
Dysregulation of the Cannabinoid System in Childhood Epilepsy: From Mechanisms to Therapy


Abstract
1. Introduction
2. Pediatric Epilepsy
2.1. Classification of Pediatric Epilepsy
2.2. Febrile Infection-Related Epilepsy Syndrome (FIRES)
2.3. Dravet and Lennox Gastaut Syndrome
Neuropsychiatric Comorbidities
2.4. Pediatric Temporal Lobe Epilepsy
2.5. Childhood Absence Epilepsy (CAE)
Neuropsychiatric Comorbidities
3. Cannabinoids
3.1. Phytocannabinoids
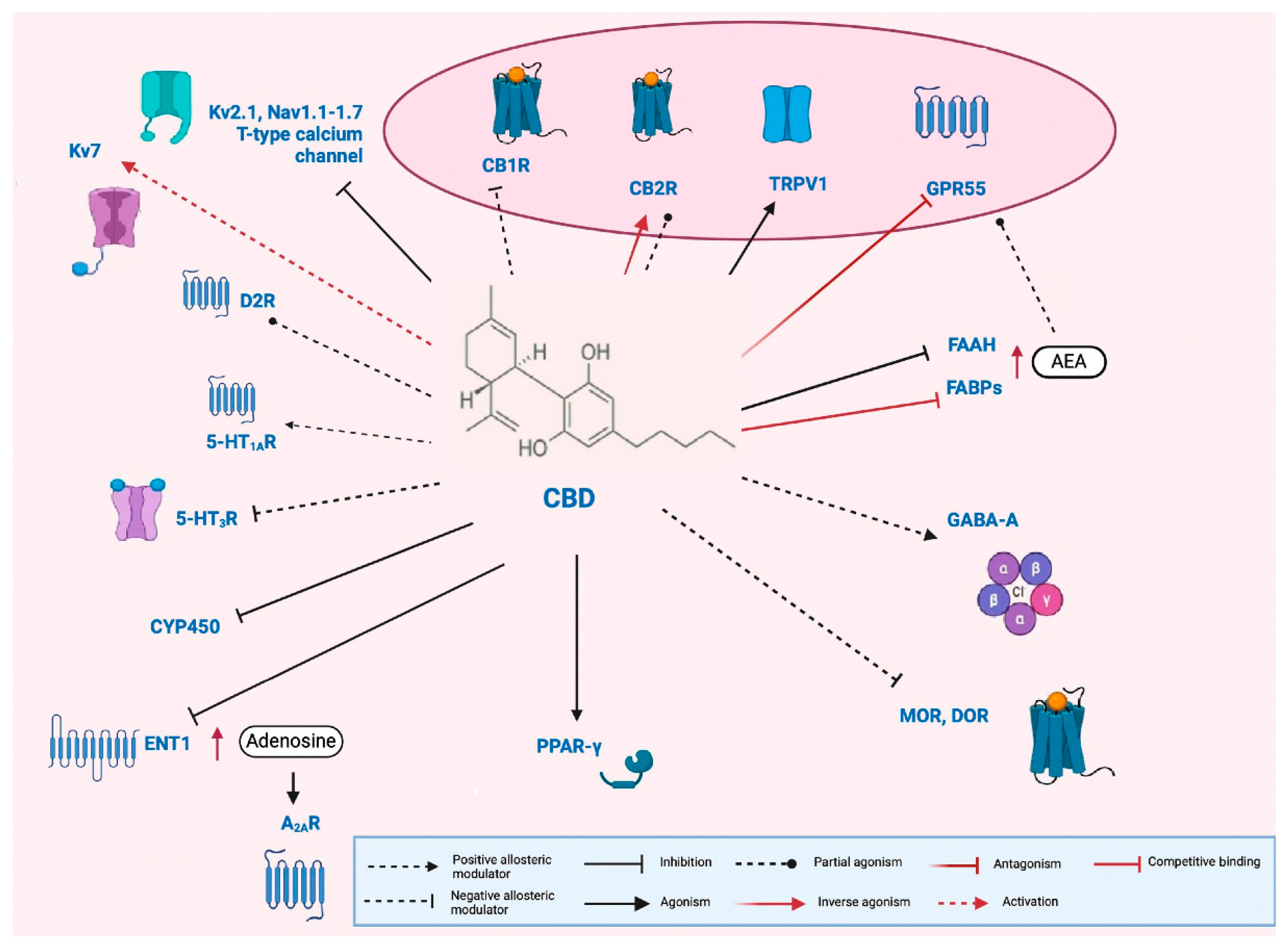
3.2. Cannabidiol (CBD)
3.3. Tetrahydrocannabinol (∆9-THC)
3.4. Endocannabinoids
4. The Endocannabinoid System in Relation to Pediatric Epilepsy
4.1. The Endocannabinoid System and Cannabinoids in Febrile Infection-Related Epilepsy Syndrome (FIRES)
4.2. The Endocannabinoid System and Cannabinoids in Dravet Syndrome (DS) and Lennox–Gastaut Syndrome (LGS)
4.2.1. Clinical Evidence of Cannabinoid Use in DS and LGS
4.2.2. Animal Evidence of Cannabinoid Use in DS and LGS
4.2.3. Cannabinoid Use for Neuropsychiatric Comorbidities in DS and LGS
4.3. The Endocannabinoid System in Other Refractory Pediatric Epilepsies
4.4. The Endocannabinoid System and Cannabinoids in Pediatric Temporal Lobe Epilepsy
4.5. The Endocannabinoid System in Childhood Absence Epilepsy (CAE)
4.5.1. Clinical Evidence of Cannabinoid Use in CAE
4.5.2. Animal Evidence of Cannabinoid Use in CAE
4.5.3. Cannabinoids Infusion in Brain Areas in CAE Animal Models
4.5.4. Cannabinoid Concentration in Brain Areas in CAE Animal Models
4.5.5. Cannabinoid Receptor Expression in CAE Animal Models
4.5.6. Modulators of Cannabinoid Synthesis and Breakdown in CAE Animal Models
4.5.7. Cannabinoids and Neuropsychiatric Comorbidities in CAE Animal Models
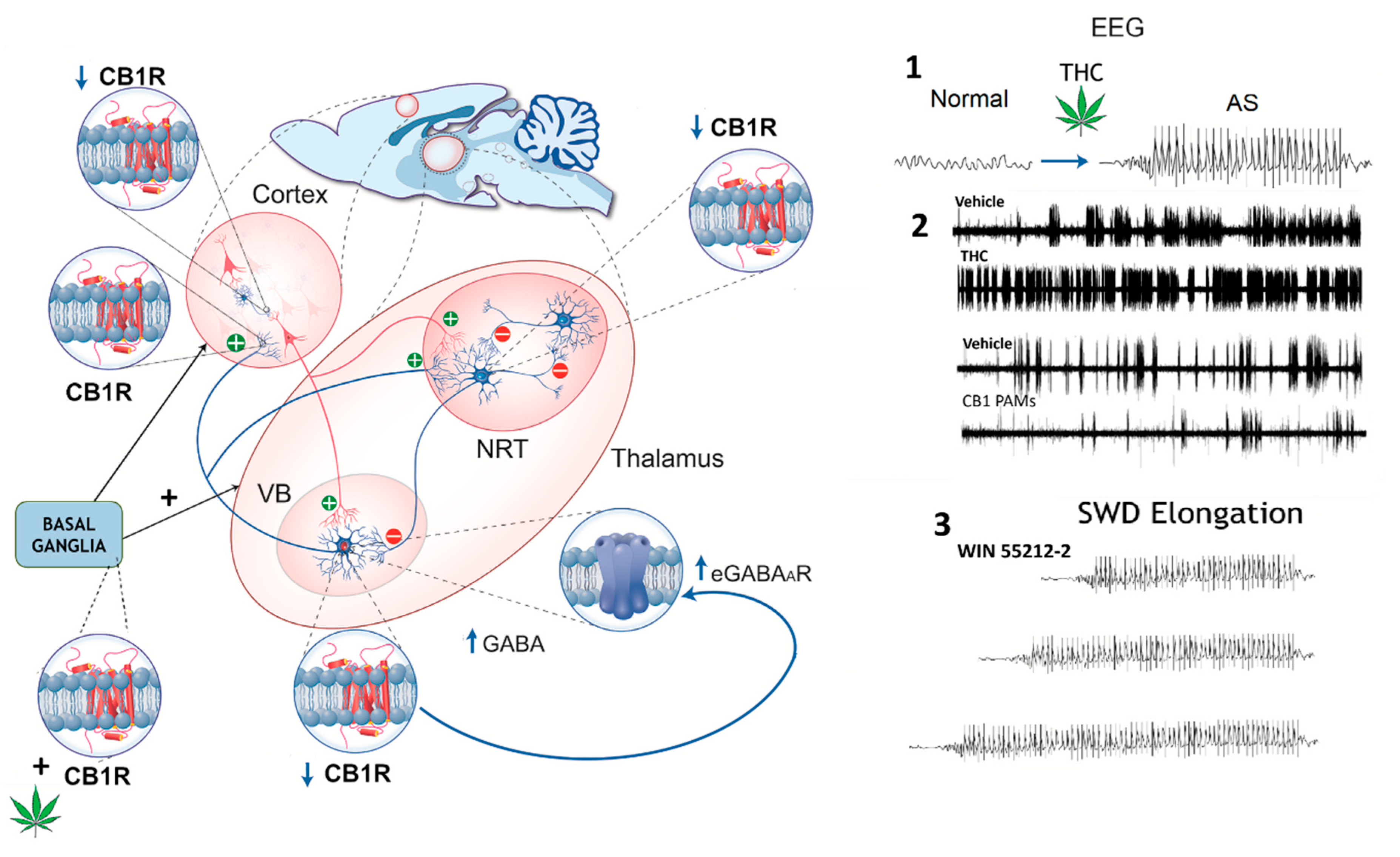
5. Limitations, Risks, and Future Directions for the Research and Treatment of Cannabinoid Use in Pediatric Epilepsy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Stafstrom, C.E.; Carmant, L. Seizures and epilepsy: An overview for neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.; Tinuper, P.; Perucca, E.; Moshé, S.L. Introduction to the epilepsy syndrome papers. Epilepsia 2022, 63, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Cross, J.H.; D’souza, C.; French, J.A.; Haut, S.R.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia 2017, 58, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Shorvon, S.D. The etiologic classification of epilepsy. Epilepsia 2011, 52, 1052–1057. [Google Scholar] [CrossRef]
- Camfield, P.; Camfield, C. Incidence, prevalence and aetiology of seizures and epilepsy in children. Epileptic Disord. 2015, 17, 117–123. [Google Scholar] [CrossRef]
- Holmes, G.L.; Ben-Ari, Y. The neurobiology and consequences of epilepsy in the developing brain. Pediatr. Res. 2001, 49, 320–325. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Holmes, G.L. Effects of seizures on developmental processes in the immature brain. Lancet. Neurol. 2006, 5, 1055–1063. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Holmes, G.L.; Ben-Ari, Y. Seizures in the developing brain: Perhaps not so benign after all. Neuron 1998, 21, 1231–1234. [Google Scholar] [CrossRef]
- Wirrell, E.C.; Grossardt, B.R.; Wong-Kisiel, L.C.; Nickels, K.C. Incidence and classification of new-onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: A population-based study. Epilepsy Res. 2011, 95, 110–118. [Google Scholar] [CrossRef]
- Wirrell, E.C. Predicting pharmacoresistance in pediatric epilepsy. Epilepsia 2013, 54, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Conway, L.; Smith, M.L.; Ferro, M.A.; Speechley, K.N.; Connoly, M.B.; Snead, O.C.; Widjaja, E. Correlates of health-related quality of life in children with drug resistant epilepsy. Epilepsia 2016, 57, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Samuelraj, S.; Lee-Archer, P.; Bowers, A.; Herbert, A. Medicinal Cannabinoids in Children: A Scoping Review. Integr. Med. Rep. 2025, 4, 9–43. [Google Scholar] [CrossRef]
- Devinsky, O.; Jones, N.A.; Cunningham, M.O.; Jayasekera, B.A.P.; Devore, S.; Whalley, B.J. Cannabinoid treatments in epilepsy and seizure disorders. Physiol. Rev. 2024, 104, 591–649. [Google Scholar] [CrossRef]
- Mechoulam, R.; Carlini, E.A. Toward drugs derived from cannabis. Naturwissenschaften 1978, 65, 174–179. [Google Scholar] [CrossRef]
- Riney, K.; Bogacz, A.; Somerville, E.; Hirsch, E.; Nabbout, R.; Scheffer, I.E.; Zuberi, S.M.; Alsaadi, T.; Jain, S.; French, J. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1443–1474. [Google Scholar] [CrossRef]
- Di Marzo, V.; De Petrocellis, L.; Bisogno, T.; Melck, D. Metabolism of anandamide and 2-arachidonoylglycerol: An historical overview and some recent developments. Lipids 1999, 34, S319–S325. [Google Scholar] [CrossRef]
- Abu-Sawwa, R.; Stehling, C. Epidiolex (Cannabidiol) Primer: Frequently Asked Questions for Patients and Caregivers. J. Pediatr. Pharmacol. Ther. JPPT Off. J. PPAG 2020, 25, 75–77. [Google Scholar] [CrossRef]
- Di Giovanni, G. Cannabis medicine offers hope for severe paediatric epilepsies. Xjenza 2018, 6, 62–64. [Google Scholar]
- European Medicines Agency. Epidyolex: Assessment Report; European Medicines Agency: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Biosciences, G. Epidiolex®(Cannabidiol) Oral Solution [Package Insert]; US Food and Drug Administration Website: Silver Spring, MD, USA, 2021. [Google Scholar]
- Blair, R.E.; Deshpande, L.S.; DeLorenzo, R.J. Cannabinoids: Is there a potential treatment role in epilepsy? Expert Opin. Pharmacother. 2015, 16, 1911–1914. [Google Scholar] [CrossRef]
- De Caro, C.; Leo, A.; Citraro, R.; De Sarro, C.; Russo, R.; Calignano, A.; Russo, E. The potential role of cannabinoids in epilepsy treatment. Expert Rev. Neurother. 2017, 17, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Billakota, S.; Devinsky, O.; Marsh, E. Cannabinoid therapy in epilepsy. Curr. Opin. Neurol. 2019, 32, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Detyniecki, K.; Hirsch, L. Marijuana Use in Epilepsy: The Myth and the Reality. Curr. Neurol. Neurosci. Rep. 2015, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.C.; Patra, P.H.; Whalley, B.J. Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav. EB 2017, 70, 319–327. [Google Scholar] [CrossRef]
- Massey, S.; Quigley, A.; Rochfort, S.; Christodoulou, J.; Van Bergen, N.J. Cannabinoids and Genetic Epilepsy Models: A Review with Focus on CDKL5 Deficiency Disorder. Int. J. Mol. Sci. 2024, 25, 10768. [Google Scholar] [CrossRef]
- DeGasperis, S.M.; Webster, R.; Pohl, D. Cannabis Treatment in Children with Epilepsy: Practices of Canadian Neurologists. Can J. Neurol. Sci. 2020, 47, 511–518. [Google Scholar] [CrossRef]
- Elliott, J.; DeJean, D.; Clifford, T.; Coyle, D.; Potter, B.; Skidmore, B.; Alexander, C.; Repetski, A.E.; McCoy, B.; Wells, G.A. Cannabis for pediatric epilepsy: Protocol for a living systematic review. Syst. Rev. 2018, 7, 95. [Google Scholar] [CrossRef]
- Filloux, F.M. Cannabinoids for pediatric epilepsy? Up in smoke or real science? Transl. Pediatr. 2015, 4, 271–282. [Google Scholar] [CrossRef]
- Huntsman, R.J.; Tang-Wai, R.; Shackelford, A.E. Cannabis for Pediatric Epilepsy. J. Clin. Neurophysiol. 2020, 37, 2–8. [Google Scholar] [CrossRef]
- Knupp, K.G.; Rice, J.D.; Helmkamp, L.J.; Galinkin, J.; Sempio, C.; Jost, K.; Chapman, K.E. Prospective evaluation of oral cannabis extracts in children with epilepsy. Seizure 2019, 72, 23–27. [Google Scholar] [CrossRef]
- Ayoub, D.; Al-Hajje, A.; Salameh, P.; Jost, J.; Hmaimess, G.; Jaafar, F.; Halabi, T.; Boumediene, F.; Beydoun, A. Beyond Seizures: Psychiatric comorbidities in children with epilepsy. Epilepsy Behav. 2025, 163, 110234. [Google Scholar] [CrossRef] [PubMed]
