; Holton, K.M.; Karhohs, K.W.; et al. High-content synaptic phenotyping in human cellular models reveals a role for BET proteins in synapse assembly. elife 2023, 12, e80168. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Chmykhalo, V.K.; Deev, R.V.; Tokarev, A.T.; Polunina, Y.A.; Xue, L.; Shidlovskii, Y.V. SWI/SNF Complex Connects Signaling and Epigenetic State in Cells of Nervous System. Mol. Neurobiol. 2025, 62, 1536–1557. [Google Scholar] [CrossRef]
- Singh, M.; Popowicz, G.M.; Krajewski, M.; Holak, T.A. Structural ramification for acetyl-lysine recognition by the bromodomain of human BRG1 protein, a central ATPase of the SWI/SNF remodeling complex. Chembiochem 2007, 8, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Limpert, A.S.; Bai, S.; Narayan, M.; Wu, J.; Yoon, S.O.; Carter, B.D.; Lu, Q.R. NF-kappaB forms a complex with the chromatin remodeler BRG1 to regulate Schwann cell differentiation. J. Neurosci. 2013, 33, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflammation 2011, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Fonken, L.K. Glial Cells Shape Pathology and Repair After Spinal Cord Injury. Neurotherapeutics 2018, 15, 554–577. [Google Scholar] [CrossRef]
- Bhatt, D.; Ghosh, S. Regulation of the NF-kappaB-Mediated Transcription of Inflammatory Genes. Front. Immunol. 2014, 5, 71. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e7. [Google Scholar] [CrossRef]
- Saccani, S.; Pantano, S.; Natoli, G. Two waves of nuclear factor kappaB recruitment to target promoters. J. Exp. Med. 2001, 193, 1351–1359. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Wahane, S.; Zhou, X.; Zhou, X.; Guo, L.; Friedl, M.-S.; Kluge, M.; Ramakrishnan, A.; Shen, L.; Friedel, C.C.; Zhang, B.; et al. Diversified transcriptional responses of myeloid and glial cells in spinal cord injury shaped by HDAC3 activity. Sci. Adv. 2021, 7, eabd8811. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Du, Y.; Wang, J.; Weng, Q.; Liu, X.; Nicholson, E.; Xin, M.; Lu, Q.R. BRG1 programs PRC2-complex repression and controls oligodendrocyte differentiation and remyelination. J. Cell Biol. 2024, 223, e202310143. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Q.; Jiang, S.; Liu, S.; Xie, Z.; Zhang, X.; Huang, H.; Zhu, S. Regulation of Glutamate Transporter Type 1 by TSA and the Antiepileptic Mechanism of TSA. Neurochem. Res. 2025, 50, 74. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.C.; Jahr, F.M.; Hawkins, E.; Kronfol, M.M.; Younis, R.M.; McClay, J.L.; Deshpande, L.S. Epigenetic histone acetylation and Bdnf dysregulation in the hippocampus of rats exposed to repeated, low-dose diisopropylfluorophosphate. Life Sci. 2021, 281, 119765. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Rudman, M.D.; Choi, J.S.; Lee, H.E.; Tan, S.K.; Ayad, N.G.; Lee, J.K. Bromodomain and extraterminal domain-containing protein inhibition attenuates acute inflammation after spinal cord injury. Exp. Neurol. 2018, 309, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Goulart, V.G.; Menezes, G.D.; Espirito-Santo, S.; Faria-Melibeu, A.D.C.; Serfaty, C.A.; Campello-Costa, P. Interleukin 4 Leads to Sustained Phosphorylation of the STAT6 and ERK Pathways in the Retina and Disrupts Subcortical Visual Circuitry in Rodents. Neuroimmunomodulation 2018, 25, 96–102. [Google Scholar] [CrossRef]
