
Therapeutic Approaches for C9ORF72-Related ALS: Current Strategies and Future Horizons

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Physiological and Pathological Roles of C9ORF72 in ALS
2.1. C9ORF72 Physiological Functions
2.2. Pathological Roles of C9ORF72 in ALS
3. Small Molecules as Therapeutic Strategies for C9ORF72-Associated ALS
3.1. Small Molecules Targeting RNA Structure
3.2. Small Molecules Acting on DPR Accumulation
3.3. Small Molecules for the Restoration of Nucleocytoplasmic Transport
3.4. Antiviral Drugs for C9ORF72-ALS
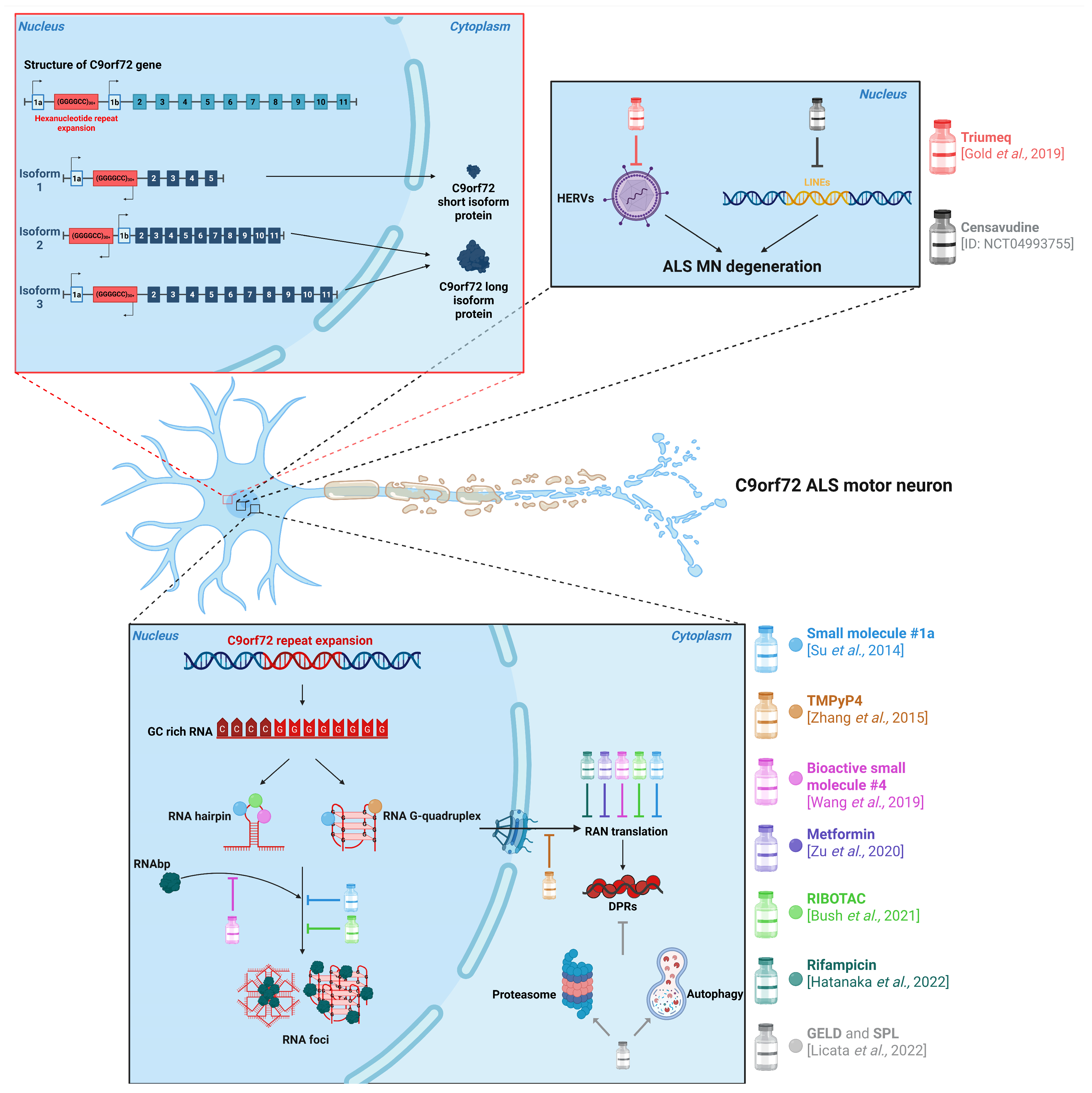
4. Biological Drugs for Treating C9ORF72-Associated ALS
4.1. RNA-Targeted Therapies
4.2. Protein-Targeted Therapies
4.3. Gene Editing
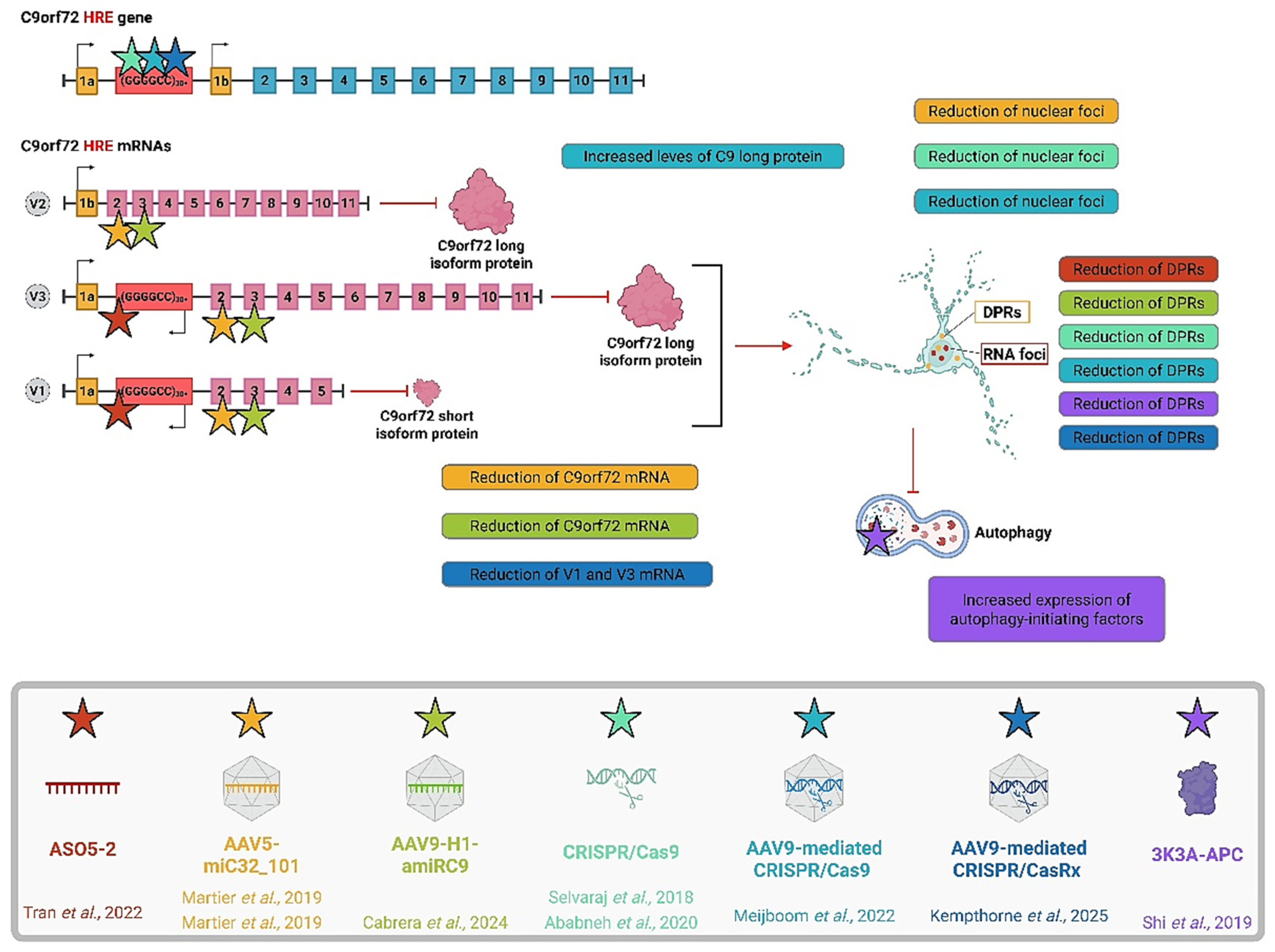
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated viral vector |
| F2R-PAR1 | Factor II thrombin receptor |
| ALS | Amyotrophic lateralsclerosis |
| ALSFRS-R | Revised ALS Functional Rating Scale |
| ART | Antiretroviral therapy |
| ASO | Antisense oligonucleotides |
| BAC | Bacterial artificial chromosome |
| C9-L | C9ORF72-Long |
| C9orf72 | Chromosome 9 open-reading frame 72 |
| C9-S | C9ORF72-Short |
| DENN | Differentially Expressed in Normal and Neoplastic Cells |
| DPR | Dipeptide repeats |
| FTD | Frontotemporal dementia |
| GELD | Geldanamycin |
| HERVs | Human endogenous retroviruses |
| LINEs | Long interspersed elements |
| PKR | RNA-dependent protein kinase |
| r(G4C2)exp | Expanded G4C2 repetition |
| RAN | Repeated-associated non-ATG |
| RanGEF | Ran guanine nucleotide exchange factor |
| RIBOTAC | RNase-targeting chimera |
| RNAbp | RNA-binding proteins |
| RNase | Endogenous ribonuclease |
| RTE | Retrotransposable element |
| SPL | Spironolactone |
References
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Zampatti, S.; Peconi, C.; Campopiano, R.; Gambardella, S.; Caltagirone, C.; Giardina, E. C9orf72-Related Neurodegenerative Diseases: From Clinical Diagnosis to Therapeutic Strategies. Front. Aging Neurosci. 2022, 14, 907122. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. [Google Scholar] [CrossRef]