- Beniczky, S.; Trinka, E.; Wirrell, E.; Abdulla, F.; Al Baradie, R.; Alonso Vanegas, M.; Auvin, S.; Singh, M.B.; Blumenfeld, H.; Bogacz Fressola, A.; et al. Updated classification of epileptic seizures: Position paper of the International League Against Epilepsy. Epilepsia 2025, 66, 1804–1823. [Google Scholar] [CrossRef]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1398–1442. [Google Scholar] [CrossRef] [PubMed]
- Kramer, U.; Chi, C.S.; Lin, K.L.; Specchio, N.; Sahin, M.; Olson, H.; Nabbout, R.; Kluger, G.; Lin, J.J.; van Baalen, A. Febrile infection–related epilepsy syndrome (FIRES): Pathogenesis, treatment, and outcome: A multicenter study on 77 children. Epilepsia 2011, 52, 1956–1965. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.B.; Katanyuwong, K.; Mackay, M.T.; Bailey, C.A.; Scheffer, I.E.; Freeman, J.L.; Berkovic, S.F.; Harvey, A.S. Long-term follow-up of febrile infection–related epilepsy syndrome. Epilepsia 2012, 53, 101–110. [Google Scholar] [CrossRef]
- Oguni, H.; Fukuyama, Y. Pathogenesis of epilepsy in childhood. Psychiatry Clin. Neurosci. 1986, 40, 293–299. [Google Scholar] [CrossRef]
- Van Baalen, A.; Häusler, M.; Boor, R.; Rohr, A.; Sperner, J.; Kurlemann, G.; Panzer, A.; Stephani, U.; Kluger, G. Febrile infection–related epilepsy syndrome (FIRES): A nonencephalitic encephalopathy in childhood. Epilepsia 2010, 51, 1323–1328. [Google Scholar] [CrossRef]
- Mikaeloff, Y.; Jambaqué, I.; Hertz-Pannier, L.; Zamfirescu, A.; Adamsbaum, C.; Plouin, P.; Dulac, O.; Chiron, C. Devastating epileptic encephalopathy in school-aged children (DESC): A pseudo encephalitis. Epilepsy Res. 2006, 69, 67–79. [Google Scholar] [CrossRef]
- Nabbout, R.; Mazzuca, M.; Hubert, P.; Peudennier, S.; Allaire, C.; Flurin, V.; Aberastury, M.; Silva, W.; Dulac, O. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010, 51, 2033–2037. [Google Scholar] [CrossRef]
- Connolly, M.B. Dravet syndrome: Diagnosis and long-term course. Can. J. Neurol. Sci. 2016, 43, S3–S8. [Google Scholar] [CrossRef]
- Dravet, C. Dravet syndrome history. Dev. Med. Child Neurol. 2011, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, R.H.; Fejerman, N. Dravet syndrome: A study of 53 patients. Epilepsy Res. 2006, 70, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Villas, N.; Meskis, M.A.; Goodliffe, S. Dravet syndrome: Characteristics, comorbidities, and caregiver concerns. Epilepsy Behav. 2017, 74, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Asadi-Pooya, A.A. Lennox-Gastaut syndrome: A comprehensive review. Neurol. Sci. 2018, 39, 403–414. [Google Scholar] [CrossRef]
- Dravet, C.; Bureau, M.; Oguni, H.; Fukuyama, Y.; Cokar, O. Severe myoclonic epilepsy in infancy (Dravet syndrome). Epileptic Syndr. Infancy Child. Adolesc. 2005, 4, 89–113. [Google Scholar]
- Asadi-Pooya, A.A.; Bazrafshan, M.; Farazdaghi, M. Long-term medical and social outcomes of patients with Lennox-Gastaut syndrome. Epilepsy Res. 2021, 178, 106813. [Google Scholar] [CrossRef]
- Griffin, A.; Hamling, K.R.; Hong, S.; Anvar, M.; Lee, L.P.; Baraban, S.C. Preclinical animal models for Dravet syndrome: Seizure phenotypes, comorbidities and drug screening. Front. Pharmacol. 2018, 9, 573. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef]
- Ogiwara, I.; Miyamoto, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K. Nav1. 1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; De La Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef]
- Anderson, L.L.; Hawkins, N.A.; Thompson, C.H.; Kearney, J.A.; George, A.L., Jr. Unexpected efficacy of a novel sodium channel modulator in Dravet syndrome. Sci. Rep. 2017, 7, 1682. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.S.; Stella, N.; Catterall, W.A.; Westenbroek, R.E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Genton, P.; Velizarova, R.; Dravet, C. Dravet syndrome: The long-term outcome. Epilepsia 2011, 52, 44–49. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef]
- Chen, B.; Choi, H.; Hirsch, L.J.; Katz, A.; Legge, A.; Buchsbaum, R.; Detyniecki, K. Psychiatric and behavioral side effects of antiepileptic drugs in adults with epilepsy. Epilepsy Behav. 2017, 76, 24–31. [Google Scholar] [CrossRef]
- Nickels, K.C.; Wong-Kisiel, L.C.; Moseley, B.D.; Wirrell, E.C. Temporal lobe epilepsy in children. Epilepsy Res. Treat. 2012, 2012, 849540. [Google Scholar] [CrossRef]
- Fogarasi, A.; Tuxhorn, I.; Janszky, J.; Janszky, I.; Rásonyi, G.; Kelemen, A.; Halász, P. Age-dependent seizure semiology in temporal lobe epilepsy. Epilepsia 2007, 48, 1697–1702. [Google Scholar] [CrossRef]
- Olbrich, A.; Urak, L.; Gröppel, G.; Serles, W.; Novak, K.; Porsche, B.; Benninger, F.; Czech, T.; Baumgartner, C.; Feucht, M. Semiology of temporal lobe epilepsy in children and adolescents value in lateralizing the seizure onset zone. Epilepsy Res. 2002, 48, 103–110. [Google Scholar] [CrossRef]
- Cascino, G.D. Surgical treatment for epilepsy. Epilepsy Res. 2004, 60, 179–186. [Google Scholar] [CrossRef]
- Skirrow, C.; Cross, J.H.; Cormack, F.; Harkness, W.; Vargha-Khadem, F.; Baldeweg, T. Long-term intellectual outcome after temporal lobe surgery in childhood. Neurology 2011, 76, 1330–1337. [Google Scholar] [CrossRef]
- Guerrini, R.; Conti, V.; Mantegazza, M.; Balestrini, S.; Galanopoulou, A.S.; Benfenati, F. Developmental and epileptic encephalopathies: From genetic heterogeneity to phenotypic continuum. Physiol. Rev. 2023, 103, 433–513. [Google Scholar] [CrossRef] [PubMed]
- Brigo, F.; Trinka, E.; Lattanzi, S.; Bragazzi, N.L.; Nardone, R.; Martini, M. A brief history of typical absence seizures—Petit mal revisited. Epilepsy Behav. 2018, 80, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Crunelli, V.; Leresche, N. Childhood absence epilepsy: Genes, channels, neurons and networks. Nat. Rev. 2002, 3, 371–382. [Google Scholar] [CrossRef]
- Reichsoellner, J.; Larch, J.; Unterberger, I.; Dobesberger, J.; Kuchukhidze, G.; Luef, G.; Bauer, G.; Trinka, E. Idiopathic generalised epilepsy of late onset: A separate nosological entity? J. Neurol. Neurosurg. Psychiatry 2010, 81, 1218–1222. [Google Scholar] [CrossRef]
- Posner, E. Absence seizures in children. BMJ Clin. Evid. 2013, 2013, 0317. [Google Scholar]
- Crunelli, V.; Lőrincz, M.L.; McCafferty, C.; Lambert, R.C.; Leresche, N.; Di Giovanni, G.; David, F. Clinical and experimental insight into pathophysiology, comorbidity and therapy of absence seizures. Brain A J. Neurol. 2020, 143, 2341–2368. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P. Idiopathic generalized epilepsies: A review and modern approach. Epilepsia 2005, 46, 1–6. [Google Scholar] [CrossRef]
- Leresche, N.; Crunelli, V. Corticothalamic network abnormalities underlying absence seizures. In The Cerebral Cortex and Thalamus; Oxford University Press: Oxford, UK, 2023. [Google Scholar]
- Cope, D.W.; Di Giovanni, G.; Fyson, S.J.; Orban, G.; Errington, A.C.; Lorincz, M.L.; Gould, T.M.; Carter, D.A.; Crunelli, V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 2009, 15, 1392–1398. [Google Scholar] [CrossRef]
- Mangan, K.P.; Nelson, A.B.; Petrou, S.; Cirelli, C.; Jones, M.V. Cortical Tonic Inhibition Gates the Expression of Spike-and-Wave Discharges Associated with Absence Epilepsy. J. Integr. Neurosci. 2024, 23, 24. [Google Scholar] [CrossRef]
- Meyer, J.; Maheshwari, A. Cortical and Thalamic PV+ Interneuron Dysfunction in the Pathogenesis of Absence Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; Avoli, M., de Curtis, M., Bernard, C., Soltesz, I., Noebels, J.L., Avoli, M., Rogawski, M.A., Vezzani, A., Delgado-Escueta, A.V., Eds.; Oxford University Press: Oxford, UK, 2024. [Google Scholar]
