
Transcriptomic Profiling of Lesional and Perilesional Skin in Atopic Dermatitis Suggests Barrier Dysfunction, Inflammatory Activation, and Alterations to Vitamin D Metabolism

 ,
, 
 , , ,
, , ,
Abstract

1. Introduction
2. Results
2.1. Patient Characteristics
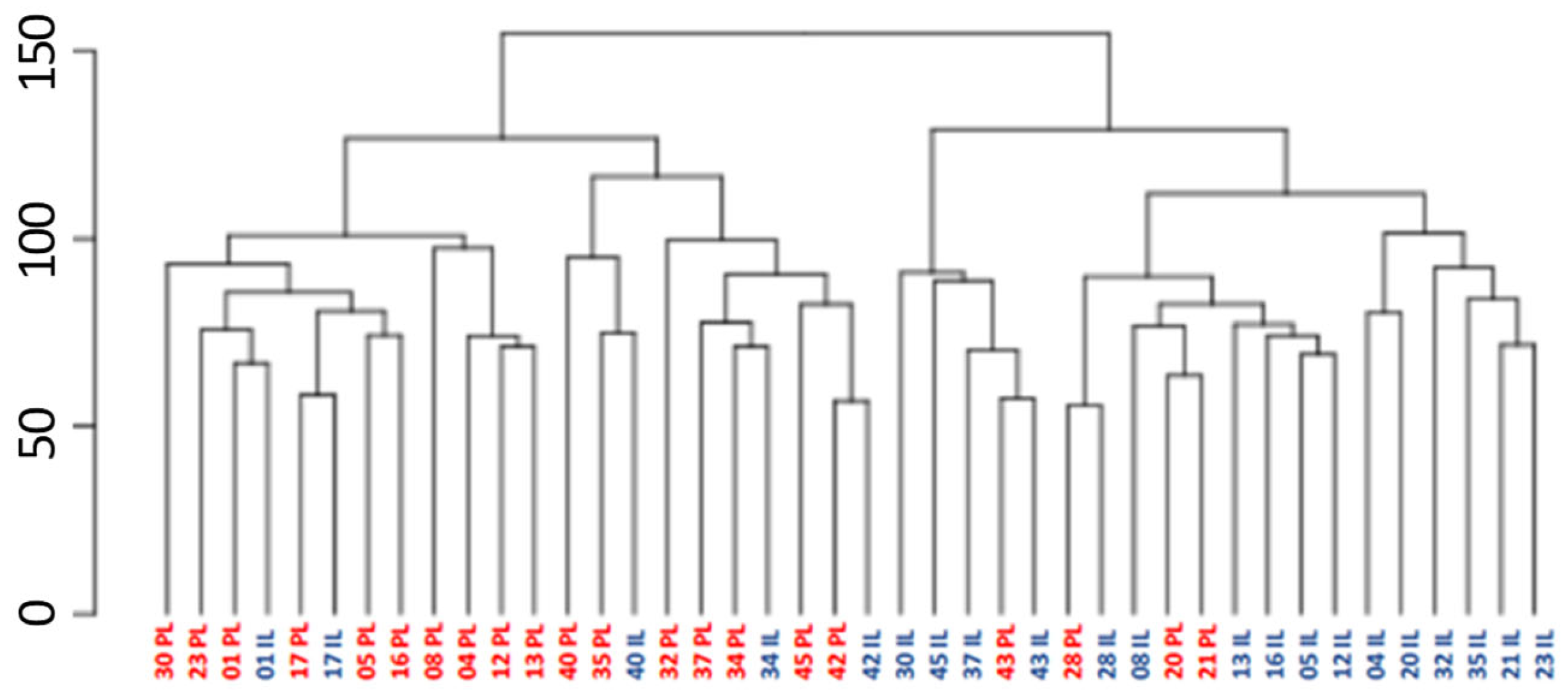
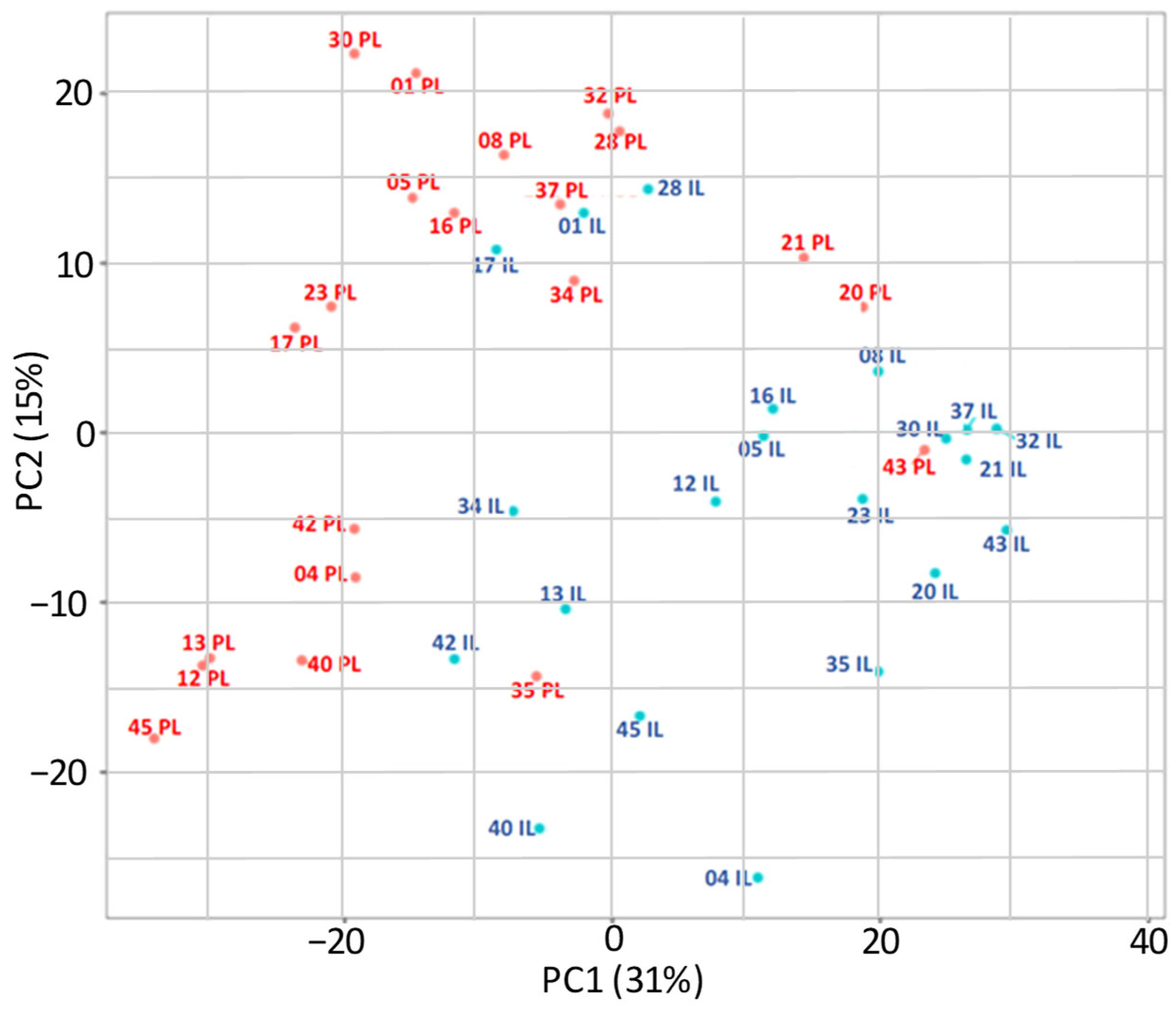
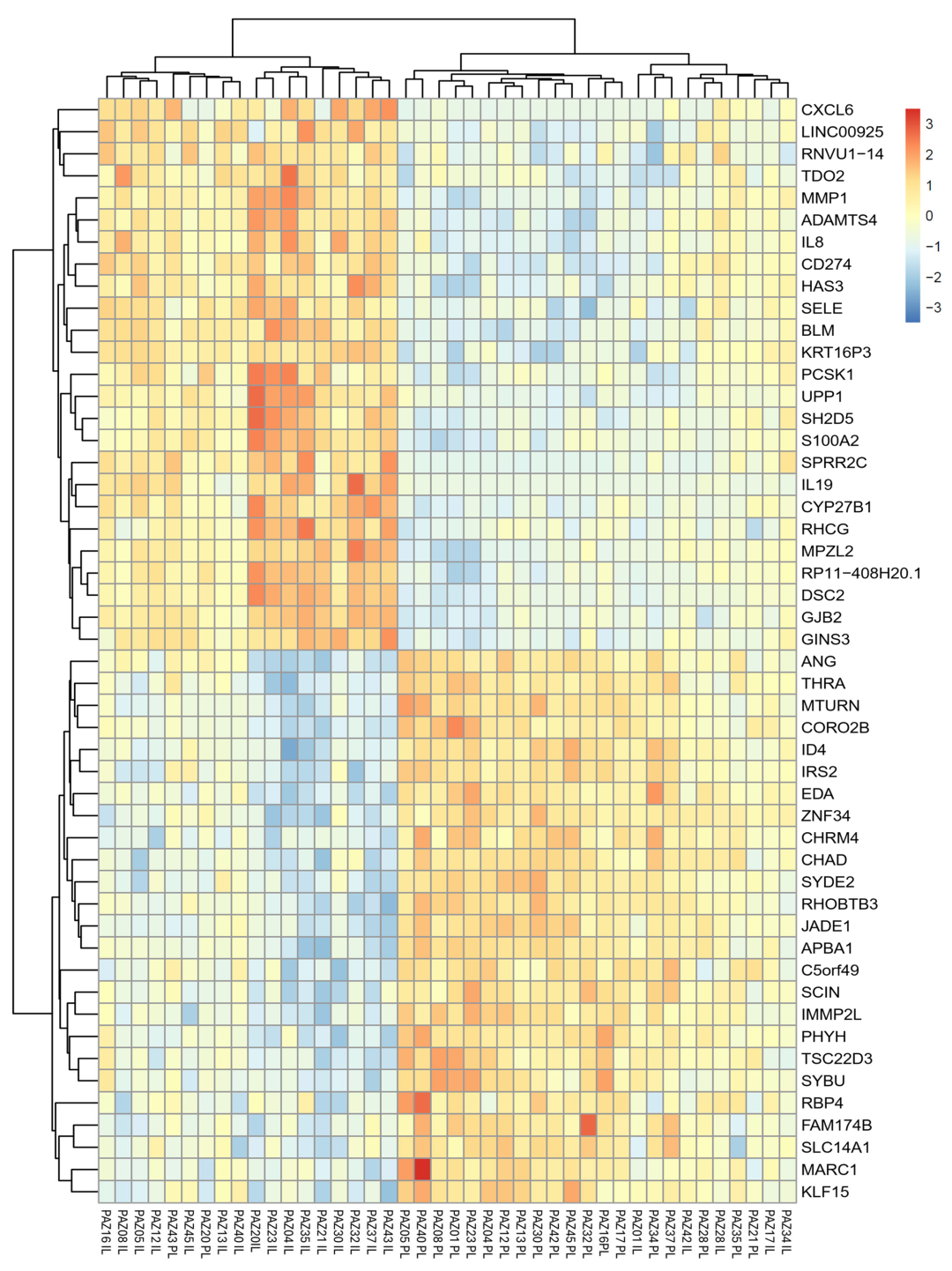
2.2. Differential Gene Expression in Lesional and Perilesional Skin Biopses
2.3. Expression Changes in Atopic Dermatitis-Related Genes
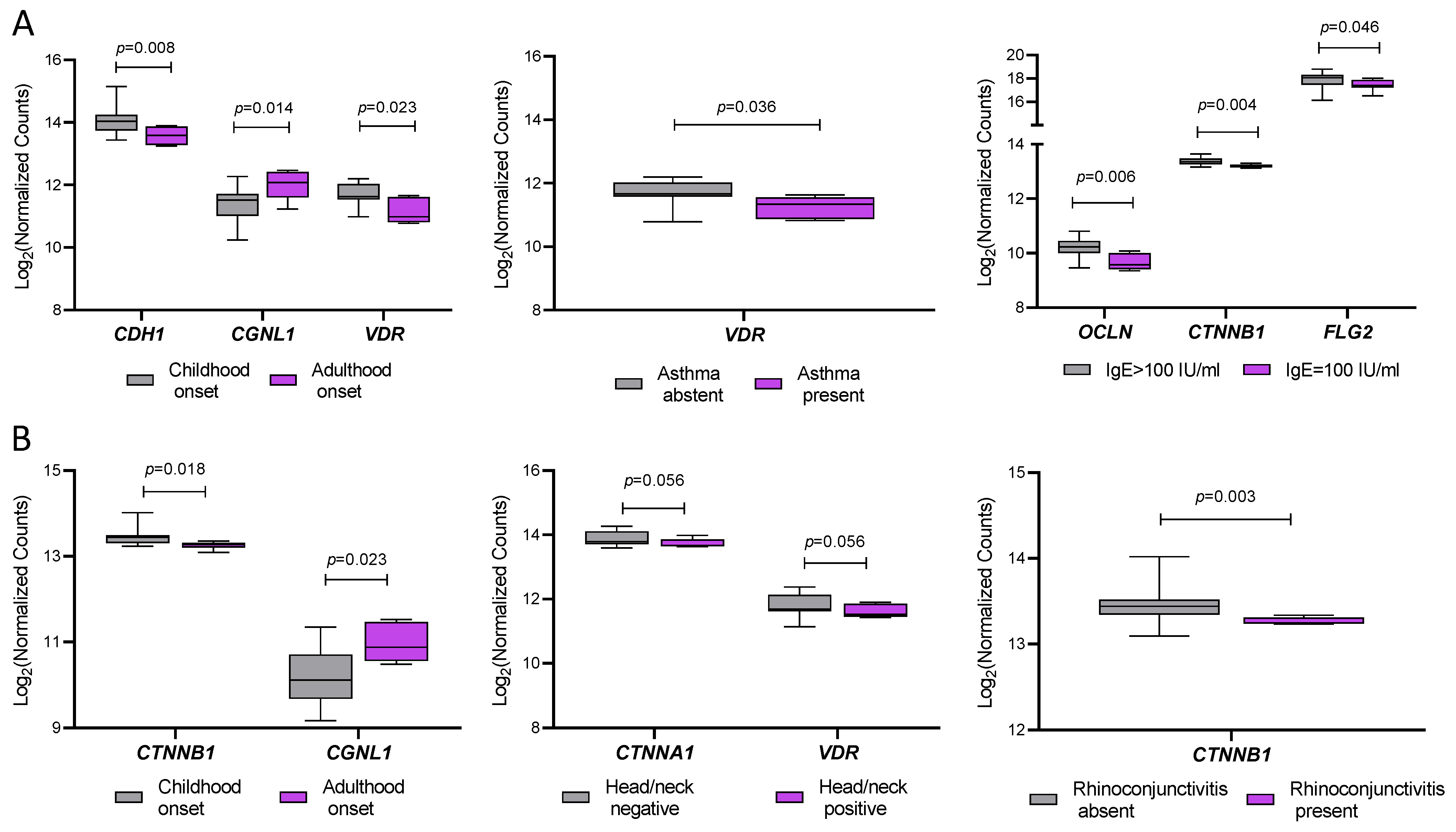
2.4. Association Between Gene Expression and Clinical Features of AD Patients
2.5. In Terminal Keratinocyte Differentiation and Epidermal Barrier Function (Table 3)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FDR | p-Value | Pathway Name |
|---|---|---|
| 6 × 10−4 | 1 × 10−6 | Keratinization |
| 6 × 10−4 | 3 × 10−6 | Interleukin-4 and Interleukin-13 signaling |
| 6.24 × 10−3 | 7.2 × 10−5 | Antimicrobial peptides |
| 9.23 × 10−3 | 1.04 × 10−4 | Chemokine receptors bind chemokines |
| 1.06 × 10−2 | 1.43 × 10−4 | Formation of the cornified envelope |
| 7.73 × 10−2 | 1.23 × 10−3 | Calcitonin-like ligand receptors |
| 8.62 × 10−2 | 1.57 × 10−3 | Neutrophil degranulation |
| 1.78 × 10−1 | 3.63 × 10−3 | Regulation of TLR by endogenous ligand |
| 2.36 × 10−1 | 5.90 × 10−3 | NR1H2 and NR1H3 regulate gene expression linked to triglyceride lipolysis in adipose |
| 2.36 × 10−1 | 5.90 × 10−3 | Muscarinic acetylcholine receptors |
| 2.78 × 10−1 | 7.51 × 10−3 | Activation of matrix metalloproteinases |
| 2.96 × 10−1 | 9.38 × 10−3 | Collagen degradation |
| 2.96 × 10−1 | 1.00 × 10−2 | Interleukin-10 signaling |
| 2.96 × 10−1 | 1.02 × 10−2 | Defective SFTP2A causes IPF |
| 3.26 × 10−1 | 1.21 × 10−2 | TRKA activation by NGF |
| 4.01 × 10−1 | 1.61 × 10−2 | Beta defensins |
| 4.01 × 10−1 | 1.67 × 10−2 | Hormone ligand-binding receptors |
| 4.47 × 10−1 | 2.03 × 10−2 | Retinoid metabolism disease events |
| 4.47 × 10−1 | 2.03 × 10−2 | Defective SLC26A4 causes Pendred syndrome (PDS) |
| 6.07 × 10−1 | 3.04 × 10−2 | Defective SLC17A8 causes autosomal dominant deafness 25 (DFNA25) |
| 6.07 × 10−1 | 3.04 × 10−2 | Defective SLC5A5 causes thyroid dyshormonogenesis 1 (TDH1) |
| 6.91 × 10−1 | 3.64 × 10−2 | NR1H2 and NR1H3 regulate gene expression linked to gluconeogenesis |
| 7.24 × 10−1 | 4.02 × 10−2 | Defensins |
| 7.73 × 10−1 | 4.88 × 10−2 | Assembly of active LPL and LIPC lipase complexes |
| Function | UniProt Id | padj | p-Value | Log2FoldChange | Gene Name | Gene_id |
|---|---|---|---|---|---|---|
| Stress-inducible keratin, structural role in epidermal repair | P04259 | 1.8 × 10−6 | <1 × 10−4 | 4.357463 | KRT6C | ENSG00000170465 |
