
Hippocampal Neurogenesis in Alzheimer’s Disease: Multimodal Therapeutics and the Neurogenic Impairment Index Framework


Abstract
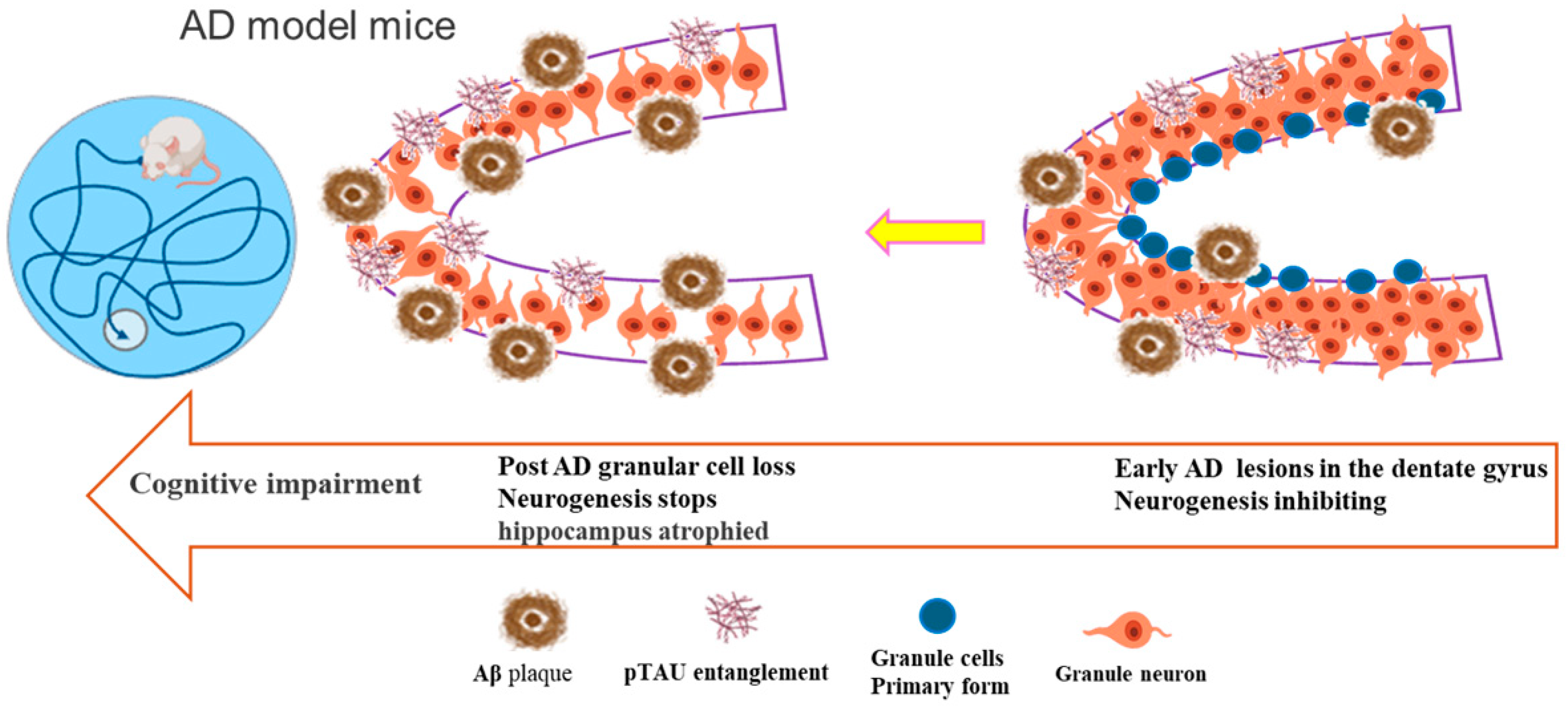
1. Introduction
2. Hippocampal Neural Stem Cells and AD Pathologic Processes
3. Research on Adult Neurogenesis in Alzheimer’s Disease
4. Neurotransmitter and Signaling Molecule Modulation of Hippocampal Neurogenesis Alleviates Alzheimer’s Disease Pathology
5. Neurotrophic Factors Promote Hippocampal Neurogenesis to Ameliorate AD Pathology
6. Natural Product Active Ingredients Promoting Neurogenesis to Improve AD Pathology
7. Non-Pharmacological Interventions and Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; Lazarov, O.; Fitzsimons, C.P.; Tanzi, R.; Lucassen, P.J.; Choi, S.H. Adult Hippocampal Neurogenesis in Alzheimer’s Disease: A Roadmap to Clinical Relevance. Cell Stem Cell 2023, 30, 120–136. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult Hippocampal Neurogenesis Is Abundant in Neurologically Healthy Subjects and Drops Sharply in Patients with Alzheimer’s Disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Terreros-Roncal, J.; Moreno-Jiménez, E.P.; Flor-García, M.; Rodríguez-Moreno, C.B.; Trinchero, M.F.; Cafini, F.; Rábano, A.; Llorens-Martín, M. Impact of Neurodegenerative Diseases on Human Adult Hippocampal Neurogenesis. Science 2021, 374, 1106–1113. [Google Scholar] [CrossRef]
- Park, T.I.H.; Waldvogel, H.J.; Montgomery, J.M.; Mee, E.W.; Bergin, P.S.; Faull, R.L.M.; Dragunow, M.; Curtis, M.A. Identifying Neural Progenitor Cells in the Adult Human Brain. Methods Mol. Biol. 2022, 2389, 125–154. [Google Scholar] [PubMed]
- Liu, X.; Ding, Y.; Jiang, C.; Xin, Y.; Ma, X.; Xu, M.; Wang, Q.; Hou, B.; Li, Y.; Zhang, S.; et al. Astragaloside Iv Mediates Radiation-Induced Neuronal Damage through Activation of BDNF-Trkb Signaling. Phytomedicine 2024, 132, 155803. [Google Scholar] [CrossRef]
- Hainmueller, T.; Bartos, M. Dentate Gyrus Circuits for Encoding, Retrieval and Discrimination of Episodic Memories. Nat. Rev. Neurosci. 2020, 21, 153–168. [Google Scholar] [CrossRef]
- Demars, M.; Hu, Y.S.; Gadadhar, A.; Lazarov, O. Impaired Neurogenesis Is an Early Event in the Etiology of Familial Alzheimer’s Disease in Transgenic Mice. J. Neurosci. Res. 2010, 88, 2103–2117. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined Adult Neurogenesis and BDNF Mimic Exercise Effects on Cognition in an Alzheimer’s Mouse Model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef]
- Moon, M.; Cha, M.Y.; Mook-Jung, I. Impaired Hippocampal Neurogenesis and Its Enhancement with Ghrelin in 5xfad Mice. J. Alzheimers Dis. 2014, 41, 233–241. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Jones, V.C.; Tabuchi, M.; Allan, S.M.; Knight, E.M.; LaFerla, F.M.; Oddo, S.; Verkhratsky, A. Impaired Adult Neurogenesis in the Dentate Gyrus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. PLoS ONE 2008, 3, e2935. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e973. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, Y.; Li, S.; Kennedy, B.C.; Zhang, D.Y.; Bond, A.M.; Sun, Y.; Jacob, F.; Lu, L.; Hu, P.; et al. Molecular Landscapes of Human Hippocampal Immature Neurons across Lifespan. Nature 2022, 607, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s Disease Patient-Derived Neurons Reveal Distinct Mutation-Specific Effects on Amyloid Beta. Mol. Psychiatry 2020, 25, 2919–2931. [Google Scholar] [CrossRef]
- Arber, C.; Lovejoy, C.; Harris, L.; Willumsen, N.; Alatza, A.; Casey, J.M.; Lines, G.; Kerins, C.; Mueller, A.K.; Zetterberg, H.; et al. Familial Alzheimer’s Disease Mutations in Psen1 Lead to Premature Human Stem Cell Neurogenesis. Cell Rep. 2021, 34, 108615. [Google Scholar] [CrossRef] [PubMed]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Téglási, A.; Bock, I.; Giudice, M.L.; Táncos, Z.; Molnár, K.; László, L.; et al. Neurons Derived from Sporadic Alzheimer’s Disease Ipscs Reveal Elevated Tau Hyperphosphorylation, Increased Amyloid Levels, and Gsk3b Activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Mertens, J.; Herdy, J.R.; Traxler, L.; Schafer, S.T.; Schlachetzki, J.C.M.; Böhnke, L.; Reid, D.A.; Lee, H.; Zangwill, D.; Fernandes, D.P.; et al. Age-Dependent Instability of Mature Neuronal Fate in Induced Neurons from Alzheimer’s Patients. Cell Stem Cell 2021, 28, 1533–1548.e6. [Google Scholar] [CrossRef]
- Culig, L.; Chu, X.; Bohr, V.A. Neurogenesis in Aging and Age-Related Neurodegenerative Diseases. Ageing Res. Rev. 2022, 78, 101636. [Google Scholar] [CrossRef]
- Gonçalves, J.T.; Schafer, S.T.; Gage, F.H. Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell 2016, 167, 897–914. [Google Scholar] [CrossRef]
- Bond, A.M.; Ming, G.L.; Song, H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef]
- Llorente, V.; Velarde, P.; Desco, M.; Gómez-Gaviro, M.V. Current Understanding of the Neural Stem Cell Niches. Cells 2022, 11, 3002. [Google Scholar] [CrossRef]
- Liang, Z.; Li, Z.; Zhang, D.; Luo, X.; Liu, Q.; Qin, D.; Wang, M.; Xu, Z.; Feng, J.; He, J.; et al. Dual recombinase-mediated intersectional genetics defines the functional heterogeneity of neural stem cells in adult hippocampus. Mol. Psychiatry, 2025; Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Winkler, J. Adult Neurogenesis in Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2015, 7, a021287. [Google Scholar] [CrossRef]
