
Silibinin Anticancer Effects Through the Modulation of the Tumor Immune Microenvironment in Triple-Negative Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Breast Cancer
2.1. Triple-Negative Breast Cancer (TNBC)
2.2. Racial Disparities in TNBC Incidence and Outcomes
2.3. Current Therapeutic Strategies for TNBC
2.3.1. Chemotherapy and Radiotherapy
2.3.2. Immunotherapy
2.4. Resistance in TNBC Therapies
3. Flavonoids Effects on Breast Cancer Tumor Immune Microenvironment
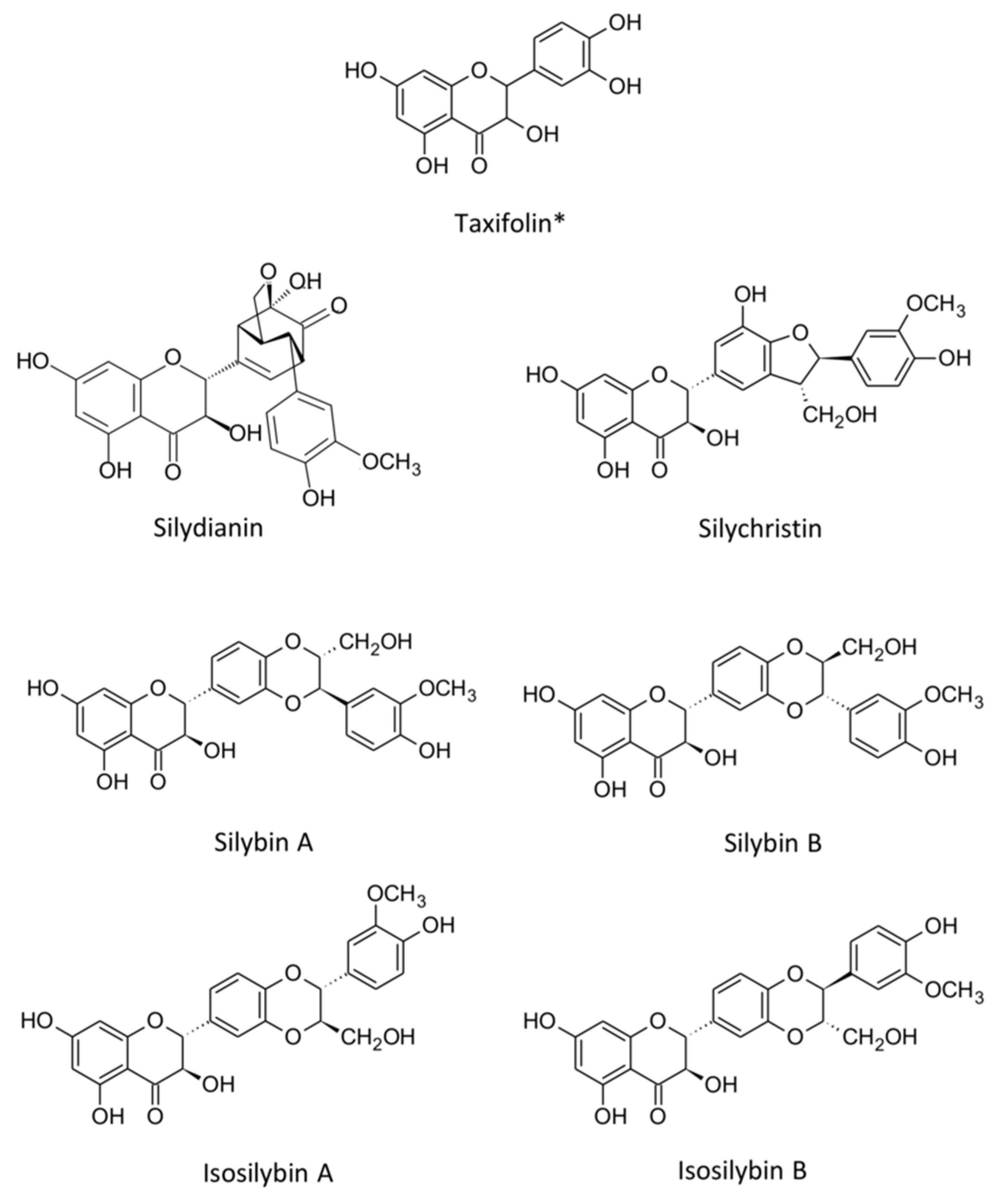
3.1. Silibinin: Chemical Structure and Classification
3.2. Silibinin Bioavailability
3.3. Silibinin Toxicity
3.4. Silibinin Pharmacological Effects on Multiple Cancer Types
3.5. Silibinin Chemopreventive Effect on Triple-Negative Breast Cancer
4. Silibinin Modulatory Effects on the TIME of Triple-Negative Breast Cancer
4.1. Silibinin Modulation of PD-L1 Expression
Silibinin Effects on JAK/STAT and MUC-1 Levels, Modulates PD-L1 Expression
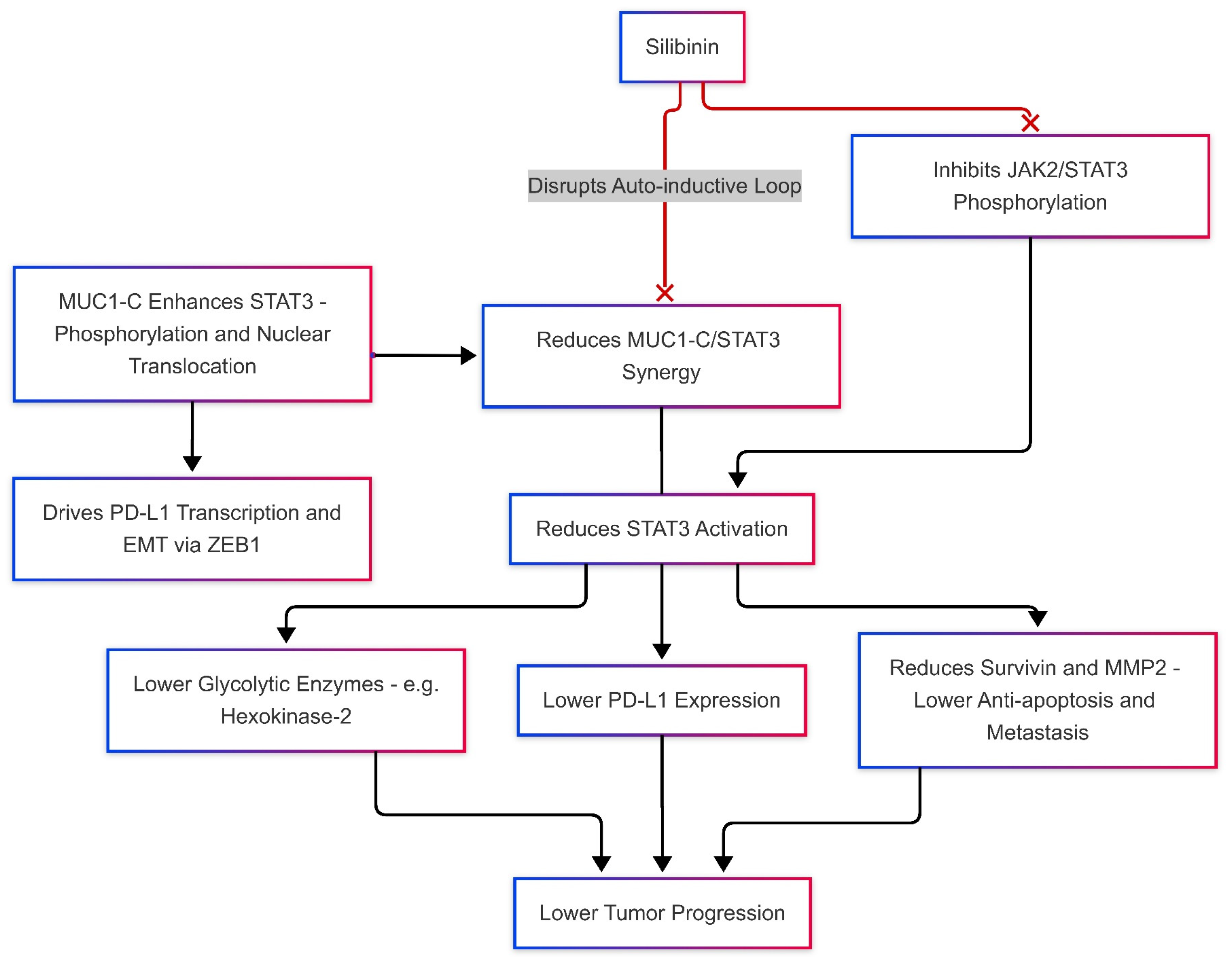
4.2. Silibinin Effects on Nrf2
Silibinin Effects on Nrf2-Mediated Antioxidant Defense System
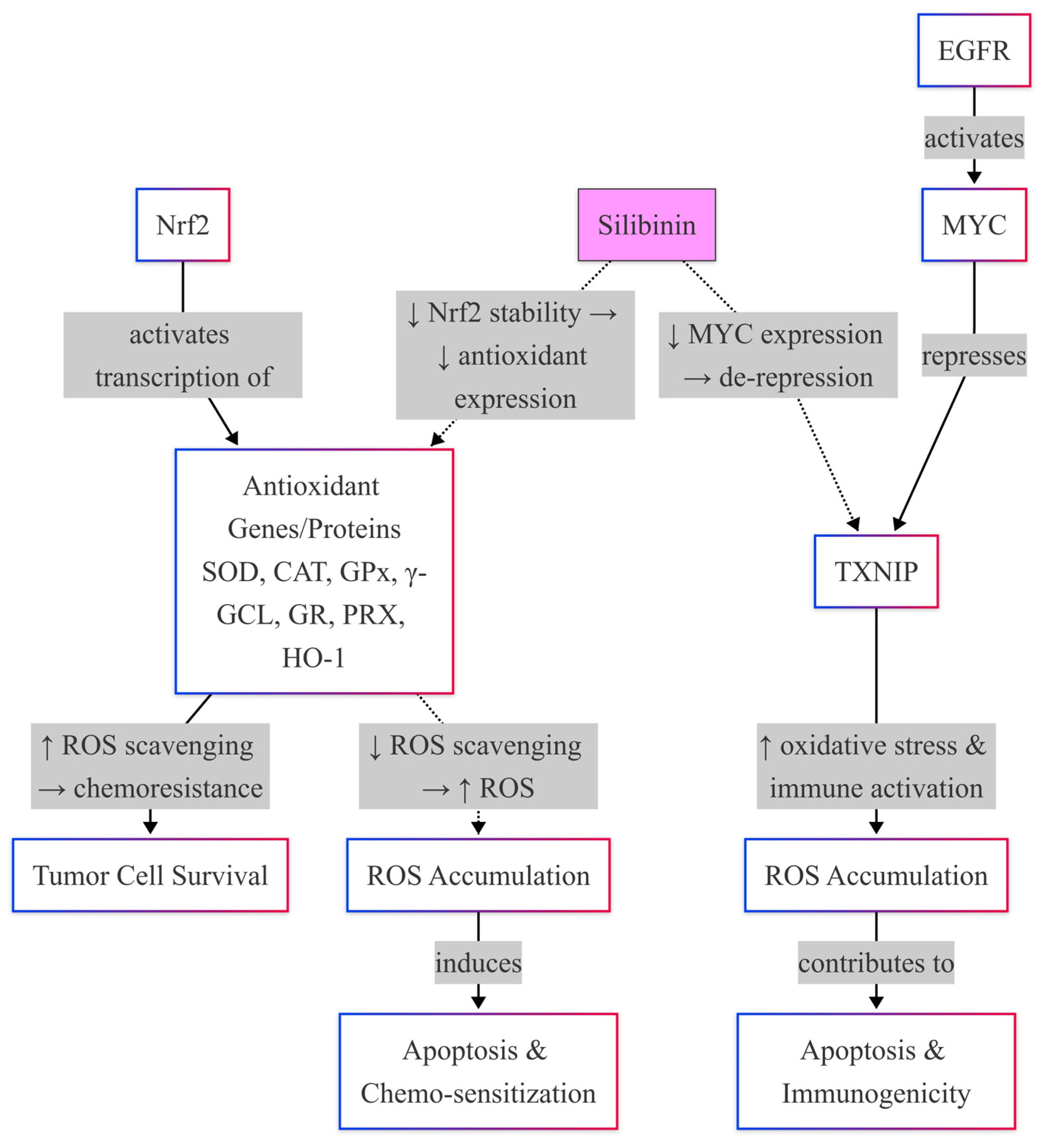
4.3. Silibinin Interaction with the NF-κB Signaling Pathway
Silibinin Inhibitory Effects on CCL2 Expression
5. The Role of Silibinin on PD-L1 Inhibition Through Nrf2 and NF-κB Signaling Modulation to Avoid Drug Resistance
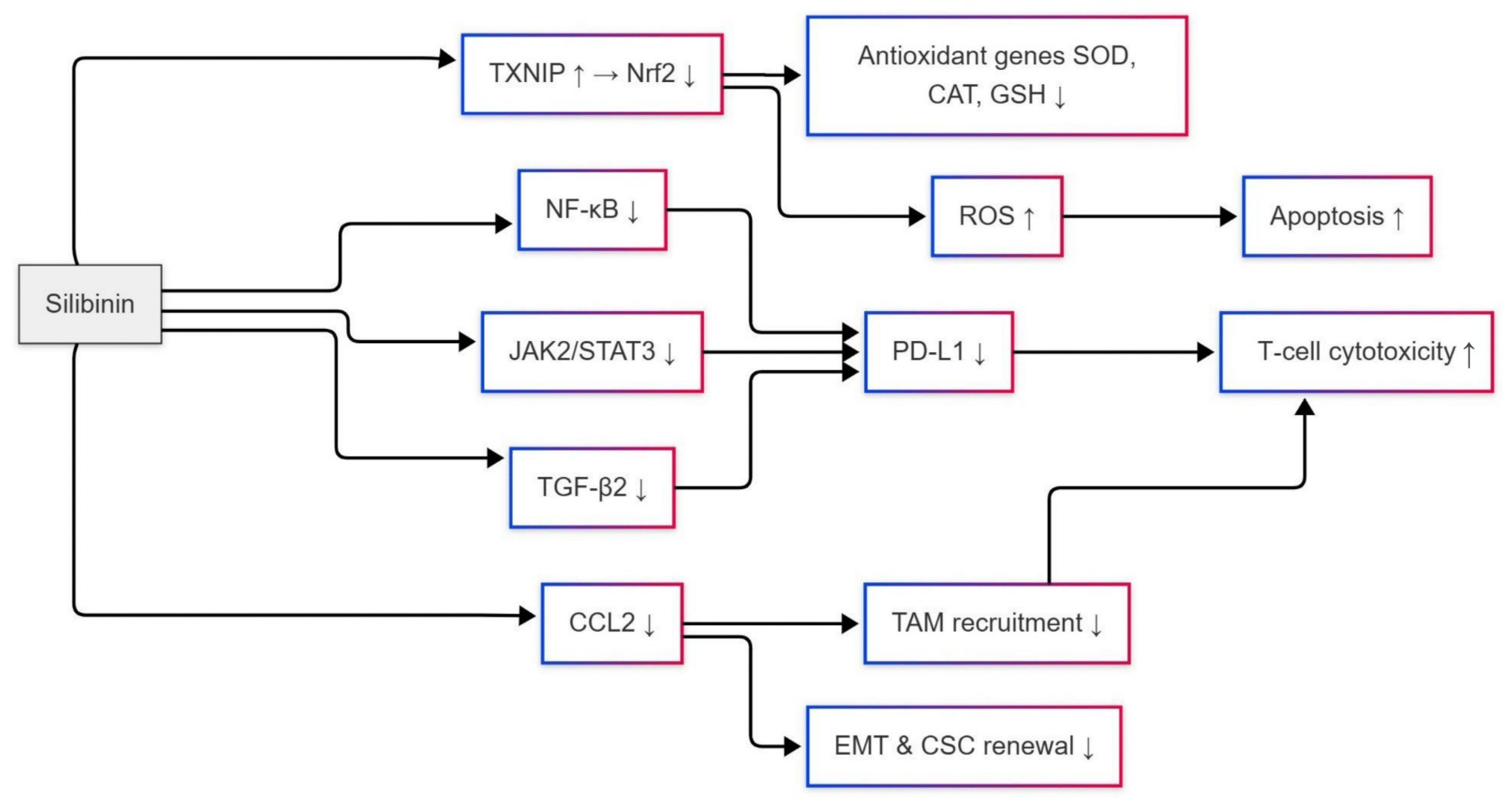
6. Translational Directions and Future Perspectives
7. Conclusions

Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-Negative Breast Cancer: Challenges and Opportunities of a Heterogeneous Disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Cancer Facts & Figures 2024. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2024-cancer-facts-figures.html (accessed on 31 May 2025).
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Treatment of Triple-Negative Breast Cancer|Treatment of TNBC. Available online: https://www.cancer.org/cancer/types/breast-cancer/treatment/treatment-of-triple-negative.html (accessed on 19 June 2025).
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhu, Z.; Lin, X.; Wang, S.; Wen, Y.; Wang, L.; Zhi, L.; Zhou, J. Tumor Microenvironment and Immunotherapy for Triple-Negative Breast Cancer. Biomark. Res. 2024, 12, 166. [Google Scholar] [CrossRef]
- Li, Q.; Ye, Z.; Wang, G.; Chen, Y.; Deng, J.; Wang, D.; Wang, Y. Natural Products as Novel Therapeutic Agents for Triple-Negative Breast Cancer: Current Evidence, Mechanisms, Challenges, and Opportunities. Molecules 2025, 30, 1201. [Google Scholar] [CrossRef] [PubMed]
- Kaleem, M.; Thool, M.; Dumore, N.G.; Abdulrahman, A.O.; Ahmad, W.; Almostadi, A.; Alhashmi, M.H.; Kamal, M.A.; Tabrez, S. Management of Triple-Negative Breast Cancer by Natural Compounds through Different Mechanistic Pathways. Front. Genet. 2024, 15, 1440430. [Google Scholar] [CrossRef]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as Anticancer Agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An Overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The Effects of Plant Flavonoids on Mammalian Cells: Implications for Inflammation, Heart Disease, and Cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.N.; Mumtaz, S. Prunin: An Emerging Anticancer Flavonoid. Int. J. Mol. Sci. 2025, 26, 2678. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Chattopadhyay, S.; Siddiqui, F.A.; Ur Rehman, A.; Siddiqui, S.; Prakasam, G.; Khan, A.; Sultana, S.; Bamezai, R.N. Silibinin Induces Metabolic Crisis in Triple-Negative Breast Cancer Cells by Modulating EGFR-MYC-TXNIP Axis: Potential Therapeutic Implications. FEBS J. 2021, 288, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Sharma, Y.; Agarwal, C.; Agarwal, R. Silibinin Impairs Constitutively Active TGFα-EGFR Autocrine Loop in Advanced Human Prostate Carcinoma Cells. Pharm. Res. 2008, 25, 2143–2150. [Google Scholar] [CrossRef]
- Varghese, L.; Agarwal, C.; Tyagi, A.; Singh, R.P.; Agarwal, R. Silibinin Efficacy against Human Hepatocellular Carcinoma. Clin. Cancer Res. 2005, 11, 8441–8448. [Google Scholar] [CrossRef]