- Boros, B.D.; Schoch, K.M.; Kreple, C.J.; Miller, T.M. Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 2022, 19, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Xiao, S.; MacNair, L.; McLean, J.; McGoldrick, P.; McKeever, P.; Soleimani, S.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. C9orf72 isoforms in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Brain Res. 2016, 1647, 43–49. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e539. [Google Scholar] [CrossRef]
- Zhang, S.; Tong, M.; Zheng, D.; Huang, H.; Li, L.; Ungermann, C.; Pan, Y.; Luo, H.; Lei, M.; Tang, Z.; et al. C9orf72-catalyzed GTP loading of Rab39A enables HOPS-mediated membrane tethering and fusion in mammalian autophagy. Nat. Commun. 2023, 14, 6360. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; De Vos, K.J. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408. [Google Scholar] [CrossRef]
- Beckers, J.; Tharkeshwar, A.K.; Van Damme, P. C9orf72 ALS-FTD: Recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef]
- Van Daele, S.H.; Moisse, M.; van Vugt, J.J.; Zwamborn, R.A.; van der Spek, R.; van Rheenen, W.; Van Damme, P. Genetic variability in sporadic amyotrophic lateral sclerosis. Brain 2023, 146, 3760–3769. [Google Scholar] [CrossRef] [PubMed]
- Devenney, E.; Hornberger, M.; Irish, M.; Mioshi, E.; Burrell, J.; Tan, R.; Hodges, J.R. Frontotemporal dementia associated with the C9ORF72 mutation: A unique clinical profile. JAMA Neurol. 2014, 71, 331–339. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Hautbergue, G.M.; Gendron, T.F.; van Blitterswijk, M.; Hardiman, O.; Ravits, J.; Isaacs, A.M.; Rademakers, R. Amyotrophic lateral sclerosis caused by hexanucleotide repeat expansions in C9orf72: From genetics to therapeutics. Lancet Neurol. 2025, 24, 261–274. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.-R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef]
- Mehta, A.R.; Selvaraj, B.T.; Barton, S.K.; McDade, K.; Abrahams, S.; Chandran, S.; Smith, C.; Gregory, J.M. Improved detection of RNA foci in C9orf72 amyotrophic lateral sclerosis post-mortem tissue using BaseScope™ shows a lack of association with cognitive dysfunction. Brain Commun. 2020, 2, fcaa009. [Google Scholar] [CrossRef]