- Yi, L.X.; Zeng, L.; Wang, Q.; Tan, E.K.; Zhou, Z.D. Reelin Links Apolipoprotein E4, Tau, and Amyloid-Β in Alzheimer’s Disease. Ageing Res. Rev. 2024, 98, 102339. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wang, B.; Cui, Y.; Tian, S.; Zhang, Y.; You, L.; Chang, Y.Z.; Gao, G. Hepcidin Deficiency Impairs Hippocampal Neurogenesis and Mediates Brain Atrophy and Memory Decline in Mice. J. Neuroinflamm. 2024, 21, 15. [Google Scholar] [CrossRef]
- Babcock, K.R.; Page, J.S.; Fallon, J.R.; Webb, A.E. Adult Hippocampal Neurogenesis in Aging and Alzheimer’s Disease. Stem Cell Rep. 2021, 16, 681–693. [Google Scholar] [CrossRef]
- Hanspal, M.A.; Gillotin, S. A New Age in Understanding Adult Hippocampal Neurogenesis in Alzheimer’s Disease. Neural Regen. Res. 2022, 17, 2615–2618. [Google Scholar]
- Colussi, C.; Bertozzi, A.; Leone, L.; Rinaudo, M.; Sollazzo, R.; Conte, F.; Paccosi, E.; Nardella, L.; Aceto, G.; Puma, D.D.L.; et al. Nucleoporin 153 Deficiency in Adult Neural Stem Cells Defines a Pathological Protein-Network Signature and Defective Neurogenesis in a Mouse Model of Ad. Stem Cell Res. Ther. 2024, 15, 275. [Google Scholar] [CrossRef]
- Li, Y.D.; Luo, Y.J.; Xie, L.; Tart, D.S.; Sheehy, R.N.; Zhang, L.; Coleman, L.G., Jr.; Chen, X.; Song, J. Activation of Hypothalamic-Enhanced Adult-Born Neurons Restores Cognitive and Affective Function in Alzheimer’s Disease. Cell Stem Cell 2023, 30, 415–432.e6. [Google Scholar] [CrossRef]
- Zhang, X.; Mei, Y.; He, Y.; Wang, D.; Wang, J.; Wei, X.; Yang, E.; Zhou, D.; Shen, H.; Peng, G.; et al. Ablating Adult Neural Stem Cells Improves Synaptic and Cognitive Functions in Alzheimer Models. Stem Cell Rep. 2021, 16, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Zhang, T.; Wang, J.; Liu, C.; Shang, Q.; Wei, X.; Zhu, H.; Shen, H.; Sun, B. Hcn2 Deficiency Correlates with Memory Deficits and Hyperexcitability of Dca1 Pyramidal Neurons in Alzheimer’s Disease. Alzheimers Res. Ther. 2025, 17, 55. [Google Scholar] [CrossRef]
- Zheng, J.; Li, H.L.; Tian, N.; Liu, F.; Wang, L.; Yin, Y.; Yue, L.; Ma, L.; Wan, Y.; Wang, J.Z. Interneuron Accumulation of Phosphorylated Tau Impairs Adult Hippocampal Neurogenesis by Suppressing Gabaergic Transmission. Cell Stem Cell 2020, 26, 331–345.e6. [Google Scholar] [CrossRef] [PubMed]
- Maguire, E.P.; Macpherson, T.; Swinny, J.D.; Dixon, C.I.; Herd, M.B.; Belelli, D.; Stephens, D.N.; King, S.L.; Lambert, J.J. Tonic Inhibition of Accumbal Spiny Neurons by Extrasynaptic A4βδ Gabaa Receptors Modulates the Actions of Psychostimulants. J. Neurosci. 2014, 34, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Liu, X.; Zhao, J. Electroacupuncture-Induced Activation of Gabaergic System Alleviates Airway Inflammation in Asthma Model by Suppressing Tlr4/Myd88/Nf-Κb Signaling Pathway. Chin. Med. J. 2023, 136, 451–460. [Google Scholar] [CrossRef]
- Mishra, P.; Silva, A.; Sharma, J.; Nguyen, J.; Pizzo, D.P.; Hinz, D.; Sahoo, D.; Cherqui, S. Rescue of Alzheimer’s Disease Phenotype in a Mouse Model by Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells. Cell Rep. 2023, 42, 112956. [Google Scholar] [CrossRef]
- Apodaca, L.A.; Baddour, A.A.D.; Garcia, C., Jr.; Alikhani, L.; Giedzinski, E.; Ru, N.; Agrawal, A.; Acharya, M.M.; Baulch, J.E. Human Neural Stem Cell-Derived Extracellular Vesicles Mitigate Hallmarks of Alzheimer’s Disease. Alzheimers Res. Ther. 2021, 13, 57. [Google Scholar] [CrossRef]
- Chang, J.; Li, Y.; Shan, X.; Chen, X.; Yan, X.; Liu, J.; Zhao, L. Neural Stem Cells Promote Neuroplasticity: A Promising Therapeutic Strategy for the Treatment of Alzheimer’s Disease. Neural Regen. Res. 2024, 19, 619–628. [Google Scholar] [CrossRef]
- Li, Y.; Xu, N.N.; Hao, Z.Z.; Liu, S. Adult Neurogenesis in the Primate Hippocampus. Zool. Res. 2023, 44, 315–322. [Google Scholar] [CrossRef]
- Chappelle, S.D.; Gigliotti, C.; Léger, G.C.; Peavy, G.M.; Jacobs, D.M.; Banks, S.J.; Little, E.A.; Galasko, D.; Salmon, D.P. Comparison of the Telephone-Montreal Cognitive Assessment (T-Moca) and Telephone Interview for Cognitive Status (Tics) as Screening Tests for Early Alzheimer’s Disease. Alzheimers Dement. 2023, 19, 4599–4608. [Google Scholar] [CrossRef]
- Wang, Q.; Schindler, S.E.; Chen, G.; McKay, N.S.; McCullough, A.; Flores, S.; Liu, J.; Sun, Z.; Wang, S.; Wang, W.; et al. Benzinger. Investigating White Matter Neuroinflammation in Alzheimer Disease Using Diffusion-Based Neuroinflammation Imaging. Neurology 2024, 102, e208013. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Qian, Y.; Deng, X. Triglyceride Glucose Index Is a Significant Predictor of Severe Disturbance of Consciousness and All-Cause Mortality in Critical Cerebrovascular Disease Patients. Cardiovasc. Diabetol. 2023, 22, 156. [Google Scholar] [CrossRef] [PubMed]
- Franjic, D.; Skarica, M.; Ma, S.; Arellano, J.I.; Tebbenkamp, A.T.N.; Choi, J.; Xu, C.; Li, Q.; Morozov, Y.M.; Andrijevic, D.; et al. Transcriptomic Taxonomy and Neurogenic Trajectories of Adult Human, Macaque, and Pig Hippocampal and Entorhinal Cells. Neuron 2022, 110, 452–469.e14. [Google Scholar] [CrossRef]
- Saini, V.; Kaur, T.; Kalotra, S.; Kaur, G. The Neuroplasticity Marker Psa-Ncam: Insights into New Therapeutic Avenues for Promoting Neuroregeneration. Pharmacol. Res. 2020, 160, 105186. [Google Scholar] [CrossRef]
