
BST2 and DIRAS3 Drive Immune Evasion and Tumor Progression in High-Grade Glioma

Abstract
1. Introduction
2. Results
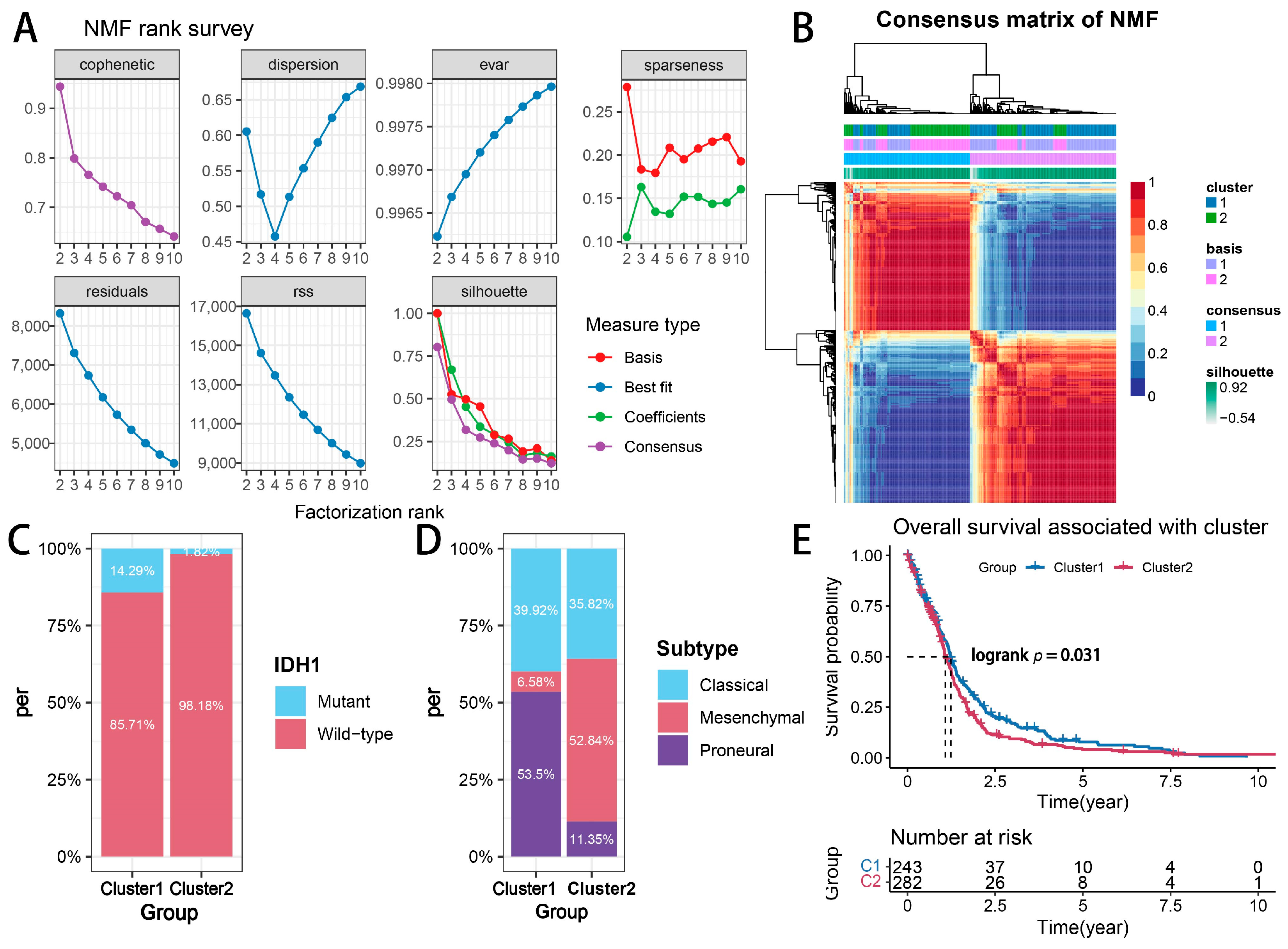
2.1. Glioma Subtypes Based on Transcriptional Signatures Associated with Cytotoxic T-Lymphocyte-Mediated Immune Escape