| Cornified envelope protein, reinforces barrier | Q9UBC9 | 1.63 × 10−3 | 1.42 × 10−4 | 3.469405 | SPRR3 | ENSG00000163209 |
| Stress-inducible keratin, part of intermediate filaments | P04259 | 8 × 10−7 | <1 × 10−4 | 3.323752 | KRT6B | ENSG00000185479 |
| Cornified envelope protein, cross-linker | Q96RM1 | 1.01 × 10−3 | 7.26 × 10−5 | 3.158956 | SPRR2F | ENSG00000244094 |
| Stress-inducible keratin, part of intermediate filaments | P08779 | 9.49 × 10−5 | 2.6 × 10−6 | 3.117222 | KRT16 | ENSG00000186832 |
| Protease inhibitor (elafin), limits inflammation/desquamation | P19957 | 5.93 × 10−5 | 1.3 × 10−6 | 3.056438 | PI3 | ENSG00000124102 |
| Stress-inducible keratin, part of intermediate filaments | P02538 | 4.46 × 10−5 | 9 × 10−7 | 2.984315 | KRT6A | ENSG00000205420 |
| Serine protease inhibitor, regulates corneodesmosome degradation | Q6UWN8 | 4.16 × 10−3 | 5.09 × 10−4 | 2.808229 | SPINK6 | ENSG00000178172 |
| Keratins in differentiating non-cornified epithelium | P12035 | 8.39 × 10−5 | 2.1 × 10−6 | 2.564823 | KRT3 | ENSG00000186442 |
| Stress-inducible keratin, part of intermediate filaments | Q04695 | 1.6 × 10−6 | <1 × 10−4 | 2.393522 | KRT17 | ENSG00000128422 |
| Keratin-associated protein, stabilizes keratin filaments | P60370 | 4.67 × 10−2 | 1.3 × 10−2 | −2.199704 | KRTAP10-5 | ENSG00000241123 |
| Keratin-associated protein, stabilizes keratin filaments | Q6L8G5 | 2.66 × 10−3 | 2.72 × 10−4 | −2.257585 | KRTAP5-10 | ENSG00000204572 |
| Keratin-associated protein, stabilizes keratin filaments | Q6L8H1 | 1.7 × 10−2 | 3.38 × 10−3 | −2.299814 | KRTAP5-4 | ENSG00000241598 |
| Keratin-associated protein, stabilizes keratin filaments | P60014 | 3.08 × 10−2 | 7.48 × 10−3 | −2.405668 | KRTAP10-10 | ENSG00000221859 |
| Keratin-associated protein, stabilizes keratin filaments | Q9BYR3 | 1.87 × 10−2 | 3.84 × 10−3 | −2.601125 | KRTAP4-4 | ENSG00000171396 |
3. Discussion
3.1. Study Limitations
3.2. Conclusions
4. Materials and Methods
4.1. Sample Collection and Ethics Approval
4.2. Biopsy Sampling
4.3. RNA Extraction and Quality Control
4.4. RNA Sequencing and Data Processing
4.5. Differential Gene Expression Analysis
4.6. Data Visualization and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic Dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Renert-Yuval, Y.; Brunner, P.M. Atopic Dermatitis. Lancet 2025, 405, 583–596. [Google Scholar] [CrossRef]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y.M. The Immunology of Atopic Dermatitis and Its Reversibility with Broad-Spectrum and Targeted Therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef]
- Afshari, M.; Kolackova, M.; Rosecka, M.; Čelakovská, J.; Krejsek, J. Unraveling the Skin; a Comprehensive Review of Atopic Dermatitis, Current Understanding, and Approaches. Front. Immunol. 2024, 15, 1361005. [Google Scholar] [CrossRef]