- Zoungrana, L.I.; Krause-Hauch, M.; Wang, H.; Fatmi, M.K.; Bates, L.; Li, Z.; Kulkarni, P.; Ren, D.; Li, J. The Interaction of Mtor and Nrf2 in Neurogenesis and Its Implication in Neurodegenerative Diseases. Cells 2022, 11, 2048. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of Ferroportin Induces Memory Impairment by Promoting Ferroptosis in Alzheimer’s Disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Karikari, T.K.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood Phosphorylated Tau 181 as a Biomarker for Alzheimer’s Disease: A Diagnostic Performance and Prediction Modelling Study Using Data from Four Prospective Cohorts. Lancet Neurol. 2020, 19, 422–433. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Fu, C.H.; Iascone, D.M.; Petrof, I.; Hazra, A.; Zhang, X.; Pyfer, M.S.; Tosi, U.; Corbett, B.F.; Cai, J.; Lee, J.; et al. Early Seizure Activity Accelerates Depletion of Hippocampal Neural Stem Cells and Impairs Spatial Discrimination in an Alzheimer’s Disease Model. Cell Rep. 2019, 27, 3741–3751.e4. [Google Scholar] [CrossRef]
- Maltsev, D.I.; Aniol, V.A.; Golden, M.A.; Petrina, A.D.; Belousov, V.V.; Gulyaeva, N.V.; Podgorny, O.V. Aging Modulates the Ability of Quiescent Radial Glia-Like Stem Cells in the Hippocampal Dentate Gyrus to Be Recruited into Division by Pro-Neurogenic Stimuli. Mol. Neurobiol. 2024, 61, 3461–3476. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Ding, W.; Sun, L.; Lu, Y.; Dong, H.; Fan, X.; Liu, Z.; Chen, R.; Zhang, S.; Ma, Q.; et al. Decoding the Development of the Human Hippocampus. Nature 2020, 577, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, X.; Xiong, W.; Chen, J. In Vivo Reprogramming Reactive Glia into Ipscs to Produce New Neurons in the Cortex Following Traumatic Brain Injury. Sci. Rep. 2016, 6, 22490. [Google Scholar] [CrossRef]
- Kang, E.M.; Jia, Y.B.; Wang, J.Y.; Wang, G.Y.; Chen, H.J.; Chen, X.Y.; Ye, Y.Q.; Zhang, X.; Su, X.H.; Wang, J.Y.; et al. Downregulation of Microrna-124-3p Promotes Subventricular Zone Neural Stem Cell Activation by Enhancing the Function of BDNF Downstream Pathways after Traumatic Brain Injury in Adult Rats. CNS Neurosci. Ther. 2022, 28, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Ha, S.; Shin, K.Y.; Kim, S.; Lee, K.J.; Chong, Y.H.; Chang, K.A.; Suh, Y.H. Neural Stem Cell Transplantation at Critical Period Improves Learning and Memory through Restoring Synaptic Impairment in Alzheimer’s Disease Mouse Model. Cell Death Dis. 2015, 6, e1789. [Google Scholar] [CrossRef]
- Katta, M.; Mathew, B.A.; Chaturvedi, P.; Ludhiadch, A.; Munshi, A. Advanced Molecular Therapies for Neurological Diseases: Focus on Stroke, Alzheimer’s Disease, and Parkinson’s Disease. Neurol. Sci. 2023, 44, 19–36. [Google Scholar] [CrossRef]
- Wang, Z.B.; Wang, Z.T.; Sun, Y.; Tan, L.; Yu, J.T. The Future of Stem Cell Therapies of Alzheimer’s Disease. Ageing Res. Rev. 2022, 80, 101655. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of Hippocampal Neurogenesis in Adult Humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef]
- Fujishima, M.; Kawasaki, Y.; Mitsuhashi, T.; Matsuda, H. Impact of Amyloid and Tau Positivity on Longitudinal Brain Atrophy in Cognitively Normal Individuals. Alzheimers Res. Ther. 2013, 153, 1219–1227. [Google Scholar] [CrossRef]
- Igarashi, K.M. Entorhinal Cortex Dysfunction in Alzheimer’s Disease. Trends Neurosci. 2023, 46, 124–136. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s Disease and Senile Dementia: Loss of Neurons in the Basal Forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human Neural Stem Cells Improve Cognition and Promote Synaptic Growth in Two Complementary Transgenic Models of Alzheimer’s Disease and Neuronal Loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Karvelas, N.; Bennett, S.; Politis, G.; Kouris, N.I.; Kole, C. Advances in Stem Cell Therapy in Alzheimer’s Disease: A Comprehensive Clinical Trial Review. Stem Cell Investig. 2022, 9, 2. [Google Scholar] [CrossRef]
- Yun, H.M.; Kim, H.S.; Park, K.R.; Shin, J.M.; Kang, A.R.; Lee, K.I.; Song, S.; Kim, Y.B.; Han, S.B.; Chung, H.M.; et al. Placenta-Derived Mesenchymal Stem Cells Improve Memory Dysfunction in an Aβ1-42-Infused Mouse Model of Alzheimer’s Disease. Cell Death Dis. 2013, 4, e958. [Google Scholar] [CrossRef]
- Silva, R.O.; Haddad, M.; Counil, H.; Zaouter, C.; Patten, S.A.; Fulop, T.; Ramassamy, C. Exploring the Potential of Plasma and Adipose Mesenchymal Stem Cell-Derived Extracellular Vesicles as Novel Platforms for Neuroinflammation Therapy. J. Control. Release 2025, 377, 880–898. [Google Scholar] [CrossRef] [PubMed]
- Nabil, M.; Kassem, D.H.; Ali, A.A.; El-Mesallamy, H.O. Adipose Tissue-Derived Mesenchymal Stem Cells Ameliorate Cognitive Impairment in Alzheimer’s Disease Rat Model: Emerging Role of Sirt1. Biofactors 2023, 49, 1121–1142. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, K.R.; Jang, H.; Lee, N.K.; Jung, Y.H.; Kim, J.P.; Lee, J.I.; Chang, J.W.; Park, S.; Kim, S.T.; et al. Intracerebroventricular Injection of Human Umbilical Cord Blood Mesenchymal Stem Cells in Patients with Alzheimer’s Disease Dementia: A Phase I Clinical Trial. Alzheimers Res. Ther. 2021, 13, 154. [Google Scholar] [CrossRef]
- Liu, S.; Fan, M.; Xu, J.X.; Yang, L.J.; Qi, C.C.; Xia, Q.R.; Ge, J.F. Exosomes Derived from Bone-Marrow Mesenchymal Stem Cells Alleviate Cognitive Decline in Ad-Like Mice by Improving BDNF-Related Neuropathology. J. Neuroinflamm. 2022, 19, 35. [Google Scholar] [CrossRef]