- Mateen, S.; Raina, K.; Agarwal, R. Chemopreventive and Anti-Cancer Efficacy of Silibinin Against Growth and Progression of Lung Cancer. Nutr. Cancer 2013, 65, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.; Kohli, K.; Ali, M. Reassessing Bioavailability of Silymarin. Altern. Med. Rev. J. Clin. Ther. 2011, 16, 239–249. [Google Scholar]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling Triple-Negative Breast Cancer Molecular Heterogeneity Using an Integrative Multiomic Analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef]
- Chen, X.; Yang, M.; Yin, J.; Li, P.; Zeng, S.; Zheng, G.; He, Z.; Liu, H.; Wang, Q.; Zhang, F.; et al. Tumor-Associated Macrophages Promote Epithelial–Mesenchymal Transition and the Cancer Stem Cell Properties in Triple-Negative Breast Cancer through CCL2/AKT/β-Catenin Signaling. Cell Commun. Signal. 2022, 20, 92. [Google Scholar] [CrossRef]
- Rogic, A.; Pant, I.; Grumolato, L.; Fernandez-Rodriguez, R.; Edwards, A.; Das, S.; Sun, A.; Yao, S.; Qiao, R.; Jaffer, S.; et al. High Endogenous CCL2 Expression Promotes the Aggressive Phenotype of Human Inflammatory Breast Cancer. Nat. Commun. 2021, 12, 6889. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Feng, L.; Huang, Y.; Wu, Y.; Xie, N. Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer. Cancers 2023, 15, 104. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Whelan, T.; Levine, M.; Sussman, J. Hypofractionated Breast Irradiation: What’s Next? J. Clin. Oncol. 2020, 38, 3245–3247. [Google Scholar] [CrossRef]
- Dhanalakshmi, S.; Singh, R.P.; Agarwal, C.; Agarwal, R. Silibinin Inhibits Constitutive and TNFα-Induced Activation of NF-κB and Sensitizes Human Prostate Carcinoma DU145 Cells to TNFα-Induced Apoptosis. Oncogene 2002, 21, 1759–1767. [Google Scholar] [CrossRef]
- Huseni, M.A.; Wang, L.; Klementowicz, J.E.; Yuen, K.; Breart, B.; Orr, C.; Liu, L.; Li, Y.; Gupta, V.; Li, C.; et al. CD8+ T Cell-Intrinsic IL-6 Signaling Promotes Resistance to Anti-PD-L1 Immunotherapy. Cell Rep. Med. 2023, 4, 100878. [Google Scholar] [CrossRef]
- Choi, J.; Lee, H.J.; Yoon, S.; Ryu, H.-M.; Lee, E.; Jo, Y.; Seo, S.; Kim, D.; Lee, C.H.; Kim, W.; et al. Blockade of CCL2 Expression Overcomes Intrinsic PD-1/PD-L1 Inhibitor-Resistance in Transglutaminase 2-Induced PD-L1 Positive Triple Negative Breast Cancer. Am. J. Cancer Res. 2020, 10, 2878–2894. [Google Scholar] [PubMed]
- Qin, J.-J.; Yan, L.; Zhang, J.; Zhang, W.-D. STAT3 as a Potential Therapeutic Target in Triple Negative Breast Cancer: A Systematic Review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted Gene Silencing of CCL2 Inhibits Triple Negative Breast Cancer Progression by Blocking Cancer Stem Cell Renewal and M2 Macrophage Recruitment. Oncotarget 2016, 7, 49349–49367. [Google Scholar] [CrossRef]
- Zhou, Y.; Eppenberger-Castori, S.; Marx, C.; Yau, C.; Scott, G.K.; Eppenberger, U.; Benz, C.C. Activation of Nuclear Factor-κB (NFκB) Identifies a High-Risk Subset of Hormone-Dependent Breast Cancers. Int. J. Biochem. Cell Biol. 2005, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Stern, S.; Liang, D.; Li, L.; Kurian, R.; Lynch, C.; Sakamuru, S.; Heyward, S.; Zhang, J.; Kareem, K.A.; Chun, Y.W.; et al. Targeting CAR and Nrf2 Improves Cyclophosphamide Bioactivation While Reducing Doxorubicin-Induced Cardiotoxicity in Triple-Negative Breast Cancer Treatment. JCI Insight 2022, 7, e153868. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhao, X.; Zhang, X.; Li, Q.; Jiang, X.; Huang, S.; Chen, X.; Chen, X.; Jia, W.; Zou, H.; et al. PTPN20 Promotes Metastasis through Activating NF-κB Signaling in Triple-Negative Breast Cancer. Breast Cancer Res. 2024, 26, 155. [Google Scholar] [CrossRef] [PubMed]
- Won, K.-A.; Spruck, C. Triple-negative Breast Cancer Therapy: Current and Future Perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Hong, R.; Xu, B. Breast Cancer: An up-to-Date Review and Future Perspectives. Cancer Commun. 2022, 42, 913–936. [Google Scholar] [CrossRef]
- Vranic, S.; Cyprian, F.S.; Gatalica, Z.; Palazzo, J. PD-L1 Status in Breast Cancer: Current View and Perspectives. Semin. Cancer Biol. 2021, 72, 146–154. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, X.; Zhu, J.; Chai, Y.; Cong, B.; Li, B.; Gao, W.; Hu, Y.; Wen, M.; Liu, Y.; et al. Inhibiting Intracellular CD28 in Cancer Cells Enhances Antitumor Immunity and Overcomes Anti-PD-1 Resistance via Targeting PD-L1. Cancer Cell 2025, 43, 86–102.e10. [Google Scholar] [CrossRef]