- Pavel, A.B.; Zhou, L.; Diaz, A.; Ungar, B.; Dan, J.; He, H.; Estrada, Y.D.; Xu, H.; Fernandes, M.; Renert-Yuval, Y.; et al. The Proteomic Skin Profile of Moderate-to-Severe Atopic Dermatitis Patients Shows an Inflammatory Signature. J. Am. Acad. Dermatol. 2020, 82, 690–699. [Google Scholar] [CrossRef]
- Nakajima, S.; Nakamizo, S.; Nomura, T.; Ishida, Y.; Sawada, Y.; Kabashima, K. Integrating Multi-Omics Approaches in Deciphering Atopic Dermatitis Pathogenesis and Future Therapeutic Directions. Allergy 2024, 79, 2366–2379. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, A.D.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight Junction Defects in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786.e7. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Nomura, T.; Rerknimitr, P.; Seidel, J.A.; Honda, T.; Kabashima, K. The Interplay between Genetic and Environmental Factors in the Pathogenesis of Atopic Dermatitis. Immunol. Rev. 2017, 278, 246–262. [Google Scholar] [CrossRef]
- Möbus, L.; Rodriguez, E.; Harder, I.; Stölzl, D.; Boraczynski, N.; Gerdes, S.; Kleinheinz, A.; Abraham, S.; Heratizadeh, A.; Handrick, C.; et al. Atopic Dermatitis Displays Stable and Dynamic Skin Transcriptome Signatures. J. Allergy Clin. Immunol. 2021, 147, 213–223. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Schauber, J.; Gallo, R.L. The Vitamin D Pathway: A New Target for Control of the Skin’s Immune Response? Exp. Dermatol. 2008, 17, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like Receptor Triggering of a Vitamin D-Mediated Human Antimicrobial Response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Grieco, T.; Paolino, G.; Moliterni, E.; Chello, C.; Sernicola, A.; Egan, C.G.; Morelli, M.; Nannipieri, F.; Battaglia, S.; Accoto, M.; et al. Differential Expression of Proteins Involved in Skin Barrier Maintenance and Vitamin D Metabolism in Atopic Dermatitis: A Cross-Sectional, Exploratory Study. Int. J. Mol. Sci. 2024, 26, 211. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic Dermatitis Is an IL-13 Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef]
- Sekita, A.; Kawasaki, H.; Fukushima-Nomura, A.; Yashiro, K.; Tanese, K.; Toshima, S.; Ashizaki, K.; Miyai, T.; Yazaki, J.; Kobayashi, A.; et al. Multifaceted Analysis of Cross-Tissue Transcriptomes Reveals Phenotype–Endotype Associations in Atopic Dermatitis. Nat. Commun. 2023, 14, 6133. [Google Scholar] [CrossRef]