- Chen, Z.R.; Huang, J.B.; Yang, S.L.; Hong, F.F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Yang, M.; Ke, X.; Dai, Y.; Lin, H.; Wang, S.; Chen, L.; Tao, J. Chemical Genetic Activation of the Cholinergic Basal Forebrain Hippocampal Circuit Rescues Memory Loss in Alzheimer’s Disease. Alzheimers Res. Ther. 2022, 14, 53. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Liu, R.; Huang, R.; Gao, Z.; Tian, L.; Zhu, Y.; Liu, Y.; Lu, C.; Wu, L. Lancao Decoction Alleviates Cognitive Dysfunction: A New Therapeutic Drug and Its Therapeutic Mechanism. Phytomedicine 2024, 128, 155531. [Google Scholar] [CrossRef]
- Thierry, M.; Ponce, J.; Martà-Ariza, M.; Askenazi, M.; Faustin, A.; Leitner, D.; Pires, G.; Kanshin, E.; Drummond, E.; Ueberheide, B.; et al. The Influence of Apoe(Ε4) on the Ptau Interactome in Sporadic Alzheimer’s Disease. Acta Neuropathol. 2024, 147, 91. [Google Scholar] [CrossRef]
- Lei, H.Y.; Pi, G.L.; He, T.; Xiong, R.; Lv, J.R.; Liu, J.L.; Wu, D.Q.; Li, M.Z.; Shi, K.; Li, S.H.; et al. Targeting Vulnerable Microcircuits in the Ventral Hippocampus of Male Transgenic Mice to Rescue Alzheimer-Like Social Memory Loss. Mil. Med. Res. 2024, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Asrican, B.; Li, W.; Gu, B.; Wen, Z.; Lim, S.A.; Haniff, I.; Ramakrishnan, C.; Deisseroth, K.; Philpot, B.; et al. Long-Range Gabaergic Inputs Regulate Neural Stem Cell Quiescence and Control Adult Hippocampal Neurogenesis. Cell Stem Cell 2017, 21, 604–617.e5. [Google Scholar] [CrossRef] [PubMed]
- She, L.; Tang, H.; Zeng, Y.; Li, L.; Xiong, L.; Sun, J.; Chen, F.; Ren, J.; Zhang, J.; Wang, W.; et al. Ginsenoside Rk3 Promotes Neurogenesis in Alzheimer’s Disease through Activation of the Creb/BDNF Pathway. J. Ethnopharmacol. 2024, 321, 117462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ren, X.; Liu, C.; Liu, Y.; Wang, Y. Rbm8a Regulates Neurogenesis and Reduces Alzheimer’s Disease-Associated Pathology in the Dentate Gyrus of 5×Fad Mice. Neural Regen. Res. 2024, 19, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, J.; Yu, M.; Meng, X.; Cui, X.; Zhao, Y.; Zhu, Y.; Xing, W.; Guan, Y. Disturbance of Intracellular Calcium Homeostasis and Camkii/Creb Signaling Is Associated with Learning and Memory Impairments Induced by Chronic Aluminum Exposure. Neurotox. Res. 2014, 26, 52–63. [Google Scholar] [CrossRef]
- Tropea, M.R.; Gulisano, W.; Vacanti, V.; Arancio, O.; Puzzo, D.; Palmeri, A. Nitric Oxide/Cgmp/Creb Pathway and Amyloid-Beta Crosstalk: From Physiology to Alzheimer’s Disease. Free Radic. Biol. Med. 2022, 193, 657–668. [Google Scholar] [CrossRef]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.K. The Role of Creb and BDNF in Neurobiology and Treatment of Alzheimer’s Disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Ivanova, P.; Krushovlieva, D.; Kortenska, L.; Angelova, V.T. Protective Effect of the Novel Melatonin Analogue Containing Donepezil Fragment on Memory Impairment Via Mt/Erk/Creb Signaling in the Hippocampus in a Rat Model of Pinealectomy and Subsequent Aβ(1-42) Infusion. Int. J. Mol. Sci. 2024, 25, 1867. [Google Scholar] [CrossRef]
- Chen, X.; Chen, A.; Wei, J.; Huang, Y.; Deng, J.; Chen, P.; Yan, Y.; Lin, M.; Chen, L.; Zhang, J.; et al. Dexmedetomidine Alleviates Cognitive Impairment by Promoting Hippocampal Neurogenesis Via BDNF/Trkb/Creb Signaling Pathway in Hypoxic-Ischemic Neonatal Rats. CNS Neurosci. Ther. 2024, 30, e14486. [Google Scholar] [CrossRef] [PubMed]
- Spoleti, E.; La Barbera, L.; Cauzzi, E.; De Paolis, M.L.; Saba, L.; Marino, R.; Sciamanna, G.; Di Lazzaro, V.; Keller, F.; Nobili, A.; et al. Dopamine Neuron Degeneration in the Ventral Tegmental Area Causes Hippocampal Hyperexcitability in Experimental Alzheimer’s Disease. Mol. Psychiatry 2024, 29, 1265–1280. [Google Scholar] [CrossRef]
- Ye, J.; Yin, Y.; Liu, H.; Fang, L.; Tao, X.; Wei, L.; Zuo, Y.; Yin, Y.; Ke, D.; Wang, J.Z. Tau Inhibits Pka by Nuclear Proteasome-Dependent Pkar2α Elevation with Suppressed Creb/Glua1 Phosphorylation. Aging Cell 2020, 19, e13055. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Yao, J.; Liu, Y.; Zhang, X.; Wang, Y.; Chen, X.; Liu, L.; Shi, N.; Yan, H. Tau Hyperphosphorylation and P-Creb Reduction Are Involved in Acrylamide-Induced Spatial Memory Impairment: Suppression by Curcumin. Brain Behav. Immun. 2018, 71, 66–80. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin Ameliorates Amyloid Beta-Induced Memory Deficits, Tau Hyperphosphorylation and Neurodegeneration Via Pi3/Akt/Gsk3β Pathway in the Mouse Hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.X.; Gao, A.X.; Dong, T.T.; Tsim, K.W. Flavonoids from Seabuckthorn (Hippophae rhamnoides L.) Mimic Neurotrophic Functions in Inducing Neurite Outgrowth in Cultured Neurons: Signaling Via Pi3k/Akt and Erk Pathways. Phytomedicine 2023, 115, 154832. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, F.; Zhai, M.; He, M.; Hu, Y.; Feng, L.; Li, Y.; Yang, J.; Wu, C. Hyperactive Neuronal Autophagy Depletes BDNF and Impairs Adult Hippocampal Neurogenesis in a Corticosterone-Induced Mouse Model of Depression. Theranostics 2023, 13, 1059–1075. [Google Scholar] [CrossRef]
- Wei, M.; Wu, T.; Chen, N. Bridging Neurotrophic Factors and Bioactive Peptides to Alzheimer’s Disease. Ageing Res. Rev. 2024, 94, 102177. [Google Scholar] [CrossRef]