- Chen, B.; Hu, J.; Hu, X.; Chen, H.; Bao, R.; Zhou, Y.; Ye, Y.; Zhan, M.; Cai, W.; Li, H.; et al. DENR Controls JAK2 Translation to Induce PD-L1 Expression for Tumor Immune Evasion. Nat. Commun. 2022, 13, 2059. [Google Scholar] [CrossRef]
- Sudhakaran, M.; Sardesai, S.; Doseff, A.I. Flavonoids: New Frontier for Immuno-Regulation and Breast Cancer Control. Antioxidants 2019, 8, 103. [Google Scholar] [CrossRef]
- Birt, D.F.; Hendrich, S.; Wang, W. Dietary Agents in Cancer Prevention: Flavonoids and Isoflavonoids. Pharmacol. Ther. 2001, 90, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, P. Cardamonin Anticancer Effects through the Modulation of the Tumor Immune Microenvironment in Triple-Negative Breast Cancer Cells. Am. J. Cancer Res. 2024, 14, 5644–5664. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, Y.; Zhang, J.; Zhang, Y.; He, W.; Ju, J.; Wu, Y.; Wang, Y. The Effect of Resveratrol, Curcumin and Quercetin Combination on Immuno-Suppression of Tumor Microenvironment for Breast Tumor-Bearing Mice. Sci. Rep. 2023, 13, 13278. [Google Scholar] [CrossRef]
- Han, S.; Wang, W.; Wang, S.; Yang, T.; Zhang, G.; Wang, D.; Ju, R.; Lu, Y.; Wang, H.; Wang, L. Tumor Microenvironment Remodeling and Tumor Therapy Based on M2-like Tumor Associated Macrophage-Targeting Nano-Complexes. Theranostics 2021, 11, 2892–2916. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, C.; Lu, L.; Yuan, F.; He, F. Baicalin and Baicalein in Modulating Tumor Microenvironment for Cancer Treatment: A Comprehensive Review with Future Perspectives. Pharmacol. Res. 2024, 199, 107032. [Google Scholar] [CrossRef]
- Agarwal, R.; Agarwal, C.; Ichikawa, H.; Singh, R.P.; Aggarwal, B.B. Anticancer Potential of Silymarin: From Bench to Bed Side. Anticancer Res. 2006, 26, 4457–4498. [Google Scholar]
- Deep, G.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilybin B Causes Androgen Receptor Degradation in Human Prostate Carcinoma Cells via PI3K-Akt-Mdm2-Mediated Pathway. Oncogene 2008, 27, 3986–3998. [Google Scholar] [CrossRef]
- Deep, G.; Kumar, R.; Jain, A.K.; Agarwal, C.; Agarwal, R. Silibinin Inhibits Fibronectin Induced Motility, Invasiveness and Survival in Human Prostate Carcinoma PC3 Cells via Targeting Integrin Signaling. Mutat. Res. Mol. Mech. Mutagen. 2014, 768, 35–46. [Google Scholar] [CrossRef]
- Drouet, S.; Abbasi, B.H.; Falguières, A.; Ahmad, W.; Sumaira; Ferroud, C.; Doussot, J.; Vanier, J.R.; Lainé, E.; Hano, C. Single Laboratory Validation of a Quantitative Core Shell-Based LC Separation for the Evaluation of Silymarin Variability and Associated Antioxidant Activity of Pakistani Ecotypes of Milk Thistle (Silybum marianum L.). Molecules 2018, 23, 904. [Google Scholar] [CrossRef]
- Wu, J.-W.; Lin, L.-C.; Hung, S.-C.; Chi, C.-W.; Tsai, T.-H. Analysis of Silibinin in Rat Plasma and Bile for Hepatobiliary Excretion and Oral Bioavailability Application. J. Pharm. Biomed. Anal. 2007, 45, 635–641. [Google Scholar] [CrossRef]
- Kidd, P.; Head, K. A Review of the Bioavailability and Clinical Efficacy of Milk Thistle Phytosome: A Silybin-Phosphatidylcholine Complex (Siliphos). Altern. Med. Rev. J. Clin. Ther. 2005, 10, 193–203. [Google Scholar]
- El-Samaligy, M.S.; Afifi, N.N.; Mahmoud, E.A. Evaluation of Hybrid Liposomes-Encapsulated Silymarin Regarding Physical Stability and in Vivo Performance. Int. J. Pharm. 2006, 319, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Novitasari, D.; Jenie, R.I.; Utomo, R.Y.; Kato, J.Y.; Meiyanto, E. CCA-1.1, a Novel Curcumin Analog, Exerts Cytotoxic Anti- Migratory Activity toward TNBC and HER2-Enriched Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2021, 22, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Lashgarian, H.E.; Adamii, V.; Ghorbanzadeh, V.; Chodari, L.; Kamali, F.; Akbari, S.; Dariushnejad, H. Silibinin Inhibit Cell Migration through Downregulation of RAC1 Gene Expression in Highly Metastatic Breast Cancer Cell Line. Drug Res. 2020, 70, 478–483. [Google Scholar] [CrossRef]