- McCafferty, C.; David, F.; Venzi, M.; Lorincz, M.L.; Delicata, F.; Atherton, Z.; Recchia, G.; Orban, G.; Lambert, R.C.; Di Giovanni, G.; et al. Cortical drive and thalamic feed-forward inhibition control thalamic output synchrony during absence seizures. Nat. Neurosci. 2018, 21, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Depaulis, A.; David, O.; Charpier, S. The genetic absence epilepsy rat from Strasbourg as a model to decipher the neuronal and network mechanisms of generalized idiopathic epilepsies. J. Neurosci. Methods 2016, 260, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Coenen, A.M.; Van Luijtelaar, E.L. Genetic animal models for absence epilepsy: A review of the WAG/Rij strain of rats. Behav. Genet. 2003, 33, 635–655. [Google Scholar] [CrossRef]
- Noebels, J.L.; Qiao, X.; Bronson, R.T.; Spencer, C.; Davisson, M.T. Stargazer: A new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990, 7, 129–135. [Google Scholar] [CrossRef]
- Tan, H.O.; Reid, C.A.; Single, F.N.; Davies, P.J.; Chiu, C.; Murphy, S.; Clarke, A.L.; Dibbens, L.; Krestel, H.; Mulley, J.C.; et al. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc. Natl. Acad. Sci. USA 2007, 104, 17536–17541. [Google Scholar] [CrossRef]
- Deransart, C.; Depaulis, A. The control of seizures by the basal ganglia? A review of experimental data. Epileptic Disord 2002, 4 (Suppl. S3), S61–S72. [Google Scholar] [CrossRef]
- Paz, J.T.; Chavez, M.; Saillet, S.; Deniau, J.M.; Charpier, S. Activity of ventral medial thalamic neurons during absence seizures and modulation of cortical paroxysms by the nigrothalamic pathway. J. Neurosci. 2007, 27, 929–941. [Google Scholar] [CrossRef]
- Glauser, T.A.; Cnaan, A.; Shinnar, S.; Hirtz, D.G.; Dlugos, D.; Masur, D.; Clark, P.O.; Adamson, P.C. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy: Initial monotherapy outcomes at 12 months. Epilepsia 2013, 54, 141–155. [Google Scholar] [CrossRef]
- Terra, V.C.; de Paola, L.; Silvado, C.E. Are children affected by epileptic neuropsychiatric comorbidities? Epilepsy Behav. EB 2014, 38, 8–12. [Google Scholar] [CrossRef]
- Tenney, J.R.; Glauser, T.A. The current state of absence epilepsy: Can we have your attention? Epilepsy Curr. 2013, 13, 135–140. [Google Scholar] [CrossRef]
- Cnaan, A.; Shinnar, S.; Arya, R.; Adamson, P.C.; Clark, P.O.; Dlugos, D.; Hirtz, D.G.; Masur, D.; Glauser, T.A. Second monotherapy in childhood absence epilepsy. Neurology 2017, 88, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Caplan, R.; Siddarth, P.; Stahl, L.; Lanphier, E.; Vona, P.; Gurbani, S.; Koh, S.; Sankar, R.; Shields, W.D. Childhood absence epilepsy: Behavioral, cognitive, and linguistic comorbidities. Epilepsia 2008, 49, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. 2006, 30, S13–S18. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Fratta, W. Beyond THC: The new generation of cannabinoid designer drugs. Front. Behav. Neurosci. 2011, 5, 60. [Google Scholar] [CrossRef]
- Di Marzo, V. Endocannabinoid signaling in the brain: Biosynthetic mechanisms in the limelight. Nat. Neurosci. 2011, 14, 9–15. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Slade, D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci. 2005, 78, 539–548. [Google Scholar] [CrossRef]
- Mechoulam, R.; Hanuš, L.O.; Pertwee, R.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15, 757–764. [Google Scholar] [CrossRef]
- Notcutt, W. Cannabinoids. Br. J. Hosp. Med. (Lond. Engl. 2005) 2016, 77, C78–C80. [Google Scholar] [CrossRef]
- Manzoni, O.J.; Manduca, A.; Trezza, V. Therapeutic potential of cannabidiol polypharmacology in neuropsychiatric disorders. Trends Pharmacol. Sci. 2025, 46, 145–162. [Google Scholar] [CrossRef]
- Mirlohi, S.; Bladen, C.; Santiago, M.J.; Arnold, J.C.; McGregor, I.; Connor, M. Inhibition of human recombinant T-type calcium channels by phytocannabinoids in vitro. Br. J. Pharmacol. 2022, 179, 4031–4043. [Google Scholar] [CrossRef]
- Rempel, V.; Fuchs, A.; Hinz, S.; Karcz, T.; Lehr, M.; Koetter, U.; Müller, C.E. Magnolia Extract, Magnolol, and Metabolites: Activation of Cannabinoid CB2 Receptors and Blockade of the Related GPR55. ACS Med. Chem. Lett. 2013, 4, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Lacerda, M.; Carona, A.; Castanheira, S.; Falcão, A.; Bicker, J.; Fortuna, A. Pharmacokinetics of Non-Psychotropic Phytocannabinoids. Pharmaceutics 2025, 17, 236. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, C.; Jin, S.; Wang, Y.; Peng, Y.; Wang, X. Cannabidiol alleviates neuroinflammation and attenuates neuropathic pain via targeting FKBP5. Brain Behav. Immun. 2023, 111, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, N.; Gómez-Acero, L.; Sarasola, L.I.; Argerich, J.; Chevigné, A.; Jacobson, K.A.; Ciruela, F.; Fernández-Dueñas, V.; Aso, E. Cannabidiol negatively modulates adenosine A2A receptor functioning in living cells. Acta Neuropsychiatr. 2023, 36, 320–324. [Google Scholar] [CrossRef]
- Alexander, C.; Jeon, J.; Nickerson, K.; Hassler, S.; Vasefi, M. CBD and the 5-HT1A receptor: A medicinal and pharmacological review. Biochem. Pharmacol. 2025, 233, 116742. [Google Scholar] [CrossRef]
- Lima, I.V.d.A.; Bellozi, P.M.Q.; Batista, E.M.; Vilela, L.R.; Brandão, I.L.; Ribeiro, F.M.; Moraes, M.F.D.; Moreira, F.A.; de Oliveira, A.C.P. Cannabidiol anticonvulsant effect is mediated by the PI3Kγ pathway. Neuropharmacology 2020, 176, 108156. [Google Scholar] [CrossRef]
- Starkus, J.; Jansen, C.; Shimoda, L.M.N.; Stokes, A.J.; Small-Howard, A.L.; Turner, H. Diverse TRPV1 responses to cannabinoids. Channels 2019, 13, 172–191. [Google Scholar] [CrossRef]
- Tsien, R.W.; Rosenberg, E.C. Ion channels and G protein-coupled receptors: Cannabidiol actions on disorders of excitability and synaptic excitatory-inhibitory ratio. Mol. Pharmacol. 2025, 107, 100017. [Google Scholar] [CrossRef]
- Singh, K.; Bhushan, B.; Chanchal, D.K.; Sharma, S.K.; Rani, K.; Yadav, M.K.; Porwal, P.; Kumar, S.; Sharma, A.; Virmani, T. Emerging therapeutic potential of cannabidiol (CBD) in neurological disorders: A comprehensive review. Behav. Neurol. 2023, 2023, 8825358. [Google Scholar] [CrossRef]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2019, 176, 1455–1469. [Google Scholar] [CrossRef]
- Kathmann, M.; Flau, K.; Redmer, A.; Tränkle, C.; Schlicker, E. Cannabidiol is an allosteric modulator at mu-and delta-opioid receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2006, 372, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.-H.; Galadari, S.; Isaev, D.; Petroianu, G.; Shippenberg, T.S.; Oz, M. The nonpsychoactive cannabinoid cannabidiol inhibits 5-hydroxytryptamine3A receptor-mediated currents in Xenopus laevis oocytes. J. Pharmacol. Exp. Ther. 2010, 333, 547–554. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.; Phillips, A.; Razdan, R.; Ross, R.A.; Pertwee, R. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanuš, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Khosropoor, S.; Alavi, M.S.; Etemad, L.; Roohbakhsh, A. Cannabidiol goes nuclear: The role of PPARγ. Phytomedicine 2023, 114, 154771. [Google Scholar] [CrossRef]
- Seeman, P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl. Psychiatry 2016, 6, e920. [Google Scholar] [CrossRef]
- De Gregorio, D.; McLaughlin, R.J.; Posa, L.; Ochoa-Sanchez, R.; Enns, J.; Lopez-Canul, M.; Aboud, M.; Maione, S.; Comai, S.; Gobbi, G. Cannabidiol modulates serotonergic transmission and reverses both allodynia and anxiety-like behavior in a model of neuropathic pain. Pain 2019, 160, 136–150. [Google Scholar] [CrossRef]