- Fonseca-Gomes, J.; Costa-Coelho, T.; Ferreira-Manso, M.; Inteiro-Oliveira, S.; Vaz, S.H.; Alemãn-Serrano, N.; Atalaia-Barbacena, H.; Ribeiro-Rodrigues, L.; Ramalho, R.M.; Pinto, R.; et al. A Small Tat-Trkb Peptide Prevents BDNF Receptor Cleavage and Restores Synaptic Physiology in Alzheimer’s Disease. Mol. Ther. 2024, 32, 3372–3401. [Google Scholar] [CrossRef]
- Nie, L.; Yao, D.; Chen, S.; Wang, J.; Pan, C.; Wu, D.; Liu, N.; Tang, Z. Directional Induction of Neural Stem Cells, a New Therapy for Neurodegenerative Diseases and Ischemic Stroke. Cell Death Discov. 2023, 9, 215. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease and Its Pharmaceutical Potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Huang, Y.Y.; Tsai, S.Y.; Kuo, Y.W.; Lin, J.H.; Ho, H.H.; Chen, J.F.; Hsia, K.C.; Sun, Y. Efficacy of Probiotic Supplements on Brain-Derived Neurotrophic Factor, Inflammatory Biomarkers, Oxidative Stress and Cognitive Function in Patients with Alzheimer’s Dementia: A 12-Week Randomized, Double-Blind Active-Controlled Study. Nutrients 2023, 16, 16. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; Massaro-Giordano, M.; Perez, V.L.; Hamrah, P.; Deng, S.X.; Espandar, L.; Foster, C.S.; Affeldt, J.; Seedor, J.A.; Afshari, N.A.; et al. Topical Recombinant Human Nerve Growth Factor (Cenegermin) for Neurotrophic Keratopathy: A Multicenter Randomized Vehicle-Controlled Pivotal Trial. Ophthalmology 2020, 127, 14–26. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, S.; Wang, R.; Bao, X.; Li, Y. Neural Stem Cells in the Treatment of Alzheimer’s Disease: Current Status, Challenges, and Future Prospects. J. Alzheimers Dis. 2023, 94, S173–S186. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, C.; Chen, L.; Liu, X.; Wu, J.; Sun, Y.; Liu, J.; Chen, C. Lingo1 in the Hippocampus Contributes to Cognitive Dysfunction after Anesthesia and Surgery in Aged Mice. Int. J. Biol. Sci. 2025, 21, 595–613. [Google Scholar] [CrossRef]
- Shen, L.L.; Mañucat-Tan, N.B.; Gao, S.H.; Li, W.W.; Zeng, F.; Zhu, C.; Wang, J.; Bu, X.L.; Liu, Y.H.; Gao, C.Y.; et al. The Prongf/P75ntr Pathway Induces Tau Pathology and Is a Therapeutic Target for Ftld-Tau. Mol. Psychiatry 2018, 23, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Cao, C.; Cawley, N.X.; Liu, T.T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased Peripheral Brain-Derived Neurotrophic Factor Levels in Alzheimer’s Disease: A Meta-Analysis Study (N = 7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef]
- Bharani, K.L.; Ledreux, A.; Gilmore, A.; Carroll, S.L.; Granholm, A.C. Serum Pro-BDNF Levels Correlate with Phospho-Tau Staining in Alzheimer’s Disease. Neurobiol. Aging 2020, 87, 49–59. [Google Scholar] [CrossRef]
- Ortega, A.; Chernicki, B.; Ou, G.; Parmar, M.S. From Lab Bench to Hope: Emerging Gene Therapies in Clinical Trials for Alzheimer’s Disease. Mol. Neurobiol. 2025, 62, 1112–1135. [Google Scholar] [CrossRef]
- Surya, K.; Manickam, N.; Jayachandran, K.S.; Kandasamy, M.; Anusuyadevi, M. Resveratrol Mediated Regulation of Hippocampal Neuroregenerative Plasticity Via Sirt1 Pathway in Synergy with Wnt Signaling: Neurotherapeutic Implications to Mitigate Memory Loss in Alzheimer’s Disease. J. Alzheimers Dis. 2023, 94, S125–S140. [Google Scholar] [CrossRef]
- Islam, F.; Nafady, M.H.; Islam, M.R.; Saha, S.; Rashid, S.; Akter, A.; Or-Rashid, M.H.; Akhtar, M.F.; Perveen, A.; Ashraf, G.M.; et al. Resveratrol and Neuroprotection: An Insight into Prospective Therapeutic Approaches against Alzheimer’s Disease from Bench to Bedside. Mol. Neurobiol. 2022, 59, 4384–4404. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, Y.; Li, M.; Nie, C. Curcumin Promotes Proliferation of Adult Neural Stem Cells and the Birth of Neurons in Alzheimer’s Disease Mice Via Notch Signaling Pathway. Cell Reprogram 2019, 21, 152–161. [Google Scholar] [CrossRef]
- Lou, S.; Gong, D.; Yang, M.; Qiu, Q.; Luo, J.; Chen, T. Curcumin Improves Neurogenesis in Alzheimer’s Disease Mice Via the Upregulation of Wnt/Β-Catenin and BDNF. Int. J. Mol. Sci. 2024, 25, 5123. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, S.; Kawamura, M.; Fujino, S.; Nakamura, F.; Arai, D.; Fusetani, N.; Nakao, Y. Coronarin D, a Metabolite from the Wild Turmeric, Curcuma Aromatica, Promotes the Differentiation of Neural Stem Cells into Astrocytes. J. Agric. Food Chem. 2022, 70, 3300–3309. [Google Scholar] [CrossRef]
- Masood, M.I.; Schäfer, K.H.; Naseem, M.; Weyland, M.; Meiser, P. Troxerutin Flavonoid Has Neuroprotective Properties and Increases Neurite Outgrowth and Migration of Neural Stem Cells from the Subventricular Zone. PLoS ONE 2020, 15, e0237025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Q.Q.; Ding, S.K.; Li, H.; Shang, Y.Z. Flavonoids from Stems and Leaves of Scutellaria Baicalensis Georgi Improve Composited Aβ-Induced Alzheimer’s Disease Model Rats’ Memory and Neuroplasticity Disorders. Comb. Chem. High. Throughput Screen. 2023, 26, 1519–1532. [Google Scholar] [CrossRef]
- Wang, X.F.; Xiao, H.H.; Wu, Y.T.; Kong, L.; Chen, J.C.; Yang, J.X.; Hu, X.L. Active Constituent of Polygala Tenuifolia Attenuates Cognitive Deficits by Rescuing Hippocampal Neurogenesis in App/Ps1 Transgenic Mice. BMC Complement. Med. 2021, 21, 267. [Google Scholar] [CrossRef]
- Oh, J.M.; Jeong, J.H.; Park, S.Y.; Chun, S. Ginsenoside Compound K Induces Adult Hippocampal Proliferation and Survival of Newly Generated Cells in Young and Elderly Mice. Biomolecules 2020, 10, 484. [Google Scholar] [CrossRef]