- Ali, S.A.; Hanks, J.E.; Stebbins, A.W.; Alkhalili, O.; Cohen, S.T.; Chen, J.Y.; Smith, D.R.; Dumont, C.M.; Shea, L.D.; Hogikyan, N.D.; et al. Delivery of Interleukin-4-Encoding Lentivirus Using Multiple-Channel Bridges Enhances Nerve Regeneration. Laryngoscope 2020, 130, 2802–2810. [Google Scholar] [CrossRef]
- Van Broeckhoven, J.; Erens, C.; Sommer, D.; Scheijen, E.; Sanchez, S.; Vidal, P.M.; Dooley, D.; Van Breedam, E.; Quarta, A.; Ponsaerts, P.; et al. Macrophage-based delivery of interleukin-13 improves functional and histopathological outcomes following spinal cord injury. J. Neuroinflammation 2022, 19, 102. [Google Scholar] [CrossRef]
- Büttner, R.; Schulz, A.; Reuter, M.; Akula, A.K.; Mindos, T.; Carlstedt, A.; Riecken, L.B.; Baader, S.L.; Bauer, R.; Morrison, H. Inflammaging impairs peripheral nerve maintenance and regeneration. Aging Cell 2018, 17, e12833. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Yoon, S.O.; Xu, X.; Hottiger, M.O.; Svaren, J.; Nave, K.A.; Kim, H.A.; Olson, E.N.; Lu, Q.R. HDAC-mediated deacetylation of NF-kappaB is critical for Schwann cell myelination. Nat. Neurosci. 2011, 14, 437–441. [Google Scholar] [CrossRef]
- Jacob, C.; Christen, C.N.; Pereira, J.A.; Somandin, C.; Baggiolini, A.; Lötscher, P.; Özçelik, M.; Tricaud, N.; Meijer, D.; Yamaguchi, T.; et al. HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat. Neurosci. 2011, 14, 429–436. [Google Scholar] [CrossRef]
- He, X.; Zhang, L.; Queme, L.F.; Liu, X.; Lu, A.; Waclaw, R.R.; Dong, X.; Zhou, W.; Kidd, G.; Yoon, S.-O.; et al. A histone deacetylase 3-dependent pathway delimits peripheral myelin growth and functional regeneration. Nat. Med. 2018, 24, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L.H.; Cattin, A.-L.; Fontana, X.; Harford-Wright, E.; Burden, J.J.; White, I.J.; Smith, J.G.; Napoli, I.; Quereda, V.; Policarpi, C.; et al. HDAC3 Regulates the Transition to the Homeostatic Myelinating Schwann Cell State. Cell Rep. 2018, 25, 2755–2765.e5. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, V.A.; Ho, P.P.; Brett, J.O.; Ucar, D.; Dugas, J.C.; Pollina, E.A.; Chow, L.M.L.; Ibrahim, A.; Baker, S.J.; Barres, B.A.; et al. Expansion of oligodendrocyte progenitor cells following SIRT1 inactivation in the adult brain. Nat. Cell Biol. 2013, 15, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Asher, R.A. The glial scar and central nervous system repair. Brain Res. Bull. 1999, 49, 377–391. [Google Scholar] [CrossRef]
- Liu, D.; Xu, G.Y.; Pan, E.; McAdoo, D.J. Neurotoxicity of glutamate at the concentration released upon spinal cord injury. Neuroscience 1999, 93, 1383–1389. [Google Scholar] [CrossRef]
- Gao, X.; Zeb, S.; He, Y.-Y.; Guo, Y.; Zhu, Y.-M.; Zhou, X.-Y.; Zhang, H.-L. Valproic Acid Inhibits Glial Scar Formation after Ischemic Stroke. Pharmacology 2022, 107, 263–280. [Google Scholar] [CrossRef]
- Penas, C.; Verdú, E.; Asensio-Pinilla, E.; Guzmán-Lenis, M.; Herrando-Grabulosa, M.; Navarro, X.; Casas, C. Valproate reduces CHOP levels and preserves oligodendrocytes and axons after spinal cord injury. Neuroscience 2011, 178, 33–44. [Google Scholar] [CrossRef]