- Bakas, T.; Van Nieuwenhuijzen, P.; Devenish, S.; McGregor, I.; Arnold, J.; Chebib, M. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA receptors. Pharmacol. Res. 2017, 119, 358–370. [Google Scholar] [CrossRef]
- Ross, R.A. The enigmatic pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30, 156–163. [Google Scholar] [CrossRef]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I. Fatty acid-binding proteins (FABPs) are intracellular carriers for Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J. Biol. Chem. 2015, 290, 8711–8721. [Google Scholar] [CrossRef]
- Carrier, E.J.; Auchampach, J.A.; Hillard, C.J. Inhibition of an equilibrative nucleoside transporter by cannabidiol: A mechanism of cannabinoid immunosuppression. Proc. Natl. Acad. Sci. USA 2006, 103, 7895–7900. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Drummond-Main, C.; Greening, D.; Yao, J.; Chen, S.; Appendino, J.; Au, P.B.; Turner, R.W. Cannabidiol counters the effects of a dominant-negative pathogenic Kv7. 2 variant. iScience 2022, 25, 105092. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.R. Cannabinoid interactions with ion channels and receptors. Channels 2019, 13, 162–167. [Google Scholar] [CrossRef]
- Smith, S.A.; Le, G.H.; Teopiz, K.M.; Kwan, A.T.; Rhee, T.G.; Ho, R.C.; Wu, J.; Cao, B.; Ceban, F.; McIntyre, R.S. Effects of cannabidiol and Δ9-tetrahydrocannabinol on cytochrome P450 enzymes: A systematic review. Drug Metab. Rev. 2024, 56, 164–174. [Google Scholar] [CrossRef]
- Chen, P.X.; Rogers, M.A. Opportunities and challenges in developing orally administered cannabis edibles. Curr. Opin. Food Sci. 2019, 28, 7–13. [Google Scholar] [CrossRef]
- Johansson, E.; Halldin, M.M. Urinary excretion half-life of Δ1-tetrahydrocannabinol-7-oic acid in heavy marijuana users after smoking. J. Anal. Toxicol. 1989, 13, 218–223. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Mechoulam, R.; Parker, L.A. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef]
- Howlett, A.; Barth, F.; Bonner, T.; Cabral, G.; Casellas, P.; Devane, W.; Felder, C.; Herkenham, M.; Mackie, K.; Martin, B. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Di Marzo, V.; Gertsch, J.; Grether, U.; Howlett, A.C.; Hua, T.; Makriyannis, A.; Piomelli, D.; Ueda, N.; van der Stelt, M. Goods and Bads of the Endocannabinoid System as a Therapeutic Target: Lessons Learned after 30 Years. Pharmacol. Rev. 2023, 75, 885–958. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; Kita, Y.; Hashimoto, K.; Shimizu, T.; Watanabe, M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.-Y.; Strassle, B.W.; Lu, P. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef]
- Straub, V.M.; Barti, B.; Tandar, S.T.; Stevens, A.F.; van Egmond, N.; van der Wel, T.; Zhu, N.; Rüegger, J.; van der Horst, C.; Heitman, L.H. The endocannabinoid 2-arachidonoylglycerol is released and transported on demand via extracellular microvesicles. Proc. Natl. Acad. Sci. USA 2025, 122, e2421717122. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Regehr, W.G. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 2001, 29, 717–727. [Google Scholar] [CrossRef]
- Ohno-Shosaku, T.; Maejima, T.; Kano, M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 2001, 29, 729–738. [Google Scholar] [CrossRef]
- Martínez-Aguirre, C.; Cinar, R.; Rocha, L. Targeting Endocannabinoid System in Epilepsy: For Good or for Bad. Neuroscience 2022, 482, 172–185. [Google Scholar] [CrossRef]
- Fetta, A.; Crotti, E.; Campostrini, E.; Bergonzini, L.; Cesaroni, C.A.; Conti, F.; Di Pisa, V.; Gentile, V.; Mondardini, M.C.; Vezzoli, C. Cannabidiol in the acute phase of febrile infection-related epilepsy syndrome (FIRES). Epilepsia Open 2023, 8, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Gofshteyn, J.S.; Wilfong, A.; Devinsky, O.; Bluvstein, J.; Charuta, J.; Ciliberto, M.A.; Laux, L.; Marsh, E.D. Cannabidiol as a potential treatment for febrile infection-related epilepsy syndrome (FIRES) in the acute and chronic phases. J. Child Neurol. 2017, 32, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, R.; Taraschenko, O.; Dilena, R.; Payne, E.T.; Specchio, N.; Nabbout, R.; Koh, S.; Gaspard, N.; Hirsch, L.J.; Group, I.N.C. International consensus recommendations for management of new onset refractory status epilepticus including febrile infection-related epilepsy syndrome: Statements and supporting evidence. Epilepsia 2022, 63, 2840–2864. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, C.M.; Furlanis, G.M.; Toldo, I.; Guarrera, B.; Luisi, C.; Pettenazzo, A.; Nosadini, M.; Boniver, C.; Sartori, S.; Landi, A. Myoclonic super-refractory status epilepticus with favourable evolution in a teenager with FIRES: Is the association of vagus nerve stimulation and cannabidiol effective? Brain Dev. 2023, 45, 293–299. [Google Scholar] [CrossRef]
- Sermet, S.; Li, J.; Bach, A.; Crawford, R.B.; Kaminski, N.E. Cannabidiol selectively modulates interleukin (IL)-1β and IL-6 production in toll-like receptor activated human peripheral blood monocytes. Toxicology 2021, 464, 153016. [Google Scholar] [CrossRef]
- Yousaf, M.; Chang, D.; Liu, Y.; Liu, T.; Zhou, X. Neuroprotection of cannabidiol, its synthetic derivatives and combination preparations against microglia-mediated neuroinflammation in neurological disorders. Molecules 2022, 27, 4961. [Google Scholar] [CrossRef]
- Gray, R.A.; Whalley, B.J. The proposed mechanisms of action of CBD in epilepsy. Epileptic Disord. 2020, 22, 10–15. [Google Scholar] [CrossRef]
- Miller, I.; Scheffer, I.E.; Gunning, B.; Sanchez-Carpintero, R.; Gil-Nagel, A.; Perry, M.S.; Saneto, R.P.; Checketts, D.; Dunayevich, E.; Knappertz, V. Dose-Ranging Effect of Adjunctive Oral Cannabidiol vs Placebo on Convulsive Seizure Frequency in Dravet Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 613–621. [Google Scholar] [CrossRef]
- Devinsky, O.; Patel, A.D.; Cross, J.H.; Villanueva, V.; Wirrell, E.C.; Privitera, M.; Greenwood, S.M.; Roberts, C.; Checketts, D.; VanLandingham, K.E.; et al. Effect of Cannabidiol on Drop Seizures in the Lennox-Gastaut Syndrome. N. Engl. J. Med. 2018, 378, 1888–1897. [Google Scholar] [CrossRef]
- Thiele, E.; Marsh, E.; Mazurkiewicz-Beldzinska, M. Cannabidiol in patients with Lennox-Gastaut syndrome: Interim analysis of an open-label extension study. Epilepsia 2019, 60, 419–428. [Google Scholar] [CrossRef]
- Privitera, M.; Bhathal, H.; Wong, M.; Cross, J.H.; Wirrell, E.; Marsh, E.D.; Mazurkiewicz-Beldzinska, M.; Villanueva, V.; Checketts, D.; Knappertz, V. Time to onset of cannabidiol (CBD) treatment effect in Lennox–Gastaut syndrome: Analysis from two randomized controlled trials. Epilepsia 2021, 62, 1130–1140. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Schubert-Bast, S.; von Podewils, F.; Knake, S.; Mayer, T.; Klotz, K.A.; Buhleier, E.; Herold, L.; Immisch, I.; Kurlemann, G.; et al. Real-world experience of cannabidiol in conjunction with clobazam for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome: Results from a retrospective multicentre chart review in Germany. Epilepsy Behav. 2025, 166, 110302. [Google Scholar] [CrossRef]
- Ong, M.J.Y.; Abd Rahman, M.S.H.; Lee, V.L.L.; Lee, K.H.; Chang, C.J.Y.; Khoo, C.S.; Hod, R.; Tan, H.J.; Trinka, E. The use of cannabidiol as adjunctive therapy in adult patients with drug-resistant epilepsy: A systematic review and meta-analysis. Ther. Adv. Neurol. Disord. 2025, 18, 17562864251313914. [Google Scholar] [CrossRef]