- Yang, L.; Ran, Y.; Quan, Z.; Wang, R.; Yang, Q.; Jia, Q.; Zhang, H.; Li, Y.; Peng, Y.; Liang, J.; et al. Pterostilbene, an Active Component of the Dragon’s Blood Extract, Acts as an Antidepressant in Adult Rats. Psychopharmacology 2019, 236, 1323–1333. [Google Scholar] [CrossRef]
- Zhang, H.; Xiang, L.; Yang, L.; Wu, S.; Liu, S.; Zhao, J.; Song, D.; Ma, C.; Ni, J.; Quan, Z.; et al. Ws6 Induces Adult Hippocampal Neurogenesis in Correlation to Its Antidepressant Effect on the Alleviation of Depressive-Like Behaviors of Rats. Neuroscience 2021, 473, 119–129. [Google Scholar] [CrossRef]
- Liang, J.H.; Yang, L.; Wu, S.; Liu, S.S.; Cushman, M.; Tian, J.; Li, N.M.; Yang, Q.H.; Zhang, H.A.; Qiu, Y.J.; et al. Discovery of Efficient Stimulators for Adult Hippocampal Neurogenesis Based on Scaffolds in Dragon’s Blood. Eur. J. Med. Chem. 2017, 136, 382–392. [Google Scholar] [CrossRef]
- Liu, S.S.; Ma, C.X.; Quan, Z.Y.; Ding, J.; Yang, L.; Liu, S.M.; Zhang, H.A.; Qing, H.; Liang, J.H. Discovery of Novel Diphenyl Acrylonitrile Derivatives That Promote Adult Rats’ Hippocampal Neurogenesis. Int. J. Mol. Sci. 2024, 25, 1241. [Google Scholar] [CrossRef]
- Chen, Y.; Tai, K.; Ma, P.; Su, J.; Dong, W.; Gao, Y.; Mao, L.; Liu, J.; Yuan, F. Novel Γ-Cyclodextrin-Metal-Organic Frameworks for Encapsulation of Curcumin with Improved Loading Capacity, Physicochemical Stability and Controlled Release Properties. Food Chem. 2021, 347, 128978. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.X.; Mackie, A.R.; Ettelaie, R.; Niaz, T.; Murray, B.S. Enhancement of Curcumin Bioaccessibility: An Assessment of Possible Synergistic Effect of Γ-Cyclodextrin Metal-Organic Frameworks with Micelles. Food Res. Int. 2025, 205, 115869. [Google Scholar] [CrossRef] [PubMed]
- Kopaeva, M.Y.; Cherepov, A.B.; Zaraiskaya, I.Y. Lactoferrin Has a Protective Effect on Mouse Brain Cells after Acute Gamma Irradiation of the Head. Bull. Exp. Biol. Med. 2023, 176, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, M.; Cianciulli, A.; Calvello, R.; Lofrumento, D.D.; Saponaro, C.; Filannino, F.M.; Porro, C.; Panaro, M.A. Lactoferrin Attenuates Pro-Inflammatory Response and Promotes the Conversion into Neuronal Lineages in the Astrocytes. Int. J. Mol. Sci. 2025, 26, 405. [Google Scholar] [CrossRef]
- Huang, Z.; Li, F.; Zheng, X.; Zheng, J.; Dong, Y.; Ding, Z.; Gou, H.; Yao, M.; Liu, J. Catalpol Promotes Hippocampal Neurogenesis and Synaptogenesis in Rats after Multiple Cerebral Infarctions by Mitochondrial Regulation: Involvement of the Shh Signaling Pathway. Front. Pharmacol. 2024, 15, 1461279. [Google Scholar] [CrossRef]
- Xing, S.; Xu, S.; Wang, L.; Guo, L.; Zhou, X.; Wu, H.; Wang, W.; Liu, L. Salidroside Exerts Antidepressant-Like Action by Promoting Adult Hippocampal Neurogenesis through Sirt1/Pgc-1α Signalling. Acta Neuropsychiatr. 2024, 36, 446–456. [Google Scholar] [CrossRef]
- Zeng, J.; Xie, Z.; Chen, L.; Peng, X.; Luan, F.; Hu, J.; Xie, H.; Liu, R.; Zeng, N. Rosmarinic Acid Alleviate Cort-Induced Depressive-Like Behavior by Promoting Neurogenesis and Regulating BDNF/Trkb/Pi3k Signaling Axis. Biomed. Pharmacother. 2024, 170, 115994. [Google Scholar] [CrossRef]
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise Benefits on Alzheimer’s Disease: State-of-the-Science. Ageing Res. Rev. 2020, 62, 101108. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Sawano, T.; Fukumoto, K.; Nakatani, J.; Inoue, S.; Doe, N.; Yanagisawa, D.; Tooyama, I.; Nakagomi, T.; Matsuyama, T.; et al. Voluntary Running Exercise after Focal Cerebral Ischemia Ameliorates Dendritic Spine Loss and Promotes Functional Recovery. Brain Res. 2021, 1767, 147542. [Google Scholar] [CrossRef]
- Akers, K.G.; Martinez-Canabal, A.; Restivo, L.; Yiu, A.P.; De Cristofaro, A.; Hsiang, H.L.; Wheeler, A.L.; Guskjolen, A.; Niibori, Y.; Shoji, H.; et al. Frankland. Hippocampal Neurogenesis Regulates Forgetting During Adulthood and Infancy. Science 2014, 344, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Juliandi, B.; Tanemura, K.; Igarashi, K.; Tominaga, T.; Furukawa, Y.; Otsuka, M.; Moriyama, N.; Ikegami, D.; Abematsu, M.; Sanosaka, T.; et al. Reduced Adult Hippocampal Neurogenesis and Cognitive Impairments Following Prenatal Treatment of the Antiepileptic Drug Valproic Acid. Stem Cell Rep. 2015, 5, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF Signaling in Context: From Synaptic Regulation to Psychiatric Disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef]
- Li, W.P.; Su, X.H.; Hu, N.Y.; Hu, J.; Li, X.W.; Yang, J.M.; Gao, T.M. Astrocytes Mediate Cholinergic Regulation of Adult Hippocampal Neurogenesis and Memory through M(1) Muscarinic Receptor. Biol. Psychiatry 2022, 92, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wei, X.; Mei, Y.; Wang, D.; Wang, J.; Zhang, Y.; Li, X.; Gu, Y.; Peng, G.; Sun, B. Modulating Adult Neurogenesis Affects Synaptic Plasticity and Cognitive Functions in Mouse Models of Alzheimer’s Disease. Stem Cell Rep. 2021, 16, 3005–3019. [Google Scholar] [CrossRef]
- Liew, A.K.Y.; Teo, C.H.; Soga, T. The Molecular Effects of Environmental Enrichment on Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 7095–7118. [Google Scholar] [CrossRef]
- Kim, T.A.; Syty, M.D.; Wu, K.; Ge, S. Adult Hippocampal Neurogenesis and Its Impairment in Alzheimer’s Disease. Zool. Res. 2022, 43, 481–496. [Google Scholar] [CrossRef]