- Mitamura, Y.; Reiger, M.; Kim, J.; Xiao, Y.; Zhakparov, D.; Tan, G.; Rückert, B.; Rinaldi, A.O.; Baerenfaller, K.; Akdis, M.; et al. Spatial Transcriptomics Combined with Single-Cell RNA-Sequencing Unravels the Complex Inflammatory Cell Network in Atopic Dermatitis. Allergy 2023, 78, 2215–2231. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Stölzl, D.; Wehkamp, U.; Sun, J.; Gerdes, S.; Sarkar, M.K.; Hübenthal, M.; Zeng, C.; Uppala, R.; et al. Progression of Acute-to-Chronic Atopic Dermatitis Is Associated with Quantitative Rather than Qualitative Changes in Cytokine Responses. J. Allergy Clin. Immunol. 2020, 145, 1406–1415. [Google Scholar] [CrossRef]
- Grieco, T.; Moliterni, E.; Paolino, G.; Chello, C.; Sernicola, A.; Egan, C.G.; Nannipieri, F.; Battaglia, S.; Accoto, M.; Tirotta, E.; et al. Association between Vitamin D Receptor Polymorphisms, Tight Junction Proteins and Clinical Features of Adult Patients with Atopic Dermatitis. Dermatol. Pract. Concept. 2024, 14, e2024214. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, S.; Makris, M.; Vakirlis, E.; Gregoriou, S. The Role of Tight Junctions in Atopic Dermatitis: A Systematic Review. J. Clin. Med. 2023, 12, 1538. [Google Scholar] [CrossRef]
- Giannini, S.; Giusti, A.; Minisola, S.; Napoli, N.; Passeri, G.; Rossini, M.; Sinigaglia, L. The Immunologic Profile of Vitamin D and Its Role in Different Immune-Mediated Diseases: An Expert Opinion. Nutrients 2022, 14, 473. [Google Scholar] [CrossRef]
- González-Tarancón, R.; Goñi-Ros, N.; Salvador-Rupérez, E.; Hernández-Martín, Á.; Izquierdo-Álvarez, S.; Puzo-Foncillas, J.; Gilaberte-Calzada, Y. Association Between VDR and CYP24A1 Polymorphisms, Atopic Dermatitis, and Biochemical Lipid and Vitamin D Profiles in Spanish Population: Case-Control Study. JMIR Dermatol. 2023, 6, e39567. [Google Scholar] [CrossRef] [PubMed]
- Szabó, L.; Kapitány, A.; Somogyi, O.; Alhafez, I.; Gáspár, K.; Palatka, R.; Soltész, L.; Törőcsik, D.; Hendrik, Z.; Dajnoki, Z.; et al. Antimicrobial Peptide Loss, Except for LL-37, Is Not Characteristic of Atopic Dermatitis. Acta Derm. Venereol. 2023, 103, adv9413. [Google Scholar] [CrossRef] [PubMed]
- Plager, D.A.; Leontovich, A.A.; Henke, S.A.; Davis, M.D.P.; McEvoy, M.T.; Sciallis, G.F.; Pittelkow, M.R. Early Cutaneous Gene Transcription Changes in Adult Atopic Dermatitis and Potential Clinical Implications. Exp. Dermatol. 2007, 16, 28–36. [Google Scholar] [CrossRef]
- Knor, T.; Meholjić-Fetahović, A.; Mehmedagić, A. Stratum Corneum Hydration and Skin Surface pH in Patients with Atopic Dermatitis. Acta Dermatovenerol. Croat. 2011, 19, 242–247. [Google Scholar]
- Suárez-Fariñas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional Atopic Dermatitis Skin Is Characterized by Broad Terminal Differentiation Defects and Variable Immune Abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954-964.e1-4. [Google Scholar] [CrossRef] [PubMed]