2.2. Biological Function Analysis of Glioma Subtypes Mediated by Cytotoxic T-Lymphocyte Immune Evasion Dynamics

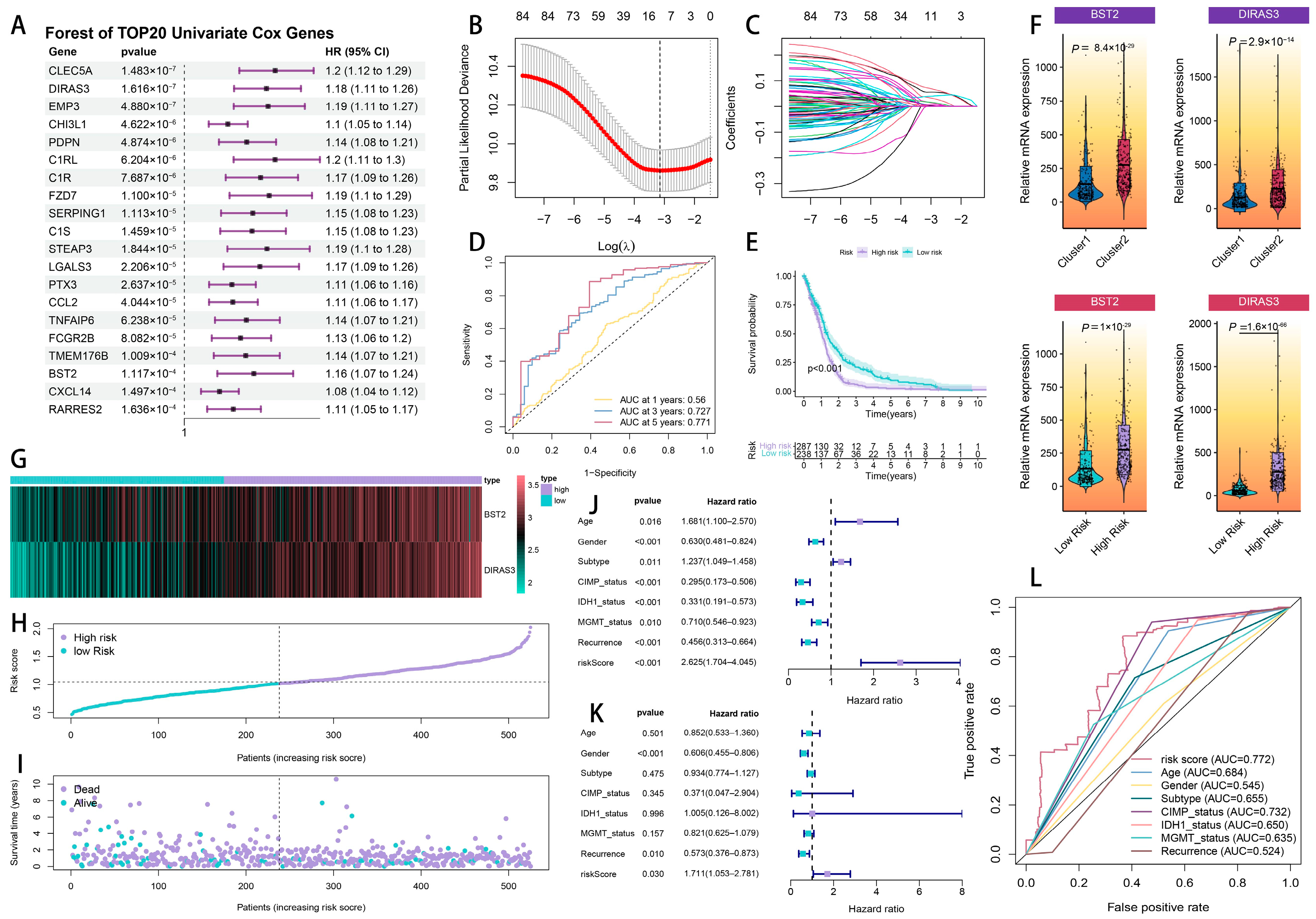
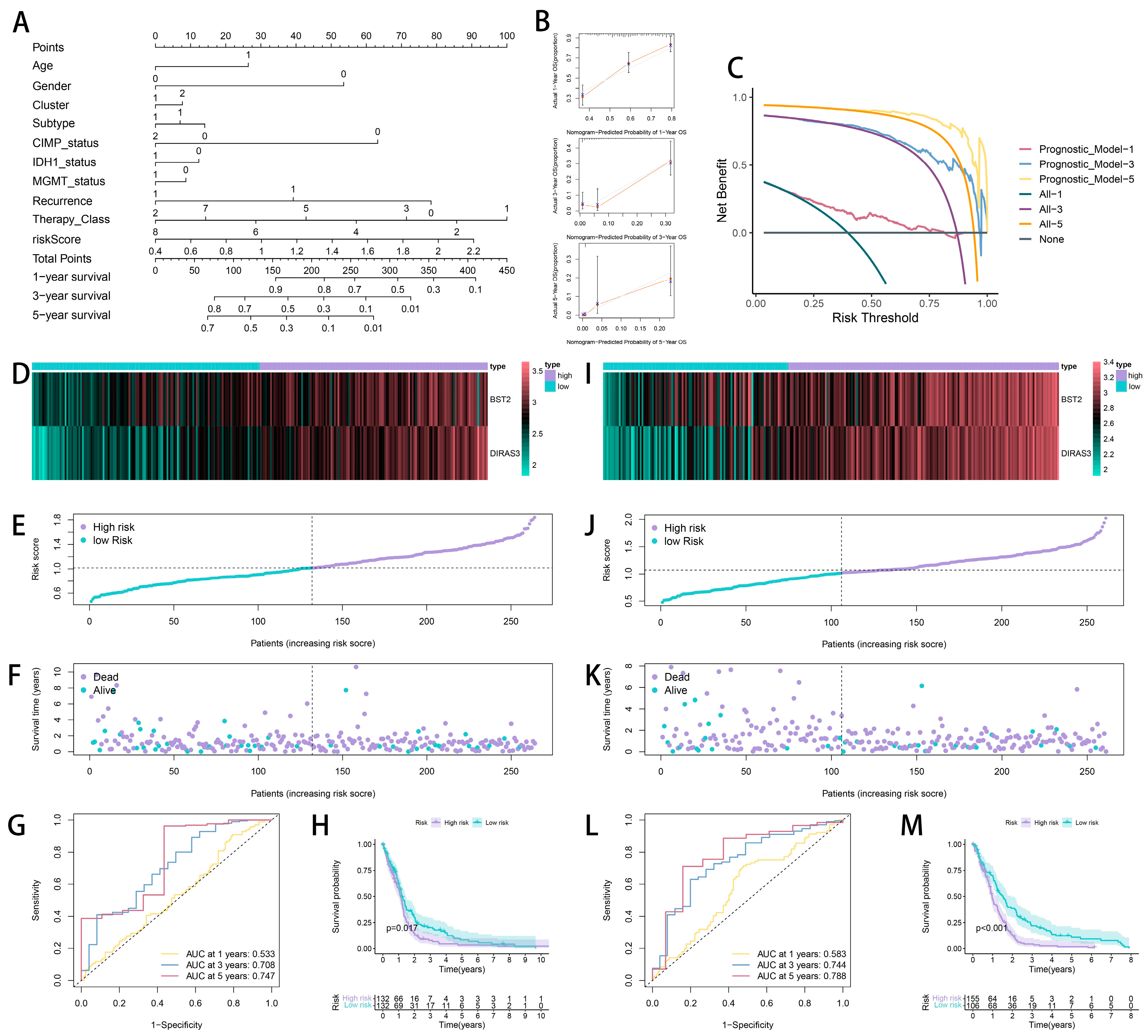
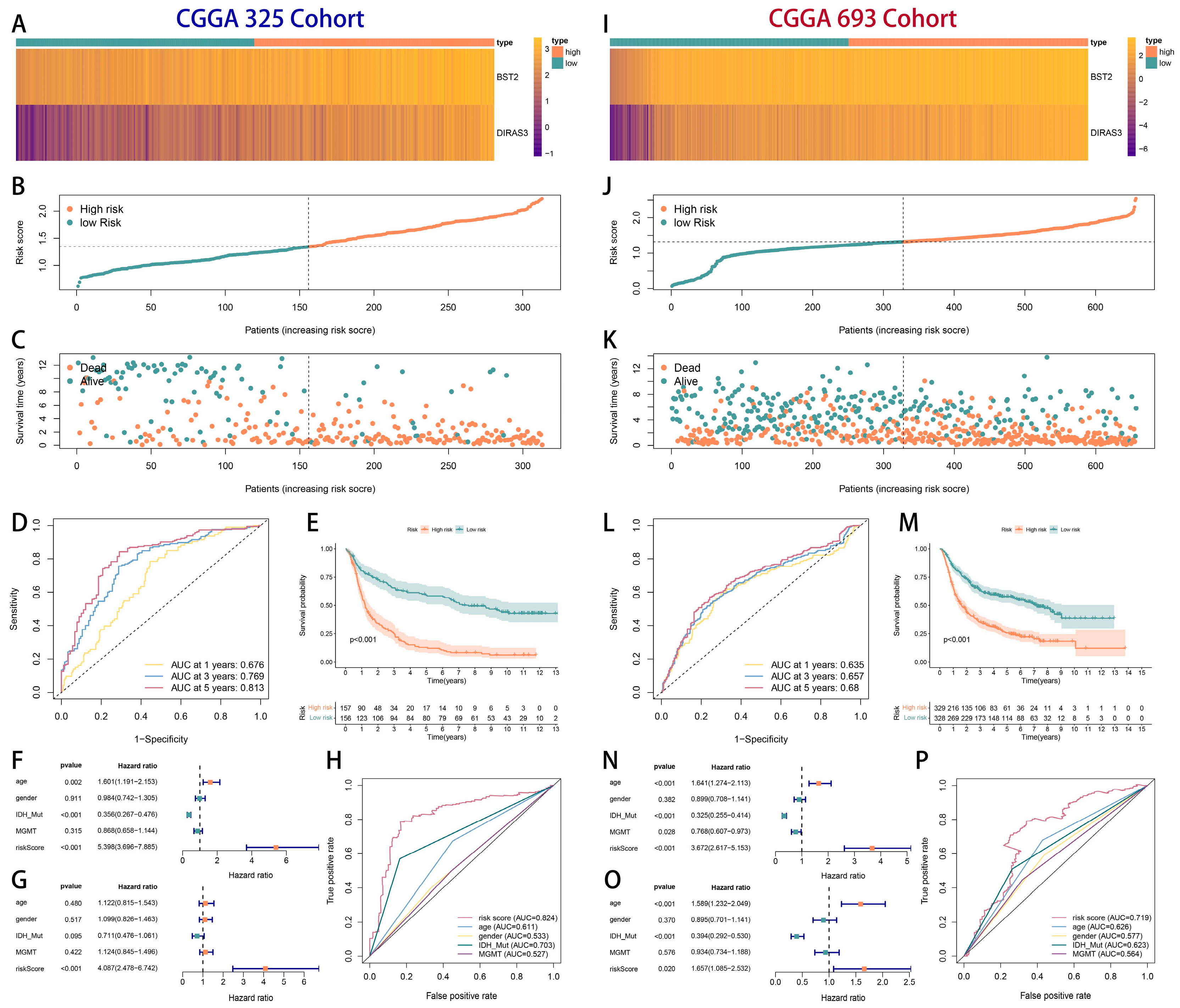
2.3. The Development and Verification of a Prognostic Model