- Litzenburger, B.C.; Brown, P.H. Advances in Preventive Therapy for Estrogen-Receptor-Negative Breast Cancer. Curr. Breast Cancer Rep. 2014, 6, 96–109. [Google Scholar] [CrossRef]
- Singh, R.P.; Agarwal, R. Mechanisms and Preclinical Efficacy of Silibinin in Preventing Skin Cancer. Eur. J. Cancer 2005, 41, 1969–1979. [Google Scholar] [CrossRef]
- Flaig, T.W.; Glodé, M.; Gustafson, D.; Van Bokhoven, A.; Tao, Y.; Wilson, S.; Su, L.; Li, Y.; Harrison, G.; Agarwal, R.; et al. A Study of High-dose Oral Silybin-phytosome Followed by Prostatectomy in Patients with Localized Prostate Cancer. Prostate 2010, 70, 848–855. [Google Scholar] [CrossRef]
- Abenavoli, L.; Capasso, R.; Milic, N.; Capasso, F. Milk Thistle in Liver Diseases: Past, Present, Future. Phytother. Res. 2010, 24, 1423–1432. [Google Scholar] [CrossRef]
- Ray, P.P.; Islam, M.A.; Islam, M.S.; Han, A.; Geng, P.; Aziz, M.A.; Mamun, A.A. A Comprehensive Evaluation of the Therapeutic Potential of Silibinin: A Ray of Hope in Cancer Treatment. Front. Pharmacol. 2024, 15, 1349745. [Google Scholar] [CrossRef]
- Ting, H.; Deep, G.; Kumar, S.; Jain, A.K.; Agarwal, C.; Agarwal, R. Beneficial Effects of the Naturally Occurring Flavonoid Silibinin on the Prostate Cancer Microenvironment: Role of Monocyte Chemotactic Protein-1 and Immune Cell Recruitment. Carcinogenesis 2016, 37, 589–599. [Google Scholar] [CrossRef]
- Si, L.; Fu, J.; Liu, W.; Hayashi, T.; Nie, Y.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Onodera, S.; Ikejima, T. Silibinin Inhibits Migration and Invasion of Breast Cancer MDA-MB-231 Cells through Induction of Mitochondrial Fusion. Mol. Cell. Biochem. 2020, 463, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, R.S.; Ishikawa, U.; Silva, E.S.; Silva-Júnior, A.A.; Araújo, A.A.; Cruz, L.J.; Chan, A.B.; de Araújo Júnior, R.F. STAT3/NF-κB Signalling Disruption in M2 Tumour-Associated Macrophages Is a Major Target of PLGA Nanocarriers/PD-L1 Antibody Immunomodulatory Therapy in Breast Cancer. Br. J. Pharmacol. 2021, 178, 2284–2304. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef]
- Gunda, V.; Souchek, J.; Abrego, J.; Shukla, S.K.; Goode, G.D.; Vernucci, E.; Dasgupta, A.; Chaika, N.V.; King, R.J.; Li, S.; et al. MUC1-Mediated Metabolic Alterations Regulate Response to Radiotherapy in Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 5881–5891. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, Z.; Pirpour Tazehkand, A.; Mansoori, B.; Khaze, V.; Asadi, M.; Baradaran, B.; Samadi, N. The Impact of Nrf2 Silencing on Nrf2-PD-L1 Axis to Overcome Oxaliplatin Resistance as Well as Migration in Colon Cancer. Avicenna J. Med. Biotechnol. 2021, 13, 116–122. [Google Scholar] [PubMed]
- Zhang, C.; Liao, Y.; Li, T.; Zhong, H.; Shan, L.; Yu, P.; Xia, C.; Xu, L. Apigenin Promotes Apoptosis of 4T1 Cells through PI3K/AKT/Nrf2 Pathway and Improves Tumor Immune Microenvironment in Vivo. Toxicol. Res. 2024, 13, tfae011. [Google Scholar] [CrossRef]
- Binienda, A.; Ziolkowska, S.; Pluciennik, E. The Anticancer Properties of Silibinin: Its Molecular Mechanism and Therapeutic Effect in Breast Cancer. Anticancer Agents Med. Chem. 2019, 20, 1787–1796. [Google Scholar] [CrossRef]
- Rabelo, A.C.S.; Guerreiro, C.d.A.; Shinzato, V.I.; Ong, T.P.; Noratto, G. Anthocyanins Reduce Cell Invasion and Migration through Akt/mTOR Downregulation and Apoptosis Activation in Triple-Negative Breast Cancer Cells: A Systematic Review and Meta-Analysis. Cancers 2023, 15, 2300. [Google Scholar] [CrossRef]
- Singh, S.; Maurya, A.K.; Meena, A.; Mishra, N.; Luqman, S. Narirutin Downregulates Lipoxygenase-5 Expression and Induces G0/G1 Arrest in Triple-Negative Breast Carcinoma Cells. Biochim. Biophys. Acta BBA-Gen. Subj. 2023, 1867, 130340. [Google Scholar] [CrossRef]
- Mallipeddi, H.; Thyagarajan, A.; Sahu, R.P. Implications of Withaferin-A for Triple-Negative Breast Cancer Chemoprevention. Biomed. Pharmacother. 2021, 134, 111124. [Google Scholar] [CrossRef]