- Dessie, E.Y.; Ding, L.; Satish, L.; Mersha, T.B. Co-Expression Network and Machine Learning Analysis of Transcriptomics Data Identifies Distinct Gene Signatures and Pathways in Lesional and Non-Lesional Atopic Dermatitis. J. Pers. Med. 2024, 14, 960. [Google Scholar] [CrossRef]
- Wongvibulsin, S.; Sutaria, N.; Kannan, S.; Alphonse, M.P.; Belzberg, M.; Williams, K.A.; Brown, I.D.; Choi, J.; Roh, Y.S.; Pritchard, T.; et al. Transcriptomic Analysis of Atopic Dermatitis in African Americans Is Characterized by Th2/Th17-Centered Cutaneous Immune Activation. Sci. Rep. 2021, 11, 11175. [Google Scholar] [CrossRef]
- Song, J.; Kim, D.; Lee, S.; Jung, J.; Joo, J.W.J.; Jang, W. Integrative Transcriptome-Wide Analysis of Atopic Dermatitis for Drug Repositioning. Commun. Biol. 2022, 5, 615. [Google Scholar] [CrossRef]
- Zhou, J.; Liang, G.; Liu, L.; Feng, S.; Zheng, Z.; Wu, Y.; Chen, X.; Li, X.; Wang, L.; Wang, L.; et al. Single-Cell RNA-Seq Reveals Abnormal Differentiation of Keratinocytes and Increased Inflammatory Differentiated Keratinocytes in Atopic Dermatitis. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 2336–2348. [Google Scholar] [CrossRef]
- Makowska, K.; Nowaczyk, J.; Blicharz, L.; Waśkiel-Burnat, A.; Czuwara, J.; Olszewska, M.; Rudnicka, L. Immunopathogenesis of Atopic Dermatitis: Focus on Interleukins as Disease Drivers and Therapeutic Targets for Novel Treatments. Int. J. Mol. Sci. 2023, 24, 781. [Google Scholar] [CrossRef]
- Vu, Y.H.; Furue, M.; Tsuji, G. The Role of Interleukin-24 in Atopic Dermatitis. Explor. Immunol. 2021, 1, 4–15. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Z.; Chen, F. Association of Key Genes and Pathways with Atopic Dermatitis by Bioinformatics Analysis. Med. Sci. Monit. 2019, 25, 4353–4361. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, Y.; Wu, X.; Li, D. MicroRNA-939 Amplifies Staphylococcus Aureus-Induced Matrix Metalloproteinase Expression in Atopic Dermatitis. Front. Immunol. 2024, 15, 1354154. [Google Scholar] [CrossRef]
- Dębińska, A. New Treatments for Atopic Dermatitis Targeting Skin Barrier Repair via the Regulation of FLG Expression. J. Clin. Med. 2021, 10, 2506. [Google Scholar] [CrossRef] [PubMed]
- Hatano, Y.; Elias, P.M. “Outside-to-inside,” “inside-to-Outside,” and “Intrinsic” Endogenous Pathogenic Mechanisms in Atopic Dermatitis: Keratinocytes as the Key Functional Cells Involved in Both Permeability Barrier Dysfunction and Immunological Alterations. Front. Immunol. 2023, 14, 1239251. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.; Mendes-Bastos, P.; Cruz, M.J.; Duarte, B.; Filipe, P.; Lopes, M.J.P.; Gonçalo, M. Interleukin-4 and Atopic Dermatitis: Why Does It Matter? A Narrative Review. Dermatol. Ther. 2025, 15, 579–597. [Google Scholar] [CrossRef]