Associated with Cytotoxic T-Lymphocyte Immune Evasion in Glioma
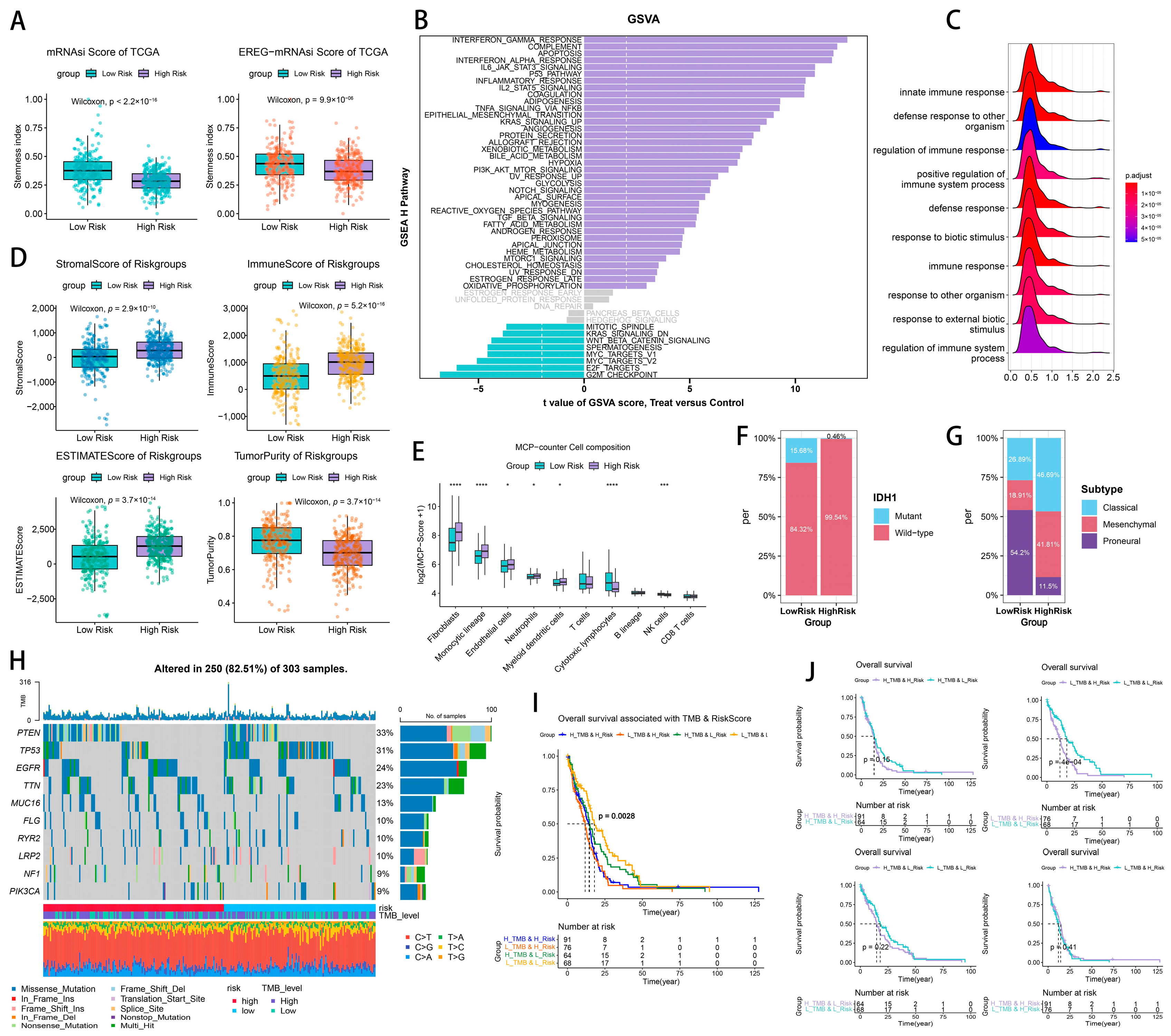
2.4. Tumor Heterogeneity and Microenvironment Landscape of CTLE-Associated Prognostic Models in Glioma
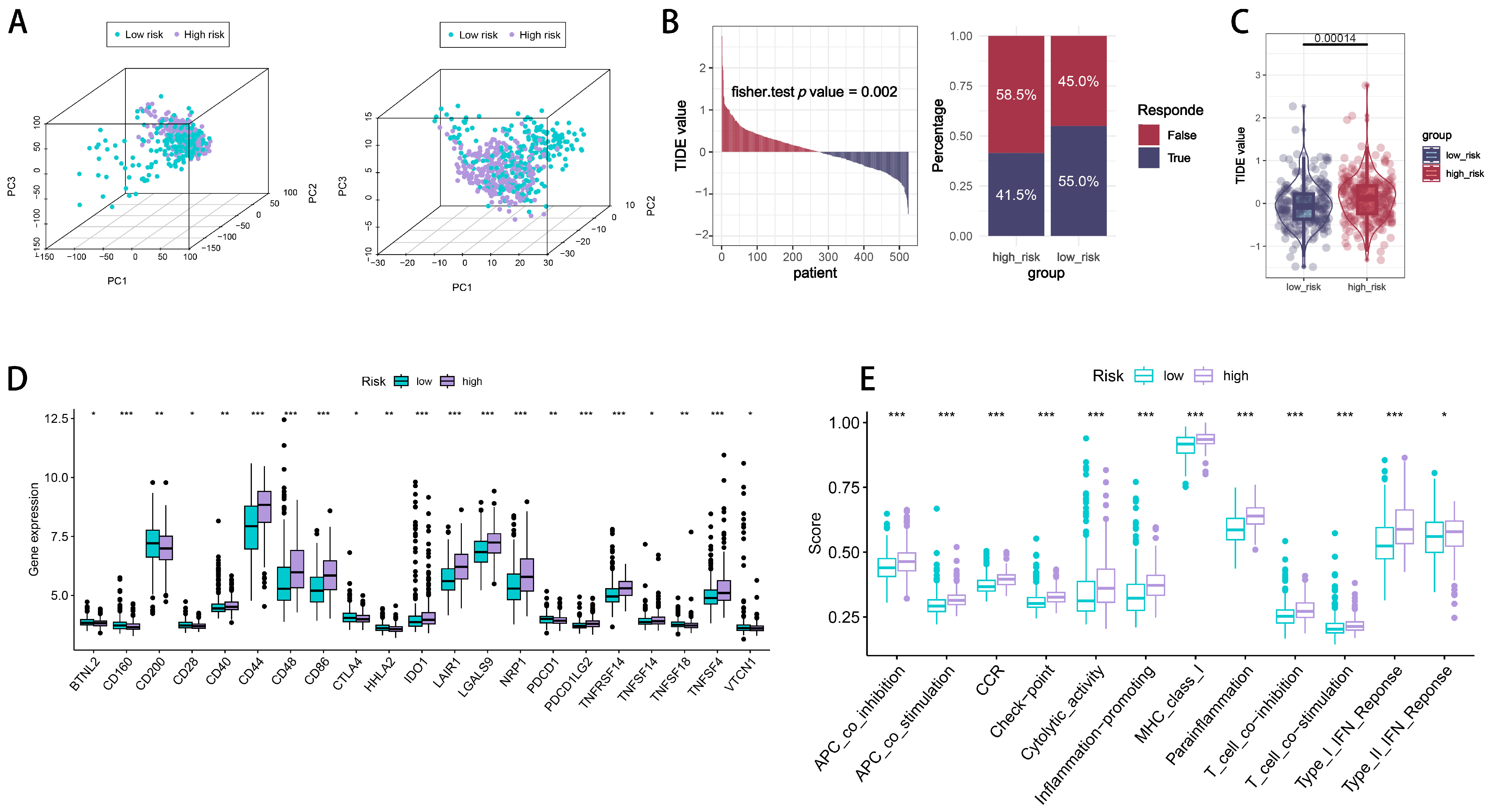