- Guha, S.; Talukdar, D.; Mandal, G.K.; Mukherjee, R.; Ghosh, S.; Naskar, R.; Saha, P.; Murmu, N.; Das, G. Crude Extract of Ruellia Tuberosa L. Flower Induces Intracellular ROS, Promotes DNA Damage and Apoptosis in Triple Negative Breast Cancer Cells. J. Ethnopharmacol. 2024, 332, 118389. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.T.; Anderson, K.S.; Lenkiewicz, E.; Andreozzi, M.; Cunliffe, H.E.; Klassen, C.L.; Dueck, A.C.; McCullough, A.E.; Reddy, S.K.; Ramanathan, R.K.; et al. Genomic Amplification of 9p24.1 Targeting JAK2, PD-L1, and PD-L2 Is Enriched in High-Risk Triple Negative Breast Cancer. Oncotarget 2015, 6, 26483–26493. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Han, J.; Jeon, M.; You, D.; Lee, J.; Kim, H.J.; Bae, S.; Nam, S.J.; Lee, J.E. Silibinin Inhibits Triple Negative Breast Cancer Cell Motility by Suppressing TGF-Β2 Expression. Tumor Biol. 2016, 37, 11397–11407. [Google Scholar] [CrossRef]
- Bahhnassy, A.; Mohanad, M.; Shaarawy, S.; Ismail, M.F.; El-Bastawisy, A.; Ashmawy, A.M.; Zekri, A.-R. Transforming Growth Factor-β, Insulin-like Growth Factor I/Insulin-like Growth Factor I Receptor and Vascular Endothelial Growth factor-A: Prognostic and Predictive Markers in Triple-negative and Non-triple-negative Breast Cancer. Mol. Med. Rep. 2015, 12, 851–864. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.; Harrell, J.C.; Fan, Z.; Edgerton, S.M.; Liu, B.; Thor, A.D. Metformin Attenuates Transforming Growth Factor Beta (TGF-β) Mediated Oncogenesis in Mesenchymal Stem-like/Claudin-Low Triple Negative Breast Cancer. Cell Cycle 2016, 15, 1046–1059. [Google Scholar] [CrossRef]
- Chang, S.-S.; Yamaguchi, H.; Xia, W.; Lim, S.-O.; Khotskaya, Y.; Wu, Y.; Chang, W.-C.; Liu, Q.; Hung, M.-C. Aurora A Kinase Activates YAP Signaling in Triple-Negative Breast Cancer. Oncogene 2017, 36, 1265–1275. [Google Scholar] [CrossRef]
- Diamond, J.R.; Eckhardt, S.G.; Pitts, T.M.; van Bokhoven, A.; Aisner, D.; Gustafson, D.L.; Capasso, A.; Sams, S.; Kabos, P.; Zolman, K.; et al. A Phase II Clinical Trial of the Aurora and Angiogenic Kinase Inhibitor ENMD-2076 for Previously Treated, Advanced, or Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2018, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Michaut, M.; Hoogstraat, M.; Mulder, L.; Besselink, N.J.; Koudijs, M.J.; Cuppen, E.; Voest, E.E.; Bernards, R.; Nederlof, P.M.; et al. Next Generation Sequencing of Triple Negative Breast Cancer to Find Predictors for Chemotherapy Response. Breast Cancer Res. 2015, 17, 134. [Google Scholar] [CrossRef]
- Rydén, L.; Jirström, K.; Haglund, M.; Stål, O.; Fernö, M. Epidermal Growth Factor Receptor and Vascular Endothelial Growth Factor Receptor 2 Are Specific Biomarkers in Triple-Negative Breast Cancer. Results from a Controlled Randomized Trial with Long-Term Follow-Up. Breast Cancer Res. Treat. 2010, 120, 491–498. [Google Scholar] [CrossRef]
- Maire, V.; Némati, F.; Richardson, M.; Vincent-Salomon, A.; Tesson, B.; Rigaill, G.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Lang, G.; et al. Polo-like Kinase 1: A Potential Therapeutic Option in Combination with Conventional Chemotherapy for the Management of Patients with Triple-Negative Breast Cancer. Cancer Res. 2013, 73, 813–823. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent Activation of Stat3 Signaling Induces Survivin Gene Expression and Confers Resistance to Apoptosis in Human Breast Cancer Cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.J.; Darvin, P.; Kang, D.Y.; Sp, N.; Joung, Y.H.; Park, J.H.; Kim, S.J.; Yang, Y.M. Silibinin Downregulates MMP2 Expression via Jak2/STAT3 Pathway and Inhibits the Migration and Invasive Potential in MDA-MB-231 Cells. Oncol. Rep. 2017, 37, 3270–3278. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E. The JAK-STAT Pathway at Twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Hattrup, C.L.; Gendler, S.J. Structure and Function of the Cell Surface (Tethered) Mucins. Annu. Rev. Physiol. 2008, 70, 431–457. [Google Scholar] [CrossRef]
- Ahmad, R.; Rajabi, H.; Kosugi, M.; Joshi, M.D.; Alam, M.; Vasir, B.; Kawano, T.; Kharbanda, S.; Kufe, D. MUC1-C Oncoprotein Promotes STAT3 Activation in an Autoinductive Regulatory Loop. Sci. Signal. 2011, 4, ra9. [Google Scholar] [CrossRef]