- Sonobe, Y.; Lee, S.; Krishnan, G.; Gu, Y.; Kwon, D.Y.; Gao, F.-B.; Roos, R.P.; Kratsios, P. Translation of dipeptide repeat proteins in C9ORF72 ALS/FTD through unique and redundant AUG initiation codons. eLife 2023, 12, e83189. [Google Scholar] [CrossRef]
- Bitetto, G.; Di Fonzo, A. Nucleo-cytoplasmic transport defects and protein aggregates in neurodegeneration. Transl. Neurodegener. 2020, 9, 25. [Google Scholar] [CrossRef]
- Yang, Q.; Jiao, B.; Shen, L. The Development of C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Disorders. Front. Genet. 2020, 11, 562758. [Google Scholar] [CrossRef]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.; Terpstra, M.L.; Zundel, C.A.C.; de Sá, R.V.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- Yuva-Aydemir, Y.; Almeida, S.; Gao, F.B. Insights into C9ORF72-Related ALS/FTD from Drosophila and iPSC Models. Trends Neurosci. 2018, 41, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Fortier, G.; Butti, Z.; Patten, S.A. Modelling C9orf72-Related Amyotrophic Lateral Sclerosis in Zebrafish. Biomedicines 2020, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, Y.; Aburas, J.; Krishnan, G.; Fleming, A.C.; Ghadge, G.; Islam, P.; Warren, E.C.; Gu, Y.; Kankel, M.W.; Brown, A.E.X.; et al. A C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role for eIF2D in RAN translation. Nat. Commun. 2021, 12, 6025. [Google Scholar] [CrossRef]
- Elmansy, M.F.; Reidl, C.T.; Rahaman, M.; Özdinler, P.H.; Silverman, R.B. Small molecules targeting different cellular pathologies for the treatment of amyotrophic lateral sclerosis. Med. Res. Rev. 2023, 43, 2260–2302. [Google Scholar] [CrossRef]
- Raguseo, F.; Wang, Y.; Li, J.; Howe, M.P.; Balendra, R.; Huyghebaert, A.; Vadukul, D.M.; Tanase, D.A.; Maher, T.E.; Malouf, L.; et al. The ALS/FTD-related C9orf72 hexanucleotide repeat expansion forms RNA condensates through multimolecular G-quadruplexes. Nat. Commun. 2023, 14, 8272. [Google Scholar] [CrossRef]