- de Oliveira, V.G.; de Almeida, N.B.; Radmann, G.C.; de Oliveira Santos, B.F. The efficacy of cannabidiol for seizures reduction in pharmacoresistant epilepsy: A systematic review and meta-analysis. Acta Epileptol. 2025, 7, 20. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Z.; Xiao, W.; Wang, C.; Liang, R. Efficacy and safety of pharmacological and non-pharmacological therapies in Lennox-Gastaut syndrome: A systematic review and network meta-analysis. Front. Pharmacol. 2025, 16, 1522543. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Wang, C. Efficacy and safety of antiseizure medication for Lennox–Gastaut syndrome: A systematic review and network meta-analysis. Dev. Med. Child Neurol. 2022, 64, 305–313. [Google Scholar] [CrossRef]
- Chesney, E.; Oliver, D.; Green, A. Adverse effects of cannabidiol: A systematic review and meta-analysis of randomized clinical trials. Neuropsychopharmacology 2020, 45, 1799–1806. [Google Scholar] [CrossRef]
- Fazlollahi, A.; Zahmatyar, M.; ZareDini, M. Adverse events of cannabidiol use in patients with epilepsy: A systematic review and meta-analysis. JAMA Netw. Open 2023, 6, e239126. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, Y.; Gozal, D.; Carney, P. Channelopathy of Dravet Syndrome and Potential Neuroprotective Effects of Cannabidiol. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211048045. [Google Scholar] [CrossRef]
- Rubio, M.; Valdeolivas, S.; Piscitelli, F.; Verde, R.; Satta, V.; Barroso, E.; Montolio, M.; Aras, L.M.; Di Marzo, V.; Sagredo, O. Analysis of endocannabinoid signaling elements and related proteins in lymphocytes of patients with Dravet syndrome. Pharmacol. Res. Perspect. 2016, 4, e00220. [Google Scholar] [CrossRef]
- Anderson, L.L.; Absalom, N.L.; Abelev, S.V.; Low, I.K.; Doohan, P.T.; Martin, L.J.; Chebib, M.; McGregor, I.S.; Arnold, J.C. Coadministered cannabidiol and clobazam: Preclinical evidence for both pharmacodynamic and pharmacokinetic interactions. Epilepsia 2019, 60, 2224–2234. [Google Scholar] [CrossRef]
- Anderson, L.L.; Ametovski, A.; Lin Luo, J.; Everett-Morgan, D.; McGregor, I.S.; Banister, S.D.; Arnold, J.C. Cannabichromene, related phytocannabinoids, and 5-fluoro-cannabichromene have anticonvulsant properties in a mouse model of Dravet Syndrome. ACS Chem. Neurosci. 2021, 12, 330–339. [Google Scholar] [CrossRef]
- Anderson, L.L.; Heblinski, M.; Absalom, N.L.; Hawkins, N.A.; Bowen, M.T.; Benson, M.J.; Zhang, F.; Bahceci, D.; Doohan, P.T.; Chebib, M. Cannabigerolic acid, a major biosynthetic precursor molecule in cannabis, exhibits divergent effects on seizures in mouse models of epilepsy. Br. J. Pharmacol. 2021, 178, 4826–4841. [Google Scholar] [CrossRef]
- Yip, K.L.; Udoh, M.; Sharman, L.A.; Harman, T.; Bedoya-Pérez, M.; Anderson, L.L.; Banister, S.D.; Arnold, J.C. Cannabinoid-like compounds found in non-cannabis plants exhibit antiseizure activity in genetic mouse models of drug-resistant epilepsy. Epilepsia 2025, 66, 303–314. [Google Scholar] [CrossRef]
- Miller, A.R.; Hawkins, N.A.; McCollom, C.E.; Kearney, J.A. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 2014, 13, 163–172. [Google Scholar] [CrossRef]
- Bahceci, D.; Anderson, L.L.; Kevin, R.C.; Doohan, P.T.; Arnold, J.C. Hyperthermia-Induced Seizures Enhance Brain Concentrations of the Endocannabinoid-Related Linoleoyl Glycerols in a Scn1a+/− Mouse Model of Dravet Syndrome. Cannabis Cannabinoid Res. 2023, 8, 495–504. [Google Scholar] [CrossRef]
- Anderson, L.L.; Doohan, P.T.; Hawkins, N.A.; Bahceci, D.; Garai, S.; Thakur, G.A.; Kearney, J.A.; Arnold, J.C. The endocannabinoid system impacts seizures in a mouse model of Dravet syndrome. Neuropharmacology 2022, 205, 108897. [Google Scholar] [CrossRef]
- Anderson, L.L.; Etchart, M.G.; Bahceci, D.; Golembiewski, T.A.; Arnold, J.C. Cannabis constituents interact at the drug efflux pump BCRP to markedly increase plasma cannabidiolic acid concentrations. Sci. Rep. 2021, 11, 14948. [Google Scholar] [CrossRef]
- Farrell, J.S.; Colangeli, R.; Dong, A.; George, A.G.; Addo-Osafo, K.; Kingsley, P.J.; Morena, M.; Wolff, M.D.; Dudok, B.; He, K. In vivo endocannabinoid dynamics at the timescale of physiological and pathological neural activity. Neuron 2021, 109, 2398–2403. e2394. [Google Scholar] [CrossRef]
- Satta, V.; Alonso, C.; Díez, P.; Martín-Suárez, S.; Rubio, M.; Encinas, J.M.; Fernández-Ruiz, J.; Sagredo, O. Neuropathological characterization of a Dravet syndrome knock-in mouse model useful for investigating cannabinoid treatments. Front. Mol. Neurosci. 2021, 13, 602801. [Google Scholar] [CrossRef]
- Qu, S.; Catron, M.; Zhou, C.; Janve, V.; Shen, W.; Howe, R.K.; Macdonald, R.L. GABAA receptor β3 subunit mutation D120N causes Lennox–Gastaut syndrome in knock-in mice. Brain Commun. 2020, 2, fcaa028. [Google Scholar] [CrossRef]
- Gaston, T.E.; Ampah, S.B.; Bebin, E.M.; Grayson, L.P.; Cutter, G.R.; Hernando, K.; Szaflarski, J.P. Long-term safety and efficacy of highly purified cannabidiol for treatment refractory epilepsy. Epilepsy Behav. 2021, 117, 107862. [Google Scholar] [CrossRef]
- Gaston, T.E.; Szaflarski, M.; Hansen, B.; Bebin, E.M.; Szaflarski, J.P. Quality of life in adults enrolled in an open-label study of cannabidiol (CBD) for treatment-resistant epilepsy. Epilepsy Behav. 2019, 95, 10–17. [Google Scholar] [CrossRef]
- Martin, R.C.; Gaston, T.E.; Thompson, M.; Ampah, S.B.; Cutter, G.; Bebin, E.M.; Szaflarski, J.P. Cognitive functioning following long-term cannabidiol use in adults with treatment-resistant epilepsy. Epilepsy Behav. 2019, 97, 105–110. [Google Scholar] [CrossRef]
- Patra, P.H.; Serafeimidou-Pouliou, E.; Bazelot, M.; Whalley, B.J.; Williams, C.M.; McNeish, A.J. Cannabidiol improves survival and behavioural co-morbidities of Dravet syndrome in mice. Br. J. Pharmacol. 2020, 177, 2779–2792. [Google Scholar] [CrossRef]
- Cheah, C.S.; Yu, F.H.; Westenbroek, R.E.; Kalume, F.K.; Oakley, J.C.; Potter, G.B.; Rubenstein, J.L.; Catterall, W.A. Specific deletion of NaV1. 1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 14646–14651. [Google Scholar] [CrossRef]
- Raucci, U.; Pietrafusa, N.; Paolino, M.C.; Di Nardo, G.; Villa, M.P.; Pavone, P.; Terrin, G.; Specchio, N.; Striano, P.; Parisi, P. Cannabidiol Treatment for Refractory Epilepsies in Pediatrics. Front. Pharmacol. 2020, 11, 586110. [Google Scholar] [CrossRef]
- Lattanzi, S.; Trinka, E.; Striano, P.; Rocchi, C.; Salvemini, S.; Silvestrini, M.; Brigo, F. Highly Purified Cannabidiol for Epilepsy Treatment: A Systematic Review of Epileptic Conditions Beyond Dravet Syndrome and Lennox-Gastaut Syndrome. CNS Drugs 2021, 35, 265–281. [Google Scholar] [CrossRef]
- Hess, E.J.; Moody, K.A.; Geffrey, A.L.; Pollack, S.F.; Skirvin, L.A.; Bruno, P.L.; Paolini, J.L.; Thiele, E.A. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia 2016, 57, 1617–1624. [Google Scholar] [CrossRef]
- Thiele, E.A.; Bebin, E.M.; Bhathal, H.; Jansen, F.E.; Kotulska, K.; Lawson, J.A.; O’Callaghan, F.J.; Wong, M.; Sahebkar, F.; Checketts, D. Add-on cannabidiol treatment for drug-resistant seizures in tuberous sclerosis complex: A placebo-controlled randomized clinical trial. JAMA Neurol. 2021, 78, 285–292. [Google Scholar] [CrossRef]
- Ong, K.S.; Carlin, J.B.; Fahey, M.; Freeman, J.L.; Scheffer, I.E.; Gillam, L.; Anderson, M.; Huque, M.H.; Legge, D.; Dirnbauer, N.; et al. Protocol for a single patient therapy plan: A randomised, double-blind, placebo-controlled N-of-1 trial to assess the efficacy of cannabidiol in patients with intractable epilepsy. J. Paediatr. Child Health 2020, 56, 1918–1923. [Google Scholar] [CrossRef]