- Ninkovic, J.; Götz, M. Signaling in Adult Neurogenesis: From Stem Cell Niche to Neuronal Networks. Curr. Opin. Neurobiol. 2007, 17, 338–344. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Ohtsuka, T.; Takao, K.; Miyakawa, T.; Yamaguchi, M.; Mori, K.; Ikeda, T.; Itohara, S.; Kageyama, R. Roles of Continuous Neurogenesis in the Structural and Functional Integrity of the Adult Forebrain. Nat. Neurosci. 2008, 11, 1153–1161. [Google Scholar] [CrossRef]
- Lazarov, O.; Hollands, C. Hippocampal Neurogenesis: Learning to Remember. Prog. Neurobiol. 2016, 138–140, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Walter, E.; Wohleb, E.; Fan, Y.; Wang, C. Atg5 (Autophagy Related 5) in Microglia Controls Hippocampal Neurogenesis in Alzheimer Disease. Autophagy 2024, 20, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lapiscina, E.H.; Clavero, P.; Toledo, E.; Estruch, R.; Salas-Salvadó, J.; Julián, B.S.; Sanchez-Tainta, A.; Ros, E.; Valls-Pedret, C.; Martinez-Gonzalez, M. Mediterranean Diet Improves Cognition: The Predimed-Navarra Randomised Trial. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-Mediated Brain Drug Delivery: Overcoming Blood-Brain Barrier to Treat Neurodegenerative Diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef]
- Niu, W.; Xiao, Q.; Wang, X.; Zhu, J.; Li, J.; Liang, X.; Peng, Y.; Wu, C.; Lu, R.; Pan, Y.; et al. A Biomimetic Drug Delivery System by Integrating Grapefruit Extracellular Vesicles and Doxorubicin-Loaded Heparin-Based Nanoparticles for Glioma Therapy. Nano Lett. 2021, 21, 1484–1492. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Molecule | References | Mechanism of Action | Impact on Neurogenesis | Role in AD Pathology |
|---|---|---|---|---|
| Acetylcholine (ACh) | [47,48,49] | Released by cholinergic neurons (regulated by ChAT/AchE). Activates AMPAR to enhance ACh levels (Lancao Decoction). | Supports synaptic plasticity and neuronal differentiation. Upregulates hippocampal neural activity. | Early AD pathology involves cholinergic neuron loss in the basal forebrain. Reduced ChAT activity and ACh levels correlate with cognitive decline. Restoring ACh signaling rescues memory deficits. |
| GABA | [17,50,51] | Inhibitory neurotransmitter regulating neural precursor quiescence. Tau accumulation in GABAergic interneurons disrupts GABAergic signaling. | Maintains NSC quiescence via long-range GABAergic inputs. Impaired GABAergic transmission suppresses AHN. | Tau pathology in GABAergic interneurons causes circuit imbalance and AHN deficits. Enhancing GABAergic signaling rescues Tau -induced AHN impairment. |
| CREB Pathway | [52,54,55,58,60,61] | Activated by NO/cGMP/PKG, BDNF/TrkB, or PKA. Regulates neuronal survival, synaptic plasticity, and memory. | Promotes neurogenesis and synaptic development. Critical for integrating new neurons into networks. | Tau inhibits PKA/CREB/BDNF signaling, impairing memory. rescue CREB phosphorylation to improve cognition. |
| Dopamine (DA) | [58] | Degeneration of VTA dopaminergic neurons reduces hippocampal DA innervation. Regulates PV interneuron activity via D2 receptors. | DA loss disrupts PV interneuron-mediated inhibition, impairing gamma oscillations. | Hippocampal overexcitation due to DA deficiency exacerbates AD pathology. L-DOPA/D2 agonists restore p-CREB levels and reduce hyperexcitability. |
| Abnormally phosphorylated Tau proteins | [17,59,60] | GABAergic neuronal injury, inhibiting synaptic transmission. Suppresses PKA/CREB/BDNF signaling. | Tau suppresses AHN by impairing GABAergic transmission/CREB activity. | Hyper-pTau disrupts synaptic plasticity and memory. Curcumin/melatonin reduce Tau phosphorylation and restore CREB signaling. |
| cAMP/ cGMP | [53,54] | NO/cGMP/CREB pathway supports synaptic plasticity. Calcium/CaMKII/CREB signaling impaired by aluminum toxicity. | Enhances neurogenesis under physiological conditions. | Aβ/Tau disrupt NO/cGMP/CREB crosstalk, contributing to cognitive decline. Restoring cAMP/CREB signaling mitigates AD pathology. |
| Neurotrophic Factor | References | Mechanism in Neurogenesis | Role in AD Pathology | Therapeutic Approaches |
|---|---|---|---|---|
| BDNF | [62,63,64,65,66,68,69,74,75,76] | Promotes neural stem cell (NSC) proliferation and differentiation. Enhances synaptic plasticity and neuronal survival via TrkB signaling. | The peripheral blood BDNF level in AD patients is significantly reduced Aβ-induced TrkB-FL cleavage impairs BDNF signaling. | TAT-TrkB peptide prevents TrkB cleavage, rescues synaptic defects. Exosomes/nanocarriers deliver BDNF to the brain. |
| NGF | [65,67,70,71,77] | Supports cholinergic neuron survival and differentiation. Enhances synaptic repair. | Cholinergic neuron degeneration in AD leads to cognitive deficits. | NGF gene therapy improves cognition in models of familial AD. |
| NT-3/NT-4 | [62] | Mimic BDNF functions in neurite outgrowth via PI3K/AKT and ERK pathways. | Limited direct evidence in AD; implied roles in neuroprotection. | Potential for flavonoid-based therapies. |
| Lingo1 | [72,73] | Inhibits EGFR/PI3K/AKT pathway, increasing neuronal apoptosis. Promotes myelin loss. | Upregulation in AD induces Tau hyperphosphorylation via RhoA/ROCK1 signaling. | Genetic reduction of Lingo1 ameliorates cognitive dysfunction and Tau pathology. |
| p75NTR | [72,73] | Interacts with proNGF to regulate neuronal survival. Modulates AKT/GSK3β pathway. | Overactivation exacerbates Tau pathology in frontotemporal dementia (FTD) and AD. | p75NTR inhibition rescues memory deficits and reduces Tau phosphorylation. |