2.5. Immune Checkpoint Signatures of CTLE-Associated Prognostic Models in Glioma
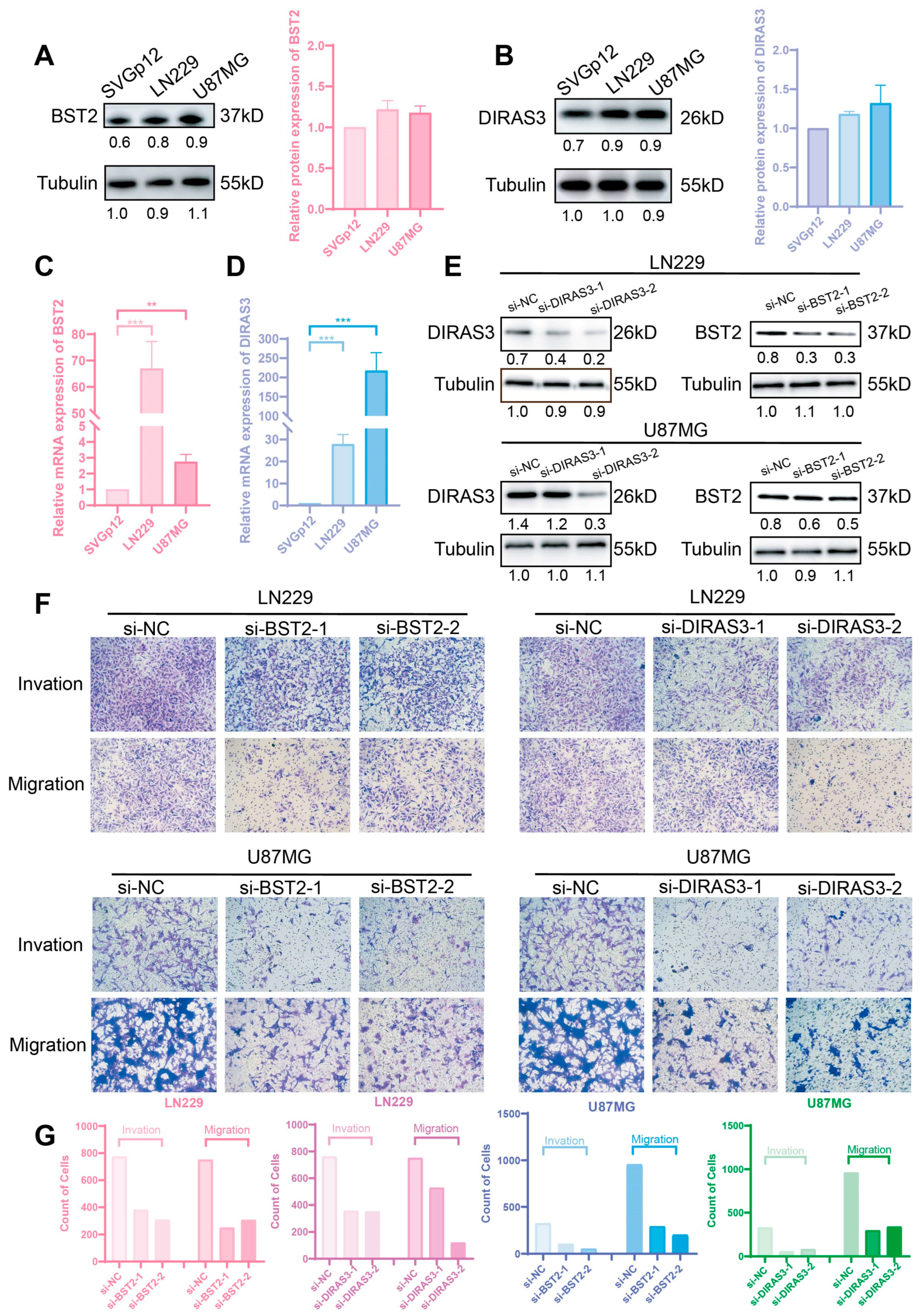
2.6. BST2/DIRAS3 Knockdown Suppresses Glioma Invasion and Migration
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Data Preprocessing
4.3. Non-Negative Matrix Factorization (NMF)
4.4. Differential Expression Analysis
4.5. Biological Function and Pathway Analysis
4.6. Immune Infiltration Analysis
4.7. Prognostic Model Construction
- Univariate Cox Regression: Applied to the 238 DEGs using the survival package (version 3.5-5), identifying 85 genes with a potential prognostic value.