- Yamashita, N.; Long, M.; Fushimi, A.; Yamamoto, M.; Hata, T.; Hagiwara, M.; Bhattacharya, A.; Hu, Q.; Wong, K.-K.; Liu, S.; et al. MUC1-C Integrates Activation of the IFN-γ Pathway with Suppression of the Tumor Immune Microenvironment in Triple-Negative Breast Cancer. J. Immunother. Cancer 2021, 9, e002115. [Google Scholar] [CrossRef]
- Rajabi, H.; Alam, M.; Takahashi, H.; Kharbanda, A.; Guha, M.; Ahmad, R.; Kufe, D. MUC1-C Oncoprotein Activates the ZEB1/miR-200c Regulatory Loop and Epithelial–Mesenchymal Transition. Oncogene 2014, 33, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Cascio, S.; Zhang, L.; Finn, O.J. MUC1 Protein Expression in Tumor Cells Regulates Transcription of Proinflammatory Cytokines by Forming a Complex with Nuclear Factor-κB P65 and Binding to Cytokine Promoters: IMPORTANCE OF EXTRACELLULAR DOMAIN *. J. Biol. Chem. 2011, 286, 42248–42256. [Google Scholar] [CrossRef]
- Turkson, J.; Jove, R. STAT Proteins: Novel Molecular Targets for Cancer Drug Discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef]
- Burdelya, L.; Kujawski, M.; Niu, G.; Zhong, B.; Wang, T.; Zhang, S.; Kortylewski, M.; Shain, K.; Kay, H.; Djeu, J.; et al. Stat3 Activity in Melanoma Cells Affects Migration of Immune Effector Cells and Nitric Oxide-Mediated Antitumor Effects1. J. Immunol. 2005, 174, 3925–3931. [Google Scholar] [CrossRef]
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 Inhibitor AZD1480 Potently Blocks Stat3 Signaling and Oncogenesis in Solid Tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-Human Trial of a STAT3 Decoy Oligonucleotide in Head and Neck Tumors: Implications for Cancer Therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Raina, D.; Uchida, Y.; Kharbanda, A.; Rajabi, H.; Panchamoorthy, G.; Jin, C.; Kharbanda, S.; Scaltriti, M.; Baselga, J.; Kufe, D. Targeting the MUC1-C Oncoprotein Downregulates HER2 Activation and Abrogates Trastuzumab Resistance in Breast Cancer Cells. Oncogene 2014, 33, 3422–3431. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Anderson, A.C.; Kuchroo, V.K. LAG-3, TIM-3, and TIGIT: Distinct Functions in Immune Regulation. Immunity 2024, 57, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Singh, P.K.; Hollingsworth, M.A. Cell Surface-Associated Mucins in Signal Transduction. Trends Cell Biol. 2006, 16, 467–476. [Google Scholar] [CrossRef]
- López-Mejía, J.A.; Mantilla-Ollarves, J.C.; Rocha-Zavaleta, L. Modulation of JAK-STAT Signaling by LNK: A Forgotten Oncogenic Pathway in Hormone Receptor-Positive Breast Cancer. Int. J. Mol. Sci. 2023, 24, 14777. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 Signaling in the Hematopoietic System Elicits Multicomponent Antitumor Immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Tascioglu Aliyev, A.; Panieri, E.; Stepanić, V.; Gurer-Orhan, H.; Saso, L. Involvement of NRF2 in Breast Cancer and Possible Therapeutical Role of Polyphenols and Melatonin. Molecules 2021, 26, 1853. [Google Scholar] [CrossRef]
- Bottoni, L.; Minetti, A.; Realini, G.; Pio, E.; Giustarini, D.; Rossi, R.; Rocchio, C.; Franci, L.; Salvini, L.; Catona, O.; et al. NRF2 Activation by Cysteine as a Survival Mechanism for Triple-Negative Breast Cancer Cells. Oncogene 2024, 43, 1701–1713. [Google Scholar] [CrossRef]
- Roca, E.; Colloca, G.; Lombardo, F.; Bellieni, A.; Cucinella, A.; Madonia, G.; Martinelli, L.; Damiani, M.E.; Zampieri, I.; Santo, A. The Importance of Integrated Therapies on Cancer: Silibinin, an Old and New Molecule. Oncotarget 2024, 15, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 Mutations in Squamous Cell Carcinomas of Oesophagus and Skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- Wang, N.; Song, L.; Xu, Y.; Zhang, L.; Wu, Y.; Guo, J.; Ji, W.; Li, L.; Zhao, J.; Zhang, X.; et al. Loss of Scribble Confers Cisplatin Resistance during NSCLC Chemotherapy via Nox2/ROS and Nrf2/PD-L1 Signaling. eBioMedicine 2019, 47, 65–77. [Google Scholar] [CrossRef]
- Wang, H.-J.; Jiang, Y.-Y.; Wei, X.-F.; Huang, H.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Silibinin Induces Protective Superoxide Generation in Human Breast Cancer MCF-7 Cells. Free Radic. Res. 2010, 44, 90–100. [Google Scholar] [CrossRef]