- Chang, P.; Williams, A.M.; Bhatti, U.F.; Biesterveld, B.E.; Liu, B.; Nikolian, V.C.; Dennahy, I.S.; Lee, J.; Li, Y.; Alam, H.B. Valproic Acid and Neural Apoptosis, Inflammation, and Degeneration 30 Days after Traumatic Brain Injury, Hemorrhagic Shock, and Polytrauma in a Swine Model. J. Am. Coll. Surg. 2019, 228, 265–275. [Google Scholar] [CrossRef]
- Baltan, S.; Murphy, S.P.; Danilov, C.A.; Bachleda, A.; Morrison, R.S. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J. Neurosci. 2011, 31, 3990–3999. [Google Scholar] [CrossRef]
- Wu, X.; Chen, P.S.; Dallas, S.; Wilson, B.; Block, M.L.; Wang, C.-C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng, Y.; et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 2008, 11, 1123–1134. [Google Scholar] [CrossRef]
- Nikkar, R.; Esmaeili-Bandboni, A.; Badrikoohi, M.; Babaei, P. Effects of inhibiting astrocytes and BET/BRD4 chromatin reader on spatial memory and synaptic proteins in rats with Alzheimer’s disease. Metab. Brain Dis. 2022, 37, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Yuan, D.; Shi, F.; Wu, X.; Luo, Z.; Gan, W. The Dose-Dependent Effects of Fluorocitrate on the Metabolism and Activity of Astrocytes and Neurons. Brain Sci. 2025, 15, 99. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef] [PubMed]
- Ghizzoni, M.; Haisma, H.J.; Maarsingh, H.; Dekker, F.J. Histone acetyltransferases are crucial regulators in NF-kappaB mediated inflammation. Drug Discov. Today 2011, 16, 504–511. [Google Scholar] [CrossRef]
- Rao, Y.; Li, J.; Qiao, R.; Luo, J.; Liu, Y. Synergistic effects of tetramethylpyrazine and astragaloside IV on spinal cord injury via alteration of astrocyte A1/A2 polarization through the Sirt1-NF-kappaB pathway. Int. Immunopharmacol. 2024, 131, 111686. [Google Scholar] [CrossRef]
- Bolivar, S.; Udina, E. Preferential regeneration and collateral dynamics of motor and sensory neurons after nerve injury in mice. Exp. Neurol. 2022, 358, 114227. [Google Scholar] [CrossRef]
- Navarro, X.; Verdu, E.; Buti, M. Comparison of regenerative and reinnervating capabilities of different functional types of nerve fibers. Exp. Neurol. 1994, 129, 217–224. [Google Scholar] [CrossRef]
- Bolivar, S.; Sanz, E.; Ovelleiro, D.; Zochodne, D.W.; Udina, E. Neuron-specific RNA-sequencing reveals different responses in peripheral neurons after nerve injury. elife 2024, 12, RP91316. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Shi, M.-Q.; Xu, Y.; Fu, X.; Pan, D.-S.; Lu, X.-P.; Xiao, Y.; Jiang, Y.-Z. Advances in targeting histone deacetylase for treatment of solid tumors. J. Hematol. Oncol. 2024, 17, 37. [Google Scholar] [CrossRef]
- McCutcheon, S.R.; Rohm, D.; Iglesias, N.; Gersbach, C.A. Epigenome editing technologies for discovery and medicine. Nat. Biotechnol. 2024, 42, 1199–1217. [Google Scholar] [CrossRef] [PubMed]
- Ueda, J.; Yamazaki, T.; Funakoshi, H. Toward the Development of Epigenome Editing-Based Therapeutics: Potentials and Challenges. Int. J. Mol. Sci. 2023, 24, 4778. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Histone Modification | Effects on Gene Expression | Location | Source |
|---|---|---|---|
| H3K9ac | Activation | Promoters, enhancers | [17] |