- Martínez-Aguirre, C.; Carmona-Cruz, F.; Velasco, A.L.; Velasco, F.; Aguado-Carrillo, G.; Cuéllar-Herrera, M.; Rocha, L. Cannabidiol Acts at 5-HT(1A) Receptors in the Human Brain: Relevance for Treating Temporal Lobe Epilepsy. Front. Behav. Neurosci. 2020, 14, 611278. [Google Scholar] [CrossRef]
- Nuñez-Lumbreras, M.d.l.Á.; Castañeda-Cabral, J.L.; Valle-Dorado, M.G.; Sánchez-Valle, V.; Orozco-Suárez, S.; Guevara-Guzmán, R.; Martínez-Juárez, I.; Alonso-Vanegas, M.; Walter, F.; Deli, M.A.; et al. Drug-Resistant Temporal Lobe Epilepsy Alters the Expression and Functional Coupling to Gαi/o Proteins of CB1 and CB2 Receptors in the Microvasculature of the Human Brain. Front. Behav. Neurosci. 2021, 14, 611780. [Google Scholar] [CrossRef]
- Rocha, L.; Cinar, R.; Guevara-Guzmán, R.; Alonso-Vanegas, M.; San-Juan, D.; Martínez-Juárez, I.; Castañeda-Cabral, J.L.; Carmona-Cruz, F. Endocannabinoid System and Cannabinoid 1 Receptors in Patients With Pharmacoresistant Temporal Lobe Epilepsy and Comorbid Mood Disorders. Front. Behav. Neurosci. 2020, 14, 52. [Google Scholar] [CrossRef]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Romigi, A.; Bari, M.; Placidi, F.; Marciani, M.G.; Malaponti, M.; Torelli, F.; Izzi, F.; Prosperetti, C.; Zannino, S.; Corte, F. Cerebrospinal fluid levels of the endocannabinoid anandamide are reduced in patients with untreated newly diagnosed temporal lobe epilepsy. Epilepsia 2010, 51, 768–772. [Google Scholar] [CrossRef]
- Ludányi, A.; Erőss, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef]
- Goffin, K.; Van Paesschen, W.; Van Laere, K. In vivo activation of endocannabinoid system in temporal lobe epilepsy with hippocampal sclerosis. Brain A J. Neurol. 2011, 134, 1033–1040. [Google Scholar] [CrossRef]
- Nearing, K.; Madhavan, D.; Devinsky, O. Temporal lobe epilepsy: A progressive disorder? Rev. Neurol. Dis. 2007, 4, 122–127. [Google Scholar]
- Colangeli, R.; Teskey, G.C.; Di Giovanni, G. Endocannabinoid-serotonin systems interaction in health and disease. Prog. Brain Res. 2021, 259, 83–134. [Google Scholar] [CrossRef]
- Nogueira, M.H.; Yasuda, C.L.; Coan, A.C.; Kanner, A.M.; Cendes, F. Concurrent mood and anxiety disorders are associated with pharmacoresistant seizures in patients with MTLE. Epilepsia 2017, 58, 1268–1276. [Google Scholar] [CrossRef]
- Vinti, V.; Dell’Isola, G.B.; Tascini, G.; Mencaroni, E.; Cara, G.D.; Striano, P.; Verrotti, A. Temporal Lobe Epilepsy and Psychiatric Comorbidity. Front. Neurol. 2021, 12, 775781. [Google Scholar] [CrossRef]
- Colangeli, R.; Pierucci, M.; Benigno, A.; Campiani, G.; Butini, S.; Di Giovanni, G. The FAAH inhibitor URB597 suppresses hippocampal maximal dentate afterdischarges and restores seizure-induced impairment of short and long-term synaptic plasticity. Sci. Rep. 2017, 7, 11152. [Google Scholar] [CrossRef]
- Colangeli, R.; Morena, M.; Pittman, Q.J.; Hill, M.N.; Teskey, G.C. Anandamide Signaling Augmentation Rescues Amygdala Synaptic Function and Comorbid Emotional Alterations in a Model of Epilepsy. J. Neurosci. 2020, 40, 6068–6081. [Google Scholar] [CrossRef]
- Colangeli, R.; Di Maio, R.; Pierucci, M.; Deidda, G.; Casarrubea, M.; Di Giovanni, G. Synergistic action of CB1 and 5-HT2B receptors in preventing pilocarpine-induced status epilepticus in rats. Neurobiol. Dis. 2019, 125, 135–145. [Google Scholar] [CrossRef]
- Di Maio, R.; Colangeli, R.; Di Giovanni, G. WIN 55,212-2 Reverted Pilocarpine-Induced Status Epilepticus Early Changes of the Interaction among 5-HT2C/NMDA/CB1 Receptors in the Rat Hippocampus. ACS Chem. Neurosci. 2019, 10, 3296–3306. [Google Scholar] [CrossRef]
- Azevedo, M.; Benbadis, S.R. Efficacy of highly purified cannabidiol (CBD) in typical absence seizures: A pilot study. Epilepsy Behav. 2023, 149, 109512. [Google Scholar] [CrossRef]
- Flamini, R.J.; Comi, A.M.; Bebin, E.M.; Chez, M.G.; Clark, G.; Devinsky, O.; Hussain, S.A.; Lyons, P.D.; Patel, A.D.; Rosengard, J.L.; et al. Efficacy of cannabidiol in convulsive and nonconvulsive seizure types associated with treatment-resistant epilepsies in the Expanded Access Program. Epilepsia 2023, 64, e156–e163. [Google Scholar] [CrossRef]
- Patel, S.; Grinspoon, R.; Fleming, B.; Skirvin, L.A.; Wade, C.; Wolper, E.; Bruno, P.L.; Thiele, E.A. The long-term efficacy of cannabidiol in the treatment of refractory epilepsy. Epilepsia 2021, 62, 1594–1603. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Cannabidiol Oral Solution in Pediatric Participants with Treatment-Resistant Childhood Absence Seizures. Available online: https://clinicaltrials.gov/study/NCT03336242 (accessed on 2 April 2025).
- Turkanis, S.A.; Chiu, P.; Borys, H.K.; Karler, R. Influence of Δ9 and cannabidiol on photically evoked after-discharge potentials. Psychopharmacology 1977, 52, 207–212. [Google Scholar] [CrossRef]
- Roebuck, A.J.; Greba, Q.; Onofrychuk, T.J.; McElroy, D.L.; Sandini, T.M.; Zagzoog, A.; Simone, J.; Cain, S.M.; Snutch, T.P.; Laprairie, R.B.; et al. Dissociable changes in spike and wave discharges following exposure to injected cannabinoids and smoked cannabis in Genetic Absence Epilepsy Rats from Strasbourg. Eur. J. Neurosci. 2022, 55, 1063–1078. [Google Scholar] [CrossRef]
- Sales-Carbonell, C.; Rueda-Orozco, P.E.; Soria-Gomez, E.; Buzsaki, G.; Marsicano, G.; Robbe, D. Striatal GABAergic and cortical glutamatergic neurons mediate contrasting effects of cannabinoids on cortical network synchrony. Proc. Natl. Acad. Sci. USA 2013, 110, 719–724. [Google Scholar] [CrossRef]
- van Rijn, C.M.; Gaetani, S.; Santolini, I.; Badura, A.; Gabova, A.; Fu, J.; Watanabe, M.; Cuomo, V.; van Luijtelaar, G.; Nicoletti, F.; et al. WAG/Rij rats show a reduced expression of CB(1) receptors in thalamic nuclei and respond to the CB(1) receptor agonist, R(+)WIN55,212-2, with a reduced incidence of spike-wave discharges. Epilepsia 2010, 51, 1511–1521. [Google Scholar] [CrossRef]
- Perescis, M.F.J.; Flipsen, N.A.R.; van Luijtelaar, G.; van Rijn, C.M. Altered SWD stopping mechanism in WAG/Rij rats subchronically treated with the cannabinoid agonist R(+)WIN55,212-2. Epilepsy Behav. EB 2020, 102, 106722. [Google Scholar] [CrossRef]
- Citraro, R.; Russo, E.; Ngomba, R.T.; Nicoletti, F.; Scicchitano, F.; Whalley, B.J.; Calignano, A.; De Sarro, G. CB1 agonists, locally applied to the cortico-thalamic circuit of rats with genetic absence epilepsy, reduce epileptic manifestations. Epilepsy Res. 2013, 106, 74–82. [Google Scholar] [CrossRef]
- Roebuck, A.J.; Greba, Q.; Smolyakova, A.M.; Alaverdashvili, M.; Marks, W.N.; Garai, S.; Baglot, S.L.; Petrie, G.; Cain, S.M.; Snutch, T.P.; et al. Positive allosteric modulation of type 1 cannabinoid receptors reduces spike-and-wave discharges in Genetic Absence Epilepsy Rats from Strasbourg. Neuropharmacology 2021, 190, 108553. [Google Scholar] [CrossRef]
- Citraro, R.; Russo, E.; Scicchitano, F.; van Rijn, C.M.; Cosco, D.; Avagliano, C.; Russo, R.; D’Agostino, G.; Petrosino, S.; Guida, F.; et al. Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacology 2013, 69, 115–126. [Google Scholar] [CrossRef]
- McElroy, D.L.; Roebuck, A.J.; Greba, Q.; Garai, S.; Brandt, A.L.; Yilmaz, O.; Cain, S.M.; Snutch, T.P.; Thakur, G.A.; Laprairie, R.B.; et al. The type 1 cannabinoid receptor positive allosteric modulators GAT591 and GAT593 reduce spike-and-wave discharges in Genetic Absence Epilepsy Rats from Strasbourg. IBRO Neurosci. Rep. 2022, 12, 121–130. [Google Scholar] [CrossRef]
- USA Controlled Substances Act. USA Controlled Substances Act of 1970. Available online: https://uscode.house.gov/view.xhtml?path=/prelim@title21/chapter13&edition=prelim (accessed on 20 June 2022).