- Umar, M.; Sastry, K.S.; Al Ali, F.; Al-Khulaifi, M.; Wang, E.; Chouchane, A.I. Vitamin D and the Pathophysiology of Inflammatory Skin Diseases. Skin Pharmacol. Physiol. 2018, 31, 74–86. [Google Scholar] [CrossRef]
- Searing, D.A.; Leung, D.Y. Vitamin D in Atopic Dermatitis, Asthma and Allergic Diseases. Immunol. Allergy Clin. N. Am. 2010, 30, 397–409. [Google Scholar] [CrossRef]
- Pfisterer, K.; Wielscher, M.; Samardzic, D.; Weinzettl, P.; Symmank, D.; Shaw, L.E.; Campana, R.; Huang, H.-J.; Farlik, M.; Bangert, C.; et al. Non-IgE-Reactive Allergen Peptides Deteriorate the Skin Barrier in House Dust Mite-Sensitized Atopic Dermatitis Patients. Front. Cell Dev. Biol. 2023, 11, 1240289. [Google Scholar] [CrossRef]
- Lu, R.; Peng, Z.; Lian, P.; Wazir, J.; Gu, C.; Ma, C.; Wei, L.; Li, L.; Pu, W.; Liu, J.; et al. Vitamin D Attenuates DNCB-Induced Atopic Dermatitis-like Skin Lesions by Inhibiting Immune Response and Restoring Skin Barrier Function. Int. Immunopharmacol. 2023, 122, 110558. [Google Scholar] [CrossRef]
- Margolis, D.J. Atopic Dermatitis: Filaggrin and Skin Barrier Dysfunction. Br. J. Dermatol. 2022, 186, 396. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, T.; van Amerongen, R. Walking the Tight Wire between Cell Adhesion and WNT Signalling: A Balancing Act for β-Catenin. Open Biol. 2020, 10, 200267. [Google Scholar] [CrossRef]
- Antonatos, C.; Mitsoudi, D.; Pontikas, A.; Akritidis, A.; Xiropotamos, P.; Georgakilas, G.K.; Georgiou, S.; Tsiogka, A.; Gregoriou, S.; Grafanaki, K.; et al. Transcriptome-Wide Analyses Delineate the Genetic Architecture of Expression Variation in Atopic Dermatitis. HGG Adv. 2025, 6, 100422. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the Regulation of Protein Abundance from Proteomic and Transcriptomic Analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-Spectrometry-Based Draft of the Human Proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Perl, K.; Ushakov, K.; Pozniak, Y.; Yizhar-Barnea, O.; Bhonker, Y.; Shivatzki, S.; Geiger, T.; Avraham, K.B.; Shamir, R. Reduced Changes in Protein Compared to mRNA Levels across Non-Proliferating Tissues. BMC Genom. 2017, 18, 305. [Google Scholar] [CrossRef]
- Guillemin, A.; Kumar, A.; Wencker, M.; Ricci, E.P. Shaping the Innate Immune Response Through Post-Transcriptional Regulation of Gene Expression Mediated by RNA-Binding Proteins. Front. Immunol. 2022, 12, 796012. [Google Scholar] [CrossRef] [PubMed]
- Goleva, E.; Calatroni, A.; LeBeau, P.; Berdyshev, E.; Taylor, P.; Kreimer, S.; Cole, R.N.; Leung, D.Y.M. Skin Tape Proteomics Identifies Pathways Associated with Transepidermal Water Loss and Allergen Polysensitization in Atopic Dermatitis. J. Allergy Clin. Immunol. 2020, 146, 1367–1378. [Google Scholar] [CrossRef]