- Deng, B.; Sun, M. Platycodin D Inhibits the Malignant Progression of Papillary Thyroid Carcinoma by NF-κB and Enhances the Therapeutic Efficacy of Pembrolizumab. Drug Dev. Res. 2022, 83, 708–720. [Google Scholar] [CrossRef]
- Poma, P.; Labbozzetta, M.; D’Alessandro, N.; Notarbartolo, M. NF-κB Is a Potential Molecular Drug Target in Triple-Negative Breast Cancers. OMICS J. Integr. Biol. 2017, 21, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, A.; Suzuki, Y.; Honda, G.; Muramatsu, S.; Matsuzaki, O.; Nagano, Y.; Doi, T.; Shimotohno, K.; Harada, T.; Nishida, E.; et al. Large-Scale Identification and Characterization of Human Genes That Activate NF-κB and MAPK Signaling Pathways. Oncogene 2003, 22, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Chinnikrishnan, P.; Aziz Ibrahim, I.A.; Alzahrani, A.R.; Shahzad, N.; Sivaprakasam, P.; Pandurangan, A.K. The Role of Selective Flavonoids on Triple-Negative Breast Cancer: An Update. Separations 2023, 10, 207. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, W.; Bao, X.; Liu, X.; Yang, M.; Yin, C. Eugenol Modulates the NOD1-NF-κB Signaling Pathway via Targeting NF-κB Protein in Triple-Negative Breast Cancer Cells. Front. Endocrinol. 2023, 14, 1136067. [Google Scholar] [CrossRef]
- Yoshimura, T.; Li, C.; Wang, Y.; Matsukawa, A. The Chemokine Monocyte Chemoattractant Protein-1/CCL2 Is a Promoter of Breast Cancer Metastasis. Cell. Mol. Immunol. 2023, 20, 714–738. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.-Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-Induced Chemokine Cascade Promotes Breast Cancer Metastasis by Enhancing Retention of Metastasis-Associated Macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef]
- Bonapace, L.; Coissieux, M.-M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 Inhibition Accelerates Breast Cancer Metastasis by Promoting Angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Jin, J.; Lin, J.; Xu, A.; Lou, J.; Qian, C.; Li, X.; Wang, Y.; Yu, W.; Tao, H. CCL2: An Important Mediator Between Tumor Cells and Host Cells in Tumor Microenvironment. Front. Oncol. 2021, 11, 722916. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 Signaling Axis in Cancer Metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 Mediates Cross-Talk between Cancer Cells and Stromal Fibroblasts That Regulates Breast Cancer Stem Cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, L.; Hu, Z.; Fang, Y.; Shen, Y.; Song, M.; Chen, Y. Regulation of CCL2 by EZH2 Affects Tumor-Associated Macrophages Polarization and Infiltration in Breast Cancer. Cell Death Dis. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Pozzi, S.; Satchi-Fainaro, R. The Role of CCL2/CCR2 Axis in Cancer and Inflammation: The next Frontier in Nanomedicine. Adv. Drug Deliv. Rev. 2024, 209, 115318. [Google Scholar] [CrossRef]
- Kanyomse, Q.; Le, X.; Tang, J.; Dai, F.; Mobet, Y.; Chen, C.; Cheng, Z.; Deng, C.; Ning, Y.; Yu, R.; et al. KLF15 Suppresses Tumor Growth and Metastasis in Triple-Negative Breast Cancer by Downregulating CCL2 and CCL7. Sci. Rep. 2022, 12, 19026. [Google Scholar] [CrossRef]
- Duan, J.; Zhang, Y.; Chen, R.; Liang, L.; Huo, Y.; Lu, S.; Zhao, J.; Hu, C.; Sun, Y.; Yang, K.; et al. Tumor-Immune Microenvironment and NRF2 Associate with Clinical Efficacy of PD-1 Blockade Combined with Chemotherapy in Lung Squamous Cell Carcinoma. Cell Rep. Med. 2023, 4, 101302. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-κB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e6. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Shen, X. NF-κB/P65 Antagonizes Nrf2-ARE Pathway by Depriving CBP from Nrf2 and Facilitating Recruitment of HDAC3 to MafK. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Adinew, G.M.; Messeha, S.S.; Taka, E.; Badisa, R.B.; Soliman, K.F.A. Anticancer Effects of Thymoquinone through the Antioxidant Activity, Upregulation of Nrf2, and Downregulation of PD-L1 in Triple-Negative Breast Cancer Cells. Nutrients 2022, 14, 4787. [Google Scholar] [CrossRef]
- Evans, J.A.; Mendonca, P.; Soliman, K.F.A. Involvement of Nrf2 Activation and NF-kB Pathway Inhibition in the Antioxidant and Anti-Inflammatory Effects of Hesperetin in Activated BV-2 Microglial Cells. Brain Sci. 2023, 13, 1144. [Google Scholar] [CrossRef]