- Simone, R.; Balendra, R.; Moens, T.G.; Preza, E.; Wilson, K.M.; Heslegrave, A.; Woodling, N.S.; Niccoli, T.; Gilbert-Jaramillo, J.; Abdelkarim, S.; et al. G-quadruplex-binding small molecules ameliorate. EMBO Mol. Med. 2018, 10, 22–31. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.; Shaw, P.G.; Kim, M.-S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Su, Z.; Zhang, Y.; Gendron, T.F.; Bauer, P.O.; Chew, J.; Yang, W.-Y.; Fostvedt, E.; Jansen-West, K.; Belzil, V.V.; Desaro, P.; et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 2014, 83, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-F.; Ursu, A.; Childs-Disney, J.L.; Guertler, R.; Yang, W.-Y.; Bernat, V.; Rzuczek, S.G.; Fuerst, R.; Zhang, Y.-J.; Gendron, T.F.; et al. The Hairpin Form of r(G4C2)exp in c9ALS/FTD Is Repeat-Associated Non-ATG Translated and a Target for Bioactive Small Molecules. Cell Chem. Biol. 2019, 26, 179–190.e112. [Google Scholar] [CrossRef]
- Bush, J.A.; Aikawa, H.; Fuerst, R.; Li, Y.; Ursu, A.; Meyer, S.M.; Benhamou, R.I.; Chen, J.L.; Khan, T.; Wagner-Griffin, S.; et al. Ribonuclease recruitment using a small molecule reduced c9ALS/FTD r(G 4 C 2) repeat expansion in vitro and in vivo ALS models. Sci. Transl. Med. 2021, 13, eabd5991. [Google Scholar] [CrossRef]
- Licata, N.V.; Cristofani, R.; Salomonsson, S.; Wilson, K.M.; Kempthorne, L.; Vaizoglu, D.; D’aGostino, V.G.; Pollini, D.; Loffredo, R.; Pancher, M.; et al. C9orf72 ALS/FTD dipeptide repeat protein levels are reduced by small molecules that inhibit PKA or enhance protein degradation. EMBO J. 2022, 41, e105026. [Google Scholar] [CrossRef] [PubMed]
- Ciesiolka, A.; Jazurek, M.; Drazkowska, K.; Krzyzosiak, W.J. Structural Characteristics of Simple RNA Repeats Associated with Disease and their Deleterious Protein Interactions. Front. Cell. Neurosci. 2017, 11, 97. [Google Scholar] [CrossRef]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Tusi, S.K.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Umeda, T.; Shigemori, K.; Takeuchi, T.; Nagai, Y.; Tomiyama, T. C9orf72 Hexanucleotide Repeat Expansion-Related Neuropathology Is Attenuated by Nasal Rifampicin in Mice. Biomedicines 2022, 10, 1080. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.-P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef]
- Gold, J.; Rowe, D.B.; Kiernan, M.C.; Vucic, S.; Mathers, S.; van Eijk, R.P.A.; Nath, A.; Montojo, M.G.; Norato, G.; Santamaria, U.A.; et al. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: The Lighthouse trial. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 595–604. [Google Scholar] [CrossRef]