- Cole, C.; Kroboth, K.; Schurch, N.J.; Sandilands, A.; Sherstnev, A.; O’Regan, G.M.; Watson, R.M.; McLean, W.H.I.; Barton, G.J.; Irvine, A.D.; et al. Filaggrin-Stratified Transcriptomic Analysis of Pediatric Skin Identifies Mechanistic Pathways in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2014, 134, 82–91. [Google Scholar] [CrossRef]
- Rusiñol, L.; Puig, L. Multi-Omics Approach to Improved Diagnosis and Treatment of Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2024, 25, 1042. [Google Scholar] [CrossRef]
- Bratu, D.; Boda, D.; Caruntu, C. Genomic, Epigenomic, Transcriptomic, Proteomic and Metabolomic Approaches in Atopic Dermatitis. Curr. Issues Mol. Biol. 2023, 45, 5215–5231. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic Dermatitis: Immune Deviation, Barrier Dysfunction, IgE Autoreactivity and New Therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Hatano, Y.; Williams, M.L. Basis for the Barrier Abnormality in Atopic Dermatitis: Outside-inside-Outside Pathogenic Mechanisms. J. Allergy Clin. Immunol. 2008, 121, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Hanifin, J.M.; Thurston, M.; Omoto, M.; Cherill, R.; Tofte, S.J.; Graeber, M. The Eczema Area and Severity Index (EASI): Assessment of Reliability in Atopic Dermatitis. EASI Evaluator Group. Exp. Dermatol. 2001, 10, 11–18. [Google Scholar] [CrossRef] [PubMed]
| Characteristic | n (%) |
|---|---|
| Gender, n (%) | |
| Male | 13 (61.9) |
| Female | 8 (38.9) |
| Age | |
| <60 years | 17 (81.0) |
| ≥60 years | 4 (19.0) |
| BMI (Kg/m2), mean ± SD | 24.1 ± 4.1 |
| EASI score | |
| Mild (EASI < 16) | 0 (0.0) |
| Moderate-to-severe (EASI ≥ 16) | 21 (100) |
| Phenotype (localisation), n (%) | |
| Flexural sites | 3 (14.3) |
| Generalised | 10 (47.6) |
| Head/neck | 7 (33.3) |
| Hands | 1 (4.8) |
| § Age of disease onset, n (%) | |
| Childhood | 15 (71.4) |
| Adulthood | 6 (28.6) |
| Asthma, n (%) | |
| Present | 15 (71.4) |
| Absent | 6 (28.6%) |
| Rhino conjunctivitis, n (%) | |
| Present | 7 (33.3) |
| Absent | 14 (66.7) |
| Skin prick test, n (%) | |
| Present | 8 (38.1) |
| Absent | 13 (61.9) |
| Total IgE (IU/mL), n (%) | |
| <100 IU/mL | 7 (33.3) |
| ≥100 IU/mL | 14 (66.7) |
| * 25(OH)D vitamin D | |
| ≥30 ng/mL | 8 (40.0) |
| <30 ng/mL | 12 (60.0) |
| Gene Name | Significance | padj | p-Value | Log2 Fold Change | Gene | Ensemble Gene id | |
|---|---|---|---|---|---|---|---|
| E-Caderin 1 | S | 1.30 × 10−3 | 1 × 10−4 | 0.4809 | CDH1 | ENSG0000039068 | Epithelial Barrier |
| Zonulin 1 | S | 3.32 × 10−2 | 8.30 × 10−3 | 0.2136 | TJP1 | ENSG00000104067 | |
| Alpha 1_Catenin | S | 4.28 × 10−2 | 1.16 × 10−2 | 0.1762 | CTNNA1 | ENSG00000044115 | |