| proBDNF | [75] | Precursor to mature BDNF; roles in synaptic pruning. | Elevated hippocampal proBDNF correlates with pTau in AD. | Targeting proBDNF/mature BDNF balance may reduce Tau burden. |
| Compound/Source | References | Mechanism in Neurogenesis | Impact on AD Pathology | Key Findings |
|---|---|---|---|---|
| Resveratrol | [78,79,99] | Activates SIRT1 and Wnt/β-catenin pathways. Stabilizes β-catenin via GSK-3β phosphorylation (Ser9), inducing NeuroD1-mediated differentiation. | Enhances hippocampal neurogenesis. Improves cognitive function in models of familial AD. | Synergizes with Wnt signaling to mitigate Aβ-induced neurogenesis deficits. |
| Curcumin | [80,81] | Upregulates Notch1/Hes1, CDK4/cyclin D1, and NICD. Activates PI3K/AKT, GSK3β/Wnt, and CREB/BDNF pathways. | Reduces Aβ plaques and Tau hyperphosphorylation. Rescues cognitive deficits in APP/PS1 mice. | Poor oral bioavailability; nano-encapsulation improves solubility and efficacy. |
| Coronarin D (Curcuma aromatica) | [82,98] | Promotes astrocyte differentiation via JAK/STAT signaling. Increases GFAP expression (mRNA and protein). | Role in AD unclear; potential for glial support in neuro regeneration. | Derived from wild turmeric; targets astrocyte lineage differentiation. |
| Troxerutin (Flavonoid) | [83] | Enhances neuronal differentiation and axon growth. Reverses Aβ42-induced axonal inhibition. | Mitigates Aβ toxicity in neural stem cells. | Rescues Aβ-induced axonal growth defects and promotes cell migration. |
| Scutellaria Flavonoids (Scutellaria baicalensis) | [84] | Activates CaM-CamkIV-CREB pathway. Restores synaptic plasticity. | Ameliorates Aβ-induced learning/memory deficits. | Improves neuroplasticity in Aβ-induced AD rat models. |
| DISS (Polygala tenuifolia) | [85,97] | Enhances NSC proliferation and neuronal differentiation. | Rescues cognitive deficits and neuronal damage in APP/PS1 mice. | neurogenesis in transgenic mice. |
| Ginsenoside CK (Ginseng) | [86] | Stimulates hippocampal neurogenesis in young (2-month) and aged (24-month) mice. | Improves cognitive function across age groups (p < 0.01–0.001). | Increased hippocampal neurogenesis (5–15 mg/kg). |
| Dragon’s Blood Extract (QLX-N) | [87,90,99,100] | Induces BDNF and activates MAPK/AKT pathway. Enhances BrdU+/Nestin+ proliferation (116% and DCX+ differentiation (2.3×). | Improves spatial learning and motor function in models of familial AD. | No reported adverse effects. |
| γ-CD MOFs (Curcumin) | [91,92] | Increases curcumin solubility (53× with bile salts + γ-CD MOFs). Enhances bioavailability (16% vs. 2% without MOFs). | Facilitates sustained delivery of curcumin for AD therapy. | In vitro studies confirm improved stability and controlled release. |
| Intervention Type | References | Mechanism in Neurogenesis | Impact on AD Pathology | Key Findings |
|---|---|---|---|---|
| Physical Exercise | [63,93,94,95,96,97,98] | Induces BDNF upregulation. Increases cell proliferation, migration, and dendritic spine remodeling. | Reduces neuroinflammation. Enhances cognitive function via BDNF-TrkB pathway activation. | Exercise improves neural network re-establishment in 3xTg-AD mice, delaying cognitive decline. |
| Environmental Enrichment (EE) | [68,91,99] | Boosts DCX+ neurons in DG. Activates MAPK/ERK and PI3k-AKT pathways to enhance synaptic plasticity. | Improves learning/memory in models of familial AD. | Six weeks of EE significantly benefits young mice but has limited applicability in severe AD cases. |
| Ex Vivo Stem Cell Therapy | [20,39] | Promotes synaptic integration and lesion recovery via transplanted stem cells. Uses allogeneic NSC transplantation. | Repairs neuronal loss and restores neural function. | Challenges include low cell survival and immune rejection; currently experimental. |
| Combined Interventions | [107,108,109] | Combines exercise with Mediterranean diet (antioxidants/anti-inflammatory). Nanoparticles enhance BBB penetration. | Synergistically reduces inflammation and improves cognition. | Nanoparticle delivery systems improve targeting and efficacy of neurogenic factors. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, L.; Wei, Q.; Jiang, M.; Wu, Y.; Liu, X.; Yang, Q.; Bai, Z.; Yang, L. Hippocampal Neurogenesis in Alzheimer’s Disease: Multimodal Therapeutics and the Neurogenic Impairment Index Framework. Int. J. Mol. Sci. 2025, 26, 6105. https://doi.org/10.3390/ijms26136105
Ma L, Wei Q, Jiang M, Wu Y, Liu X, Yang Q, Bai Z, Yang L. Hippocampal Neurogenesis in Alzheimer’s Disease: Multimodal Therapeutics and the Neurogenic Impairment Index Framework. International Journal of Molecular Sciences. 2025; 26(13):6105. https://doi.org/10.3390/ijms26136105
Chicago/Turabian StyleMa, Li, Qian Wei, Ming Jiang, Yanyan Wu, Xia Liu, Qinghu Yang, Zhantao Bai, and Liang Yang. 2025. "Hippocampal Neurogenesis in Alzheimer’s Disease: Multimodal Therapeutics and the Neurogenic Impairment Index Framework" International Journal of Molecular Sciences 26, no. 13: 6105. https://doi.org/10.3390/ijms26136105
APA StyleMa, L., Wei, Q., Jiang, M., Wu, Y., Liu, X., Yang, Q., Bai, Z., & Yang, L. (2025). Hippocampal Neurogenesis in Alzheimer’s Disease: Multimodal Therapeutics and the Neurogenic Impairment Index Framework. International Journal of Molecular Sciences, 26(13), 6105. https://doi.org/10.3390/ijms26136105