- LASSO Regression: Conducted using the glmnet package (version 4.1-8) to reduce variables to 11 prognostic DEGs, with the optimal penalty parameter (λ) selected at log(λ) = −3.2.
- Multivariate Cox Regression: Identified BST2 and DIRAS3 as independent risk factors, constructing a risk score (RS) formula: RS = (0.124 × BST2 expression) + (0.148 × DIRAS3 expression).
4.8. Tumor Heterogeneity and Microenvironment Analysis
4.9. Principal Component Analysis
4.10. Drug Sensitivity and Immune Checkpoint Analysis
4.11. Statistical Analysis
4.12. Software and Packages
- NMF (version 0.28): For NMF clustering.
- limma (version 3.54.0): For differential expression analysis.
- survival (version 3.5-5): For survival analysis and Cox regression.
- glmnet (version 4.1-8): For LASSO regression.
- timeROC (version 0.4): For time-dependent ROC analysis.
- clusterProfiler (version 4.6.2): For GO enrichment analysis.
- GSVA (version 1.46.0): For GSVA and ssGSEA.
- MCPcounter (version 1.2.0, GitHub): For immune cell infiltration estimation.
- estimate (version 1.0.13): For stromal, immune, and tumor purity scores.
- maftools (version 2.14.0): For somatic mutation analysis.
- oncoPredict (version 0.2, GitHub): For drug sensitivity prediction.
- ComplexHeatmap (version 2.14.0): For heatmap visualization.
- pheatmap (version 1.0.12): For heatmap visualization.
- rms (version 8.0-0): For nomogram construction.
- rmda (version 1.5): For decision curve analysis.
- factoextra (version 1.0.7): For PCA visualization.
4.13. Ethical Considerations
4.14. Cell Culture and Treatment
4.15. siRNAs and Transfection
4.16. RNA Extraction and qRT-PCR
4.17. Western Blotting (WB) and Antibodies
4.18. Transwell Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Horbinski, C.; Berger, T.; Packer, R.J.; Wen, P.Y. Clinical implications of the 2021 edition of the WHO classification of central nervous system tumours. Nat. Rev. Neurol. 2022, 18, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2024, 10, 33. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Pouyan, A.; Ghorbanlo, M.; Eslami, M.; Jahanshahi, M.; Ziaei, E.; Salami, A.; Mokhtari, K.; Shahpasand, K.; Farahani, N.; Meybodi, T.E.; et al. Glioblastoma multiforme: Insights into pathogenesis, key signaling pathways, and therapeutic strategies. Mol. Cancer 2025, 24, 58. [Google Scholar] [CrossRef]
- Wang, L.M.; Englander, Z.K.; Miller, M.L.; Bruce, J.N. Malignant Glioma. Adv. Exp. Med. Biol. 2023, 1405, 1–30. [Google Scholar] [PubMed]
- Richardson, T.E.; Walker, J.M.; Hambardzumyan, D.; Brem, S.; Hatanpaa, K.J.; Viapiano, M.S.; Pai, B.; Umphlett, M.; Becher, O.J.; Snuderl, M.; et al. Genetic and epigenetic instability as an underlying driver of progression and aggressive behavior in IDH-mutant astrocytoma. Acta Neuropathol. 2024, 148, 5. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Dmello, C.; McGrail, D.J.; Brat, D.J.; Lee-Chang, C.; Heimberger, A.B.; Chand, D.; Stupp, R.; Sonabend, A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Investig. 2023, 133, e163447. [Google Scholar] [CrossRef]
- Yasinjan, F.; Xing, Y.; Geng, H.; Guo, R.; Yang, L.; Liu, Z.; Wang, H. Immunotherapy: A promising approach for glioma treatment. Front. Immunol. 2023, 14, 1255611. [Google Scholar] [CrossRef]
- Watowich, M.B.; Gilbert, M.R.; Larion, M. T cell exhaustion in malignant gliomas. Trends Cancer 2023, 9, 270–292. [Google Scholar] [CrossRef]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, M.; Glorius, P.; Burger, R.; Kellner, C.; Klausz, K.; Gunther, A.; Repp, R.; Klapper, W.; Gramatzki, M.; Peipp, M. The novel immunotoxin HM1.24-ETA’ induces apoptosis in multiple myeloma cells. Blood Cancer J. 2014, 4, e219. [Google Scholar] [CrossRef] [PubMed]
- Mahauad-Fernandez, W.D.; Naushad, W.; Panzner, T.D.; Bashir, A.; Lal, G.; Okeoma, C.M. BST-2 promotes survival in circulation and pulmonary metastatic seeding of breast cancer cells. Sci. Rep. 2018, 8, 17608. [Google Scholar] [CrossRef]