- Terry, D.M.; Devine, S.E. Aberrantly High Levels of Somatic LINE-1 Expression and Retrotransposition in Human Neurological Disorders. Front. Genet. 2019, 10, 1244. [Google Scholar] [CrossRef]
- Liu, E.Y.; Russ, J.; Cali, C.P.; Phan, J.M.; Amlie-Wolf, A.; Lee, E.B. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 2019, 27, 1409–1421.e1406. [Google Scholar] [CrossRef]
- Prudencio, M.; Gonzales, P.K.; Cook, C.N.; Gendron, T.F.; Daughrity, L.M.; Song, Y.; Ebbert, M.T.; van Blitterswijk, M.; Zhang, Y.-J.; Jansen-West, K.; et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 2017, 26, 3421–3431. [Google Scholar] [CrossRef]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Moazami, M.P.; Yang, H.; McKenna-Yasek, D.; Douthwright, C.L.; Pinto, C.; Metterville, J.; Shin, M.; Sanil, N.; Dooley, C.; et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 2022, 28, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Je, G.; Pacut, P.; Keyhanian, K.; Gao, J.; Ghasemi, M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2066. [Google Scholar] [CrossRef] [PubMed]
- Martier, R.; Liefhebber, J.M.; Miniarikova, J.; van der Zon, T.; Snapper, J.; Kolder, I.; Petry, H.; van Deventer, S.J.; Evers, M.M.; Konstantinova, P. Artificial MicroRNAs Targeting C9orf72 Can Reduce Accumulation of Intra-nuclear Transcripts in ALS and FTD Patients. Mol. Ther. Nucleic Acids 2019, 14, 593–608. [Google Scholar] [CrossRef]
- Martier, R.; Liefhebber, J.M.; García-Osta, A.; Miniarikova, J.; Cuadrado-Tejedor, M.; Espelosin, M.; Ursua, S.; Petry, H.; van Deventer, S.J.; Evers, M.M.; et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol. Ther. Nucleic Acids 2019, 16, 26–37. [Google Scholar] [CrossRef]
- Cabrera, G.T.; Meijboom, K.E.; Abdallah, A.; Tran, H.; Foster, Z.; Weiss, A.; Wightman, N.; Stock, R.; Gendron, T.; Gruntman, A.; et al. Artificial microRNA suppresses C9ORF72 variants and decreases toxic dipeptide repeat proteins in vivo. Gene Ther. 2024, 31, 105–118. [Google Scholar] [CrossRef]
- Chua, J.P.; De Calbiac, H.; Kabashi, E.; Barmada, S.J. Autophagy and ALS: Mechanistic insights and therapeutic implications. Autophagy 2022, 18, 254–282. [Google Scholar] [CrossRef]