| H3K27ac | Activation | Enhancers, promoters | [17,18] |
| H4K8ac | Activation | Promoters | [19] |
| H4K16ac | Activation | Promoters, gene bodies | [19,20] |
| Histone Modification | Effects on Gene Expression | Location | Source |
|---|---|---|---|
| H3K4me1 | Activation | Enhancers | [18,21] |
| H3K4me2 | Activation/repression | Gene bodies | [21] |
| H3K4me3 | Activation or poised expression | Promoters | [21] |
| H3K9me2 | Repression | Transposable elements, satellite repeats, and gene bodies | [22] |
| H3K9me3 | Repression or poised expression | Enhancers, telomeres, transposable elements, gene bodies, and satellite repeats | [18,22,23] |
| H3K27me3 | Repression | Enhancers, promoters | [18] |
| H3K36me3 | Activation | Gene bodies | [24] |
| Enzyme | Hyperacetylated Histone | Altered Genes | Outcome In Vitro | Outcome In Vivo | Source |
|---|---|---|---|---|---|
| p300/ CBP | H3K9-14ac H4K8ac H3K27ac | Gap43, Coronin1b, Sgc10, α-tubulin, Sprr1a, Vegfr1, Vegfr2, Vegfb, Psd95, Shank2, and Shank3 | Neurite outgrowth in CGNs and DRG primary cultures | Neurite outgrowth after optic nerve crush Motor function repair after PNI Functional motor and sensory recovery after acute SCI | [25,26,27,28,29,30] |
| PCAF | H3K9ac | Gap43, Bdnf and Galanin | PCAF-AVV leads to increased neurite growth in dissociated DRG cultures | Overexpression leads to histological evidence of axonal regeneration after SCI | [31] |
| Enzyme | Altered Genes | Altered Proteins | Outcome In Vitro | Outcome In Vivo | Source |
|---|---|---|---|---|---|
| BET proteins | Gap43, Il-4, and Il-13 | IL-6, IL-10, IL-13, SOD1, CytC, HO-1, Cleaved caspase 3, Beclin, and LC3II/I | BET-inhibition improved resistance to ROS Inhibition of BET proteins prevented neurite growth | Inhibition of BET proteins improves functional outcome after SCI, but not after nerve injury | [61,62,63,64] |
| BRG1 | VGluT2, Camk4, Gap43, and Cacng3, among others | - | Deletion reduces spine density and neuronal function | Not assessed | [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomés-Borrajo, G.; Navarro, X.; Penas, C. Histone Acetylation in Central and Peripheral Nervous System Injuries and Regeneration: Epigenetic Dynamics and Therapeutic Perspectives. Int. J. Mol. Sci. 2025, 26, 6277. https://doi.org/10.3390/ijms26136277
Palomés-Borrajo G, Navarro X, Penas C. Histone Acetylation in Central and Peripheral Nervous System Injuries and Regeneration: Epigenetic Dynamics and Therapeutic Perspectives. International Journal of Molecular Sciences. 2025; 26(13):6277. https://doi.org/10.3390/ijms26136277
Chicago/Turabian StylePalomés-Borrajo, Georgina, Xavier Navarro, and Clara Penas. 2025. "Histone Acetylation in Central and Peripheral Nervous System Injuries and Regeneration: Epigenetic Dynamics and Therapeutic Perspectives" International Journal of Molecular Sciences 26, no. 13: 6277. https://doi.org/10.3390/ijms26136277
APA StylePalomés-Borrajo, G., Navarro, X., & Penas, C. (2025). Histone Acetylation in Central and Peripheral Nervous System Injuries and Regeneration: Epigenetic Dynamics and Therapeutic Perspectives. International Journal of Molecular Sciences, 26(13), 6277. https://doi.org/10.3390/ijms26136277