- Casarrubios, M.; Provencio, M.; Nadal, E.; Insa, A.; Del Rosario Garcia-Campelo, M.; Lazaro-Quintela, M.; Domine, M.; Majem, M.; Rodriguez-Abreu, D.; Martinez-Marti, A.; et al. Tumor microenvironment gene expression profiles associated to complete pathological response and disease progression in resectable NSCLC patients treated with neoadjuvant chemoimmunotherapy. J. Immunother. Cancer 2022, 10, e005320. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Chen, H.; Zhong, X.; Wang, Y.; Hu, Z.; Huang, H.; Zhao, S.; Wei, P.; Shi, D.; Li, D. BST2 induced macrophage M2 polarization to promote the progression of colorectal cancer. Int. J. Biol. Sci. 2023, 19, 331–345. [Google Scholar] [CrossRef]
- Sutton, M.N.; Lu, Z.; Li, Y.C.; Zhou, Y.; Huang, T.; Reger, A.S.; Hurwitz, A.M.; Palzkill, T.; Logsdon, C.; Liang, X.; et al. DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting RAS Clusters. Cell Rep. 2019, 29, 3448–3459.e6. [Google Scholar] [CrossRef]
- Bildik, G.; Liang, X.; Sutton, M.N.; Bast, R.C., Jr.; Lu, Z. DIRAS3: An Imprinted Tumor Suppressor Gene that Regulates RAS and PI3K-driven Cancer Growth, Motility, Autophagy, and Tumor Dormancy. Mol. Cancer Ther. 2022, 21, 25–37. [Google Scholar] [CrossRef]
- Bildik, G.; Gray, J.P.; Mao, W.; Yang, H.; Ozyurt, R.; Orellana, V.R.; De Wever, O.; Carey, M.S.; Bast, R.C.; Lu, Z. DIRAS3 induces autophagy and enhances sensitivity to anti-autophagic therapy in KRAS-driven pancreatic and ovarian carcinomas. Autophagy 2024, 20, 675–691. [Google Scholar] [CrossRef]
- Lawson, K.A.; Sousa, C.M.; Zhang, X.; Kim, E.; Akthar, R.; Caumanns, J.J.; Yao, Y.; Mikolajewicz, N.; Ross, C.; Brown, K.R.; et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 2020, 586, 120–126. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2018, 33, 42–56.e6. [Google Scholar] [CrossRef]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Warren, S.; Dennis, L.; D’Amico, L.; White, A.; Disis, M.L.; Geller, M.A.; Odunsi, K.; Beechem, J.; Fling, S.P. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Geurts, M.; French, P.J.; Smits, M.; Capper, D.; Bromberg, J.E.C.; Chang, S.M. Primary brain tumours in adults. Lancet 2023, 402, 1564–1579. [Google Scholar] [CrossRef]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Sosnovtseva, A.O.; Valikhov, M.P.; Chernysheva, A.A.; Abramova, O.V.; Pavlov, K.A.; Chekhonin, V.P. Systemic and local immunosuppression in glioblastoma and its prognostic significance. Front. Immunol. 2024, 15, 1326753. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, D.; Jiang, H.; Ye, J.; Zhang, L.; Bagley, S.J.; Winkler, J.; Gong, Y.; Fan, Y. Small-molecule toosendanin reverses macrophage-mediated immunosuppression to overcome glioblastoma resistance to immunotherapy. Sci. Transl. Med. 2023, 15, eabq3558. [Google Scholar] [CrossRef]
- Geraldo, L.H.; Xu, Y.; Jacob, L.; Pibouin-Fragner, L.; Rao, R.; Maissa, N.; Verreault, M.; Lemaire, N.; Knosp, C.; Lesaffre, C.; et al. SLIT2/ROBO signaling in tumor-associated microglia and macrophages drives glioblastoma immunosuppression and vascular dysmorphia. J. Clin. Investig. 2021, 131, e141083. [Google Scholar] [CrossRef]
- Khan, F.; Pang, L.; Dunterman, M.; Lesniak, M.S.; Heimberger, A.B.; Chen, P. Macrophages and microglia in glioblastoma: Heterogeneity, plasticity, and therapy. J. Clin. Investig. 2023, 133, e163446. [Google Scholar] [CrossRef] [PubMed]
- van Hooren, L.; Handgraaf, S.M.; Kloosterman, D.J.; Karimi, E.; van Mil, L.; Gassama, A.A.; Solsona, B.G.; de Groot, M.H.P.; Brandsma, D.; Quail, D.F.; et al. CD103(+) regulatory T cells underlie resistance to radio-immunotherapy and impair CD8(+) T cell activation in glioblastoma. Nat. Cancer 2023, 4, 665–681. [Google Scholar] [CrossRef]
- Read, R.D.; Tapp, Z.M.; Rajappa, P.; Hambardzumyan, D. Glioblastoma microenvironment-from biology to therapy. Genes Dev. 2024, 38, 360–379. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Bian, Q.; Wang, X.; Wang, X.; Lai, L.; Wu, Z.; Zhao, Z.; Ban, B. Bone marrow stromal cell antigen 2: Tumor biology, signaling pathway and therapeutic targeting (Review). Oncol. Rep. 2024, 51, 45. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; DeMali, K.A.; Olivier, A.K.; Okeoma, C.M. Bone marrow stromal antigen 2 expressed in cancer cells promotes mammary tumor growth and metastasis. Breast Cancer Res. 2014, 16, 493. [Google Scholar] [CrossRef]