- Shi, Y.; Hung, S.-T.; Rocha, G.; Lin, S.; Linares, G.R.; Staats, K.A.; Seah, C.; Wang, Y.; Chickering, M.; Lai, J.; et al. Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 2019, 5, e127736. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- A Ababneh, N.; Scaber, J.; Flynn, R.; Douglas, A.; Barbagallo, P.; Candalija, A.; Turner, M.R.; Sims, D.; Dafinca, R.; A Cowley, S.; et al. Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum. Mol. Genet. 2020, 29, 2200–2217. [Google Scholar] [CrossRef] [PubMed]
- Meijboom, K.E.; Abdallah, A.; Fordham, N.P.; Nagase, H.; Rodriguez, T.; Kraus, C.; Gendron, T.F.; Krishnan, G.; Esanov, R.; Andrade, N.S.; et al. CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat. Commun. 2022, 13, 6286. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Kempthorne, L.; Vaizoglu, D.; Cammack, A.J.; Carcolé, M.; Roberts, M.J.; Mikheenko, A.; Fisher, A.; Suklai, P.; Muralidharan, B.; Kroll, F.; et al. Dual-targeting CRISPR-CasRx reduces C9orf72 ALS/FTD sense and antisense repeat RNAs in vitro and in vivo. Nat. Commun. 2025, 16, 459. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cattaneo, M.; Giagnorio, E.; Lauria, G.; Marcuzzo, S. Therapeutic Approaches for C9ORF72-Related ALS: Current Strategies and Future Horizons. Int. J. Mol. Sci. 2025, 26, 6268. https://doi.org/10.3390/ijms26136268
Cattaneo M, Giagnorio E, Lauria G, Marcuzzo S. Therapeutic Approaches for C9ORF72-Related ALS: Current Strategies and Future Horizons. International Journal of Molecular Sciences. 2025; 26(13):6268. https://doi.org/10.3390/ijms26136268
Chicago/Turabian StyleCattaneo, Marco, Eleonora Giagnorio, Giuseppe Lauria, and Stefania Marcuzzo. 2025. "Therapeutic Approaches for C9ORF72-Related ALS: Current Strategies and Future Horizons" International Journal of Molecular Sciences 26, no. 13: 6268. https://doi.org/10.3390/ijms26136268
APA StyleCattaneo, M., Giagnorio, E., Lauria, G., & Marcuzzo, S. (2025). Therapeutic Approaches for C9ORF72-Related ALS: Current Strategies and Future Horizons. International Journal of Molecular Sciences, 26(13), 6268. https://doi.org/10.3390/ijms26136268