- Sysoeva, M.V.; Kuznetsova, G.D.; Sysoev, I.V.; Ngomba, R.T.; Vinogradova, L.V.; Grishchenko, A.A.; van Rijn, C.M.; van Luijtelaar, G. Network analysis reveals a role of the hippocampus in absence seizures: The effects of a cannabinoid agonist. Epilepsy Res. 2023, 192, 107135. [Google Scholar] [CrossRef]
- Lüttjohann, A.; Schoffelen, J.-M.; Van Luijtelaar, G. Termination of ongoing spike-wave discharges investigated by cortico–thalamic network analyses. Neurobiol. Dis. 2014, 70, 127–137. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Smith, S.E.; Liimatta, M.B.; Beidler, D.; Sadagopan, N.; Dudley, D.T.; Young, T.; Wren, P.; Zhang, Y.; Swaney, S. Mechanistic and pharmacological characterization of PF-04457845: A highly potent and selective FAAH inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011, 338, 114–124. [Google Scholar] [CrossRef] [PubMed]
- De Deurwaerdère, P.; Casarrubea, M.; Cassar, D.; Radic, M.; Puginier, E.; Chagraoui, A.; Crescimanno, G.; Crunelli, V.; Di Giovanni, G. Cannabinoid 1/2 receptor activation induces strain-dependent behavioral and neurochemical changes in genetic absence epilepsy rats from Strasbourg and non-epileptic control rats. Front. Cell. Neurosci. 2022, 16, 886033. [Google Scholar] [CrossRef]
- Neuparth-Sottomayor, M.; Pina, C.C.; Morais, T.P.; Farinha-Ferreira, M.; Abreu, D.S.; Solano, F.; Mouro, F.; Good, M.; Sebastião, A.M.; Di Giovanni, G.; et al. Cognitive comorbidities of experimental absence seizures are independent of anxiety. Neurobiol. Dis. 2023, 186, 106275. [Google Scholar] [CrossRef]
- Casarrubea, M.; Radic, M.; Morais, T.P.; Mifsud, E.; Cuboni, E.; Aiello, S.; Crescimanno, G.; Crunelli, V.; Di Giovanni, G. A quantitative and T-pattern analysis of anxiety-like behavior in male GAERS, NEC, and Wistar rats bred under the same conditions, against a commercially available Wistar control group in the hole board and elevated plus maze tests. CNS Neurosci. Ther. 2024, 30, e14443. [Google Scholar] [CrossRef]
- Cassar, D.; Radic, M.; Casarrubea, M.; Crunelli, V.; Di Giovanni, G. The effect of cannabinoid receptor agonist WIN 55,212–2 on anxiety-like behavior and locomotion in a genetic model of absence seizures in the elevated plus-maze. CNS Neurosci. Ther. 2022, 28, 1268. [Google Scholar] [CrossRef]
- Roebuck, A.J. Modulation of the Endocannabinoid System in Genetic Absence Epilepsy Rats from Strasbourg: New Avenues for Treatment of Absence Seizures and Behavioural Comorbidities. Ph.D. Thesis, University of Saskatchewan, Saskatoon, SK, Canada, 2021. [Google Scholar]
- Depaulis, A.; Charpier, S. Pathophysiology of absence epilepsy: Insights from genetic models. Neurosci. Lett. 2018, 667, 53–65. [Google Scholar] [CrossRef]
- Sugaya, Y.; Kano, M. Control of excessive neural circuit excitability and prevention of epileptic seizures by endocannabinoid signaling. Cell. Mol. Life Sci. CMLS 2018, 75, 2793–2811. [Google Scholar] [CrossRef]
- Suárez, J.; Ortíz, O.; Puente, N.; Bermúdez-Silva, F.J.; Blanco, E.; Fernández-Llebrez, P.; Grandes, P.; de Fonseca, F.R.; Moratalla, R. Distribution of diacylglycerol lipase alpha, an endocannabinoid synthesizing enzyme, in the rat forebrain. Neuroscience 2011, 192, 112–131. [Google Scholar] [CrossRef]
- Sun, Y.G.; Wu, C.S.; Lu, H.C.; Beierlein, M. Target-dependent control of synaptic inhibition by endocannabinoids in the thalamus. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 9222–9230. [Google Scholar] [CrossRef]
- Aguirre-Rodríguez, C.A.; Delgado, A.; Alatorre, A.; Oviedo-Chávez, A.; Martínez-Escudero, J.R.; Barrientos, R.; Querejeta, E. Local activation of CB1 receptors by synthetic and endogenous cannabinoids dampens burst firing mode of reticular thalamic nucleus neurons in rats under ketamine anesthesia. Exp. Brain Res. 2024, 242, 2137–2157. [Google Scholar] [CrossRef]
- Zhang, L.; Kolaj, M.; Renaud, L.P. Endocannabinoid 2-AG and intracellular cannabinoid receptors modulate a low-threshold calcium spike-induced slow depolarizing afterpotential in rat thalamic paraventricular nucleus neurons. Neuroscience 2016, 322, 308–319. [Google Scholar] [CrossRef]
- Marks, W.N.; Cavanagh, M.E.; Greba, Q.; Cain, S.M.; Snutch, T.P.; Howland, J.G. The G enetic A bsence E pilepsy R ats from S trasbourg model of absence epilepsy exhibits alterations in fear conditioning and latent inhibition consistent with psychiatric comorbidities in humans. Eur. J. Neurosci. 2016, 43, 25–40. [Google Scholar] [CrossRef]
- Marks, W.N.; Cain, S.M.; Snutch, T.P.; Howland, J.G. The T-type calcium channel antagonist Z944 rescues impairments in crossmodal and visual recognition memory in Genetic Absence Epilepsy Rats from Strasbourg. Neurobiol. Dis. 2016, 94, 106–115. [Google Scholar] [CrossRef]
- Marks, W.N.; Zabder, N.K.; Greba, Q.; Cain, S.M.; Snutch, T.P.; Howland, J.G. The T-type calcium channel blocker Z944 reduces conditioned fear in Genetic Absence Epilepsy Rats from Strasbourg and the non-epileptic control strain. Eur. J. Neurosci. 2019, 50, 3046–3059. [Google Scholar] [CrossRef]
- Roebuck, A.J.; An, L.; Marks, W.N.; Sun, N.; Snutch, T.P.; Howland, J.G. Cognitive impairments in touchscreen-based visual discrimination and reversal learning in genetic absence epilepsy rats from Strasbourg. Neuroscience 2020, 430, 105–112. [Google Scholar] [CrossRef]
- Chemin, J.; Monteil, A.; Perez-Reyes, E.; Nargeot, J.; Lory, P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001, 20, 7033–7040. [Google Scholar] [CrossRef]
- van der Stelt, M.; Trevisani, M.; Vellani, V.; De Petrocellis, L.; Schiano Moriello, A.; Campi, B.; McNaughton, P.; Geppetti, P.; Di Marzo, V. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J. 2005, 24, 3026–3037. [Google Scholar] [CrossRef]
- Sigel, E.; Baur, R.; Rácz, I.; Marazzi, J.; Smart, T.G.; Zimmer, A.; Gertsch, J. The major central endocannabinoid directly acts at GABAA receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 18150–18155. [Google Scholar] [CrossRef]
- Golovko, T.; Min, R.; Lozovaya, N.; Falconer, C.; Yatsenko, N.; Tsintsadze, T.; Tsintsadze, V.; Ledent, C.; Harvey, R.J.; Belelli, D.; et al. Control of Inhibition by the Direct Action of Cannabinoids on GABAA Receptors. Cereb. Cortex 2014, 25, 2440–2455. [Google Scholar] [CrossRef]
- Baur, R.; Kielar, M.; Richter, L.; Ernst, M.; Ecker, G.F.; Sigel, E. Molecular analysis of the site for 2-arachidonylglycerol (2-AG) on the β2 subunit of GABAA receptors. J. Neurochem. 2013, 126, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, G.; Chagraoui, A.; Bharatiya, R.; De Deurwaerdère, P. Chapter 10—Serotonergic control of excitability: From neuron to networks. In Handbook of Behavioral Neuroscience; Müller, C.P., Cunningham, K.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 31, pp. 197–215. [Google Scholar]
- Schonhofen, P.; Bristot, I.J.; Crippa, J.A.; Hallak, J.E.C.; Zuardi, A.W.; Parsons, R.B.; Klamt, F. Cannabinoid-Based Therapies and Brain Development: Potential Harmful Effect of Early Modulation of the Endocannabinoid System. CNS Drugs 2018, 32, 697–712. [Google Scholar] [CrossRef]
- Harkany, T.; Guzmán, M.; Galve-Roperh, I.; Berghuis, P.; Devi, L.A.; Mackie, K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol. Sci. 2007, 28, 83–92. [Google Scholar] [CrossRef]
- Dow-Edwards, D.; Silva, L. Endocannabinoids in brain plasticity: Cortical maturation, HPA axis function and behavior. Brain Res. 2017, 1654, 157–164. [Google Scholar] [CrossRef]
- Meyer, H.C.; Lee, F.S.; Gee, D.G. The role of the endocannabinoid system and genetic variation in adolescent brain development. Neuropsychopharmacology 2018, 43, 21–33. [Google Scholar] [CrossRef]
- de Salas-Quiroga, A.; Díaz-Alonso, J.; García-Rincón, D.; Remmers, F.; Vega, D.; Gómez-Cañas, M.; Lutz, B.; Guzmán, M.; Galve-Roperh, I. Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 13693–13698. [Google Scholar] [CrossRef]
- Lovelace, J.W.; Corches, A.; Vieira, P.A.; Hiroto, A.S.; Mackie, K.; Korzus, E. An animal model of female adolescent cannabinoid exposure elicits a long-lasting deficit in presynaptic long-term plasticity. Neuropharmacology 2015, 99, 242–255. [Google Scholar] [CrossRef]