- Xie, C.; Zhou, X.; Liang, C.; Li, X.; Ge, M.; Chen, Y.; Yin, J.; Zhu, J.; Zhong, C. Correction to: Apatinib Triggers Autophagic and Apoptotic Cell Death via VEGFR2/STAT3/PD-L1 and ROS/Nrf2/P62 Signaling in Lung Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 349. [Google Scholar] [CrossRef]
- Lucas, J.; Hsieh, T.-C.; Halicka, H.D.; Darzynkiewicz, Z.; Wu, J.M. Upregulation of PD-L1 Expression by Resveratrol and Piceatannol in Breast and Colorectal Cancer Cells Occurs via HDAC3/P300-mediated NF-κB Signaling. Int. J. Oncol. 2018, 53, 1469–1480. [Google Scholar] [CrossRef]
- Iseda, N.; Itoh, S.; Yoshizumi, T.; Tomiyama, T.; Morinaga, A.; Yugawa, K.; Shimokawa, M.; Shimagaki, T.; Wang, H.; Kurihara, T.; et al. Impact of Nuclear Factor Erythroid 2–Related Factor 2 in Hepatocellular Carcinoma: Cancer Metabolism and Immune Status. Hepatol. Commun. 2022, 6, 665. [Google Scholar] [CrossRef]
- Morrissey, S.M.; Zhang, F.; Ding, C.; Montoya-Durango, D.E.; Hu, X.; Yang, C.; Wang, Z.; Yuan, F.; Fox, M.; Zhang, H.; et al. Tumor-Derived Exosomes Drive Immunosuppressive Macrophages in a Pre-Metastatic Niche through Glycolytic Dominant Metabolic Reprogramming. Cell Metab. 2021, 33, 2040–2058.e10. [Google Scholar] [CrossRef] [PubMed]
- Härkönen, J.; Pölönen, P.; Deen, A.J.; Selvarajan, I.; Teppo, H.-R.; Dimova, E.Y.; Kietzmann, T.; Ahtiainen, M.; Väyrynen, J.P.; Väyrynen, S.A.; et al. A Pan-Cancer Analysis Shows Immunoevasive Characteristics in NRF2 Hyperactive Squamous Malignancies. Redox Biol. 2023, 61, 102644. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhu, P.; Li, J.; Xu, L.; Tang, Y.; Liu, X.; Guo, S.; Xia, J. Fusobacterium nucleatum Promotes PD-L1 Expression in Cancer Cells to Evade CD8+ T Cell Killing in Breast Cancer. Hum. Immunol. 2024, 85, 111168. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-H.; Choi, Y.; Kim, N.; Nam, R.H.; Kim, J.W.; Jang, J.Y.; Kim, E.H.; Ha, S.; Lee, H.-N. Sex-Specific Molecular Markers NRF2 and PD-L1 in Colon Carcinogenesis: Implications for Right-Sided Colon Cancer. Cancer Res. Treat. 2024. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Z.; Liu, Z. Formononetin Restrains Tumorigenesis of Breast Tumor by Restraining STING-NF-κB and Interfering with the Activation of PD-L1. Discov. Med. 2024, 36, 613–620. [Google Scholar] [CrossRef]
- Vasiyani, H.; Mane, M.; Rana, K.; Shinde, A.; Roy, M.; Singh, J.; Gohel, D.; Currim, F.; Srivastava, R.; Singh, R. DNA Damage Induces STING Mediated IL-6-STAT3 Survival Pathway in Triple-Negative Breast Cancer Cells and Decreased Survival of Breast Cancer Patients. Apoptosis 2022, 27, 961–978. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Zhao, H.; Tian, Z.; Cao, Q.; Li, Y.; Gu, Y.; Song, Q.; Hu, X.; Jin, M.; et al. Correlation of PD-L1 Expression with CD8+ T Cells and Oxidative Stress-Related Molecules NRF2 and NQO1 in Esophageal Squamous Cell Carcinoma. J. Pathol. Clin. Res. 2024, 10, e12390. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, S.D.; Mendonca, P.; Kaur, S.; Soliman, K.F.A. Silibinin Anticancer Effects Through the Modulation of the Tumor Immune Microenvironment in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2025, 26, 6265. https://doi.org/10.3390/ijms26136265
Mishra SD, Mendonca P, Kaur S, Soliman KFA. Silibinin Anticancer Effects Through the Modulation of the Tumor Immune Microenvironment in Triple-Negative Breast Cancer. International Journal of Molecular Sciences. 2025; 26(13):6265. https://doi.org/10.3390/ijms26136265
Chicago/Turabian StyleMishra, Shubham D., Patricia Mendonca, Sukhmandeep Kaur, and Karam F. A. Soliman. 2025. "Silibinin Anticancer Effects Through the Modulation of the Tumor Immune Microenvironment in Triple-Negative Breast Cancer" International Journal of Molecular Sciences 26, no. 13: 6265. https://doi.org/10.3390/ijms26136265
APA StyleMishra, S. D., Mendonca, P., Kaur, S., & Soliman, K. F. A. (2025). Silibinin Anticancer Effects Through the Modulation of the Tumor Immune Microenvironment in Triple-Negative Breast Cancer. International Journal of Molecular Sciences, 26(13), 6265. https://doi.org/10.3390/ijms26136265