- Zheng, C.; Wang, J.; Zhou, Y.; Duan, Y.; Zheng, R.; Xie, Y.; Wei, X.; Wu, J.; Shen, H.; Ye, M.; et al. IFNalpha-induced BST2+ tumor-associated macrophages facilitate immunosuppression and tumor growth in pancreatic cancer by ERK-CXCL7 signaling. Cell Rep. 2024, 43, 114088. [Google Scholar] [CrossRef] [PubMed]
- Shan, F.; Shen, S.; Wang, X.; Chen, G. BST2 regulated by the transcription factor STAT1 can promote metastasis, invasion and proliferation of oral squamous cell carcinoma via the AKT/ERK1/2 signaling pathway. Int. J. Oncol. 2023, 62, 54. [Google Scholar] [CrossRef]
- Xia, H.; Green, D.R.; Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 2021, 21, 281–297. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Lu, Z.; Baquero, M.T.; Yang, H.; Yang, M.; Reger, A.S.; Kim, C.; Levine, D.A.; Clarke, C.H.; Liao, W.S.; Bast, R.C., Jr. DIRAS3 regulates the autophagosome initiation complex in dormant ovarian cancer cells. Autophagy 2014, 10, 1071–1092. [Google Scholar] [CrossRef]
- Mahgoub, E.; Taneera, J.; Sulaiman, N.; Saber-Ayad, M. The role of autophagy in colorectal cancer: Impact on pathogenesis and implications in therapy. Front. Med. 2022, 9, 959348. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; Gestal-Mato, U.; Eapen, V.V.; Macinkovic, I.; Bailey, H.J.; Prieto-Garcia, C.; Misra, M.; Jacomin, A.C.; Ammanath, A.V.; Bagaric, I.; et al. IRGQ-mediated autophagy in MHC class I quality control promotes tumor immune evasion. Cell 2024, 187, 7285–7302.e29. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | |||||
|---|---|---|---|---|---|
| Characteristic | CGGA_325, N = 325 1 | CGGA_693, N = 693 1 | TCGA-Test, N = 261 1 | TCGA-Train, N = 264 1 | p-Value 2 |
| Survival | |||||
| Mean (SD) | 3.98 (4.03) | 3.29 (2.74) | 16.78 (17.00) | 16.60 (18.50) | |
| Median (IQR) | 1.93 (0.79, 7.04) | 2.37 (0.95, 5.29) | 12.20 (5.50, 20.70) | 12.20 (6.00, 19.35) | |
| Range | 0.05, 13.18 | 0.07, 13.78 | 0.10, 94.80 | 0.10, 127.60 | |
| Missing | 12 | 36 | 0 | 0 | |
| Status | 220 (70%) | 397 (60%) | 227 (87%) | 220 (83%) | <0.001 |
| Missing | 9 | 30 | 0 | 0 | |
| Patient age | |||||
| Mean (SD) | 42.94 (11.95) | 43.28 (12.39) | 57.36 (14.34) | 59.00 (14.53) | |
| Median (IQR) | 42.00 (36.00, 51.00) | 43.00 (34.00, 51.00) | 58.30 (49.13, 67.98) | 60.30 (50.30, 69.40) | |
| Range | 8.00, 79.00 | 11.00, 76.00 | 10.90, 86.60 | 14.50, 89.30 | |
| Missing | 0 | 1 | 3 | 3 | |
| Gender | 0.411 | ||||
| Female | 122 (38%) | 295 (43%) | 103 (40%) | 100 (38%) | |
| Male | 203 (62%) | 398 (57%) | 154 (60%) | 160 (62%) | |
| Missing | 0 | 0 | 4 | 4 | |
| IDH1_status | <0.001 | ||||
| Mutant | 175 (54%) | 356 (55%) | 16 (8.4%) | 14 (6.6%) | |
| Wildtype | 149 (46%) | 286 (45%) | 174 (92%) | 198 (93%) | |
| Missing | 1 | 51 | 71 | 52 | |
| MGMT_status | 0.031 | ||||
| Methylated | 157 (51%) | 315 (58%) | 90 (51%) | 80 (47%) | |
| Unmethylated | 149 (49%) | 227 (42%) | 85 (49%) | 92 (53%) | |
| Missing | 19 | 151 | 86 | 92 | |
| Recurrence | <0.001 | ||||
| Primary | 229 (71%) | 422 (61%) | 248 (96%) | 249 (95%) | |
| Recurrent | 59 (18%) | 271 (39%) | 7 (2.7%) | 9 (3.4%) | |
| Secondary | 33 (10%) | 0 (0%) | 4 (1.5%) | 3 (1.1%) | |
| Missing | 4 | 0 | 2 | 3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Z.; Wu, S.; Shi, Z.; Chen, D.; Chen, J.; Zhang, H. BST2 and DIRAS3 Drive Immune Evasion and Tumor Progression in High-Grade Glioma. Int. J. Mol. Sci. 2025, 26, 6205. https://doi.org/10.3390/ijms26136205
Liao Z, Wu S, Shi Z, Chen D, Chen J, Zhang H. BST2 and DIRAS3 Drive Immune Evasion and Tumor Progression in High-Grade Glioma. International Journal of Molecular Sciences. 2025; 26(13):6205. https://doi.org/10.3390/ijms26136205
Chicago/Turabian StyleLiao, Zhangjun, Shuyi Wu, Zhenyi Shi, Donghui Chen, Jinrui Chen, and Hua Zhang. 2025. "BST2 and DIRAS3 Drive Immune Evasion and Tumor Progression in High-Grade Glioma" International Journal of Molecular Sciences 26, no. 13: 6205. https://doi.org/10.3390/ijms26136205
APA StyleLiao, Z., Wu, S., Shi, Z., Chen, D., Chen, J., & Zhang, H. (2025). BST2 and DIRAS3 Drive Immune Evasion and Tumor Progression in High-Grade Glioma. International Journal of Molecular Sciences, 26(13), 6205. https://doi.org/10.3390/ijms26136205