- Green, H.M.; Manning, J.J.; Greig, I.R.; Ross, R.A.; Finlay, D.B.; Glass, M. Positive allosteric modulation of the cannabinoid CB1 receptor potentiates endocannabinoid signalling and changes ERK1/2 phosphorylation kinetics. Br. J. Pharmacol. 2024, 181, 3642–3662. [Google Scholar] [CrossRef]
- Papa, A.A.-O.; Pasquini, S.A.-O.; Contri, C.A.-O.; Gemma, S.A.-O.; Campiani, G.; Butini, S.A.-O.; Varani, K.A.-O.; Vincenzi, F.A.-O. Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition. Cells 2022, 11, 471. [Google Scholar] [CrossRef]
- Grillo, A.; Fezza, F.; Chemi, G.A.-O.; Colangeli, R.; Brogi, S.A.-O.; Fazio, D.; Federico, S.A.-O.; Papa, A.; Relitti, N.A.-O.; Di Maio, R.; et al. Selective Fatty Acid Amide Hydrolase Inhibitors as Potential Novel Antiepileptic Agents. ACS Chem. Neurosci. 2021, 12, 1716–1736. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Seizure Classes | Seizure Types |
|---|---|
| Focal (F) Seizures | Focal-to-bilateral tonic–clonic (FBTC) seizures Focal preserved consciousness (FPC) seizures Focal impaired consciousness (FIC) seizures Focal epilepsy, temporal lobe epilepsy (TLE), Dravet syndrome |
| Generalized (G) Seizures | Generalized tonic–clonic seizures Absence seizures, other generalized seizures Absence seizure (AS) Typical absence seizure (TA) Atypical absence seizure (AA) Myoclonic absence seizure (MA) Eyelid myoclonia with/without absence (EMA) Generalized tonic–clonic (GTC) seizure Myoclonic tonic–clonic seizure Absence-to-tonic–clonic seizure Generalized myoclonic (GM) seizure Generalized clonic (GC) seizure Generalized negative myoclonic (GNM) seizure Generalized epileptic spasms (GESs) Generalized tonic (GT) seizure Generalized atonic (GA) seizure Generalized myoclonic–atonic (GMA) seizure |
| Unknown (U) Seizures | Focal or generalized–preserved consciousness (PC) seizure Focal or generalized–impaired consciousness (IC) seizure Focal or generalized–bilateral tonic–clonic (BTC) seizure Refractory status epilepsy, febrile infection-related epilepsy syndrome (FIRES) |
| Unclassified |
| Category | Syndromes |
|---|---|
| Self-Limited Focal Epilepsies | - Self-limited epilepsy with centrotemporal spikes |
| - Self-limited epilepsy with autonomic seizures | |
| - Childhood occipital visual epilepsy | |
| Generalized Epilepsies | - Photosensitive occipital lobe epilepsy |
| - Childhood absence epilepsy | |
| Developmental and/or Epileptic Encephalopathies | - Epilepsy with myoclonic absence |
| - Epilepsy with eyelid myoclonia | |
| - Epilepsy with myoclonic–atonic seizures | |
| - Lennox–Gastaut syndrome | |
| - Developmental and/or epileptic encephalopathy with spike-and-wave activation in sleep | |
| - Hemiconvulsion–hemiplegia–epilepsy syndrome |
| Model | Cannabinoid | Dose | ROA | Primary Finding | Reference |
|---|---|---|---|---|---|
| Sprague Dawley Rats—Photically Evoked After-Discharge Potentials | ∆9-THC | 5 mg/kg | i.p. | ↑ Seizure Incidence | [195] |
| GAERS | 0.3, 1, 3, 10 mg/kg 10 mg/kg = ~80 ng/mL ∆9-THC plasma concentration | i.p. | ↑ Seizure Incidence (200% of Baseline); ↑ Total Time Spent in Seizures; ↑ Seizure Length; ↓ Seizure Frequency | [196] | |
| GAERS | ∆9-THC ~3 ng/mL plasma concentration | Smoke high-THC cannabis (Mohawk) | ↑ Seizure Incidence (>50% Increase); ↑ Total Time Spent in Seizures; ↑ Seizure Length (>100% Increase); ↓ Seizure Frequency | [196] | |
| Sprague Dawley rats—Photically Evoked After-Discharge Potentials | CBD | 50 mg/kg | i.p. | ↨ Seizure Incidence | [195] |
| GAERS | 30–100 mg/kg 100 mg = ~4000 ng/mL CBD plasma concentration | i.p. | ↓ Seizure Incidence (50% Reduction) | [196] | |
| GAERS | ~20 ng/mL CBD plasma concentration | Smoke high-CBD cannabis (Treasure Island) | ↨ Seizure Incidence | [196] | |
| C57BL/6N Mice | CB1/2R agonist CP55940 | 0.3 mg/kg | i.p. | ↑ High Voltage Spindles Incidence (HVSs) | [197] |
| WAG/Rij | CB1/2R agonist WIN 55,212-2 | 3–6–12 mg/kg ED50 value = 4.9 mg/kg | s.c. | First 2 h ↓ Seizure Incidence (80% Reduction after 12 mg/kg) Last 2 h: ↑ Seizure Length (>100 s) ↓ Motor Activity | [198] |
| WAG/Rij | 6 mg/kg | s.c. | ↨ Seizures Incidence in the First 3 h and in 24 h Recording; ↑ Seizure Length (>11 s) ↨ Motor Activity | [199] | |
| WAG/R | 6 mg/kg (subchronic = 3 times a week for 2 weeks) | s.c. | ↑ Seizure Length (>11 s) ↨ Motor Activity | [199] | |
| WAG/R | 0.1–0.3–1–2 μg/2 μL | i.c.v. | ↓ Seizure Incidence ↓ Seizure Total Time | [200] | |
| WAG/R | 0.1–0.3–1 μL/0.5 μL | NRT/VB/S1po Bilateral Brain Infusion | ↓ Seizure Incidence ↓ Seizure Total Time | [200] | |
| WAG/Rij | CB1R antagonist AM251 | 6–12 mg/kg | s.c. | ↨ Seizures Incidence | [198] |
| C57BL/6N Mice | CP55940 + AM251 | 0.3 mg/kg = 3 mg/kg | i.p. | ↨ High Voltage Spindles Incidence (HVSs) | [197] |
| WAG/Rij | WIN 55,212-2 +AM251 | WIN 6 mg/kg + AM 12 mg | s.c. | ↨ Seizures Incidence in the First 3 h ↑ Seizure Incidence in 4th and 5th h ↨ Last 2 h Seizure Length | [198] |
| GAERS (♂) | GAT211 (Ago-PAM) | 3, 10 mg/kg | i.p. | ↓ Seizure Incidence ↓ Total Time Spent in Seizures (~40%); ↨ Seizure Length ↨ Seizure Frequency | [201] |
| GAERS (♂♀) | GAT229 (AGO-PAM) | 1, 3, 10 mg/kg | i.p. | ↓ Seizure Incidence ↓ Total Time Spent in Seizures (~40%); ↨ Seizure Length ↨ Seizure Frequency ↨ Motor Activity (Open-Field Test) | [201] |
| GAERS (♂) | 125, 250, 500, 1000 μM | Cortical (motor Cx) infusion | ↓ Seizure Incidence ↓ Total Time Spent in Seizures ↨ Seizure length ↨ Seizure Frequency | [201] | |
| GAERS (♂) | SR141716A (CB1R antagonist) | 3 mg/kg | i.p. | ↨ Seizure Incidence ↨ Total Time Spent in Seizures | [201] |
| WAG/Rij | 0.5, 1, and 2 μg/2 μL | i.c.v. | ↑ Seizure Incidence ↑ Total Time Spent in Seizures (>40% Increase); | [202] | |
| WAG/Rij | 0.5–1–2.5 μg/0.5 μL | VB Bilateral Brain Infusion | ↑ Seizure Incidence (>50% Increase); ↑ Total Time Spent in Seizures | [200] | |
| WAG/Rij | 0.5–1–2.5 μg/0.5 μL | NRT/S1po Bilateral Brain Infusion | ↨ Seizure Incidence (>50% Increase); ↨ Total Time Spent in Seizures | [200] | |
| GAERS (♂) | GAT229 + SR141716A | 1000 μM 3 mg/kg | Cortical infusion i.p. | ↨ Seizure Incidence ↨ Total Time Spent in Seizures | [201] |
| WAG/Rij | N-palmitoylethanolamine (PEA) + SR141716 | 40 mg/kg + 0.5 μg/2 μL | i.p. + i.c.v. | ↨ Seizure Incidence ↨ Total Time Spent in Seizures | [202] |
| WAG/Rij | GW6471 (PPAR-α antagonist) | 1, 2 μg/2 μL | i.c.v. | ↨ Seizure Incidence ↨ Total Time Spent in Seizures | [202] |
| WAG/Rij | PEA + GW6471 | 3 μg/2 μL + 2 μg/2 μL | i.c.v. + i.c.v. | ↨ Seizure Incidence ↨ Total Time Spent in Seizures | [202] |
| GAERS (♂) | GAT591 (AGO-PAM) | 1, 3, 10 mg/kg | i.p. | ↨ Seizure Incidence ↓ Total Time Spent in Seizures ↨ Seizure Length Seizure Frequency | [203] |
| GAERS (♂) | GAT593 (AGO-PAM) | 1, 3, 10 mg/kg | i.p. | ↨ Seizure Incidence ↓ Total Time Spent in Seizures ↨ Seizure Length ↨ Seizure Frequency | [203] |
| WAG/Rij | PEA | 0.5, 1,3 and 10 μg/2 μL | i.c.v. | ↑ Seizure Incidence ↓ Total Time Spent in Seizures | [202] |
| 10, 20, 40, 60 mg/kg | i.p. | ||||
| WAG/Rij | anandamide (N-arachidonylethanolamine, AEA) | 1, 3 and 10 μg/2 μL | i.c.v. | ↑ Seizure Incidence ↓ Total Time Spent in Seizures | [202] |
| WAG/Rij | AEA + SR141716 | 3 μg/2 μL + 0.5 μg/2 μL | i.c.v. + i.c.v. | ↨ Seizure Incidence ↨ Total Time Spent in Seizures | [202] |
| WAG/Rij | AEA + GW6471 | 3 μg/2 μL + 2 μg/2 μL | i.c.v. + i.c.v. | ↑ Seizure Incidence ↓ Total Time Spent in Seizures | [202] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montebello, G.; Di Giovanni, G. Dysregulation of the Cannabinoid System in Childhood Epilepsy: From Mechanisms to Therapy. Int. J. Mol. Sci. 2025, 26, 6234. https://doi.org/10.3390/ijms26136234
Montebello G, Di Giovanni G. Dysregulation of the Cannabinoid System in Childhood Epilepsy: From Mechanisms to Therapy. International Journal of Molecular Sciences. 2025; 26(13):6234. https://doi.org/10.3390/ijms26136234
Chicago/Turabian StyleMontebello, Gloria, and Giuseppe Di Giovanni. 2025. "Dysregulation of the Cannabinoid System in Childhood Epilepsy: From Mechanisms to Therapy" International Journal of Molecular Sciences 26, no. 13: 6234. https://doi.org/10.3390/ijms26136234
APA StyleMontebello, G., & Di Giovanni, G. (2025). Dysregulation of the Cannabinoid System in Childhood Epilepsy: From Mechanisms to Therapy. International Journal of Molecular Sciences, 26(13), 6234. https://doi.org/10.3390/ijms26136234