| Beta 1_Catenin | NS | 2.88 × 10−1 | 1.54 × 10−1 | 0.0797 | CTNNB1 | ENSG00000168036 | |
| Claudin | S | 2.17 × 10−2 | 4.70 × 10−3 | −0.4409 | CLDN1 | ENSG00000163347 | |
| Cingulin like | S | <1 × 10−4 | <1 × 10−4 | −1.0219 | CGNL1 | ENSG00000128849 | |
| Filaggrin 2 | S | 2 × 10−4 | <1 × 10−4 | −1.1688 | FLG2 | ENSG00000143520 | |
| Occludin | NS | 6.5 × 10−1 | 5 × 10−1 | −0.0853 | OCLN | ENSG00000197822 | |
| Cytochrome P450 Family 27 Subfamily B Member 1 | S | <1 × 10−4 | <1 × 10−4 | 1.8934 | CYP27B1 | ENSG00000111012 | Vitamin D metabolism |
| Cytochrome P450 Family 24 Subfamily A Member 1 | S | 3 × 10−4 | <1 × 10−4 | 1.7887 | CYP24A1 | ENSG0000019186 | |
| Vitamin D Receptor | NS | 2.37 × 10−1 | 1.17 × 10−1 | 0.1863 | VDR | ENSG00000111424 | |
| Cathelicidin Antimicrobial Peptide | S | 2 × 10−3 | 2 × 10−4 | 2.3195 | CAMP | ENSG00000164047 | Immune response and inflammation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grieco, T.; Paolino, G.; Moliterni, E.; Chello, C.; Sernicola, A.; Egan, C.G.; Morelli, M.; Nannipieri, F.; Battaglia, S.; Accoto, M.; et al. Transcriptomic Profiling of Lesional and Perilesional Skin in Atopic Dermatitis Suggests Barrier Dysfunction, Inflammatory Activation, and Alterations to Vitamin D Metabolism. Int. J. Mol. Sci. 2025, 26, 6152. https://doi.org/10.3390/ijms26136152
Grieco T, Paolino G, Moliterni E, Chello C, Sernicola A, Egan CG, Morelli M, Nannipieri F, Battaglia S, Accoto M, et al. Transcriptomic Profiling of Lesional and Perilesional Skin in Atopic Dermatitis Suggests Barrier Dysfunction, Inflammatory Activation, and Alterations to Vitamin D Metabolism. International Journal of Molecular Sciences. 2025; 26(13):6152. https://doi.org/10.3390/ijms26136152
Chicago/Turabian StyleGrieco, Teresa, Giovanni Paolino, Elisa Moliterni, Camilla Chello, Alvise Sernicola, Colin Gerard Egan, Mariangela Morelli, Fabrizio Nannipieri, Santina Battaglia, Marina Accoto, and et al. 2025. "Transcriptomic Profiling of Lesional and Perilesional Skin in Atopic Dermatitis Suggests Barrier Dysfunction, Inflammatory Activation, and Alterations to Vitamin D Metabolism" International Journal of Molecular Sciences 26, no. 13: 6152. https://doi.org/10.3390/ijms26136152
APA StyleGrieco, T., Paolino, G., Moliterni, E., Chello, C., Sernicola, A., Egan, C. G., Morelli, M., Nannipieri, F., Battaglia, S., Accoto, M., Tirotta, E., Trasciatti, S., Bonaretti, S., Pellacani, G., & Calvieri, S. (2025). Transcriptomic Profiling of Lesional and Perilesional Skin in Atopic Dermatitis Suggests Barrier Dysfunction, Inflammatory Activation, and Alterations to Vitamin D Metabolism. International Journal of Molecular Sciences, 26(13), 6152. https://doi.org/10.3390/ijms26136152