
Isolation and Characterization of Tissue-Derived Extracellular Vesicles from Mouse Lymph Nodes

 , , , , ,
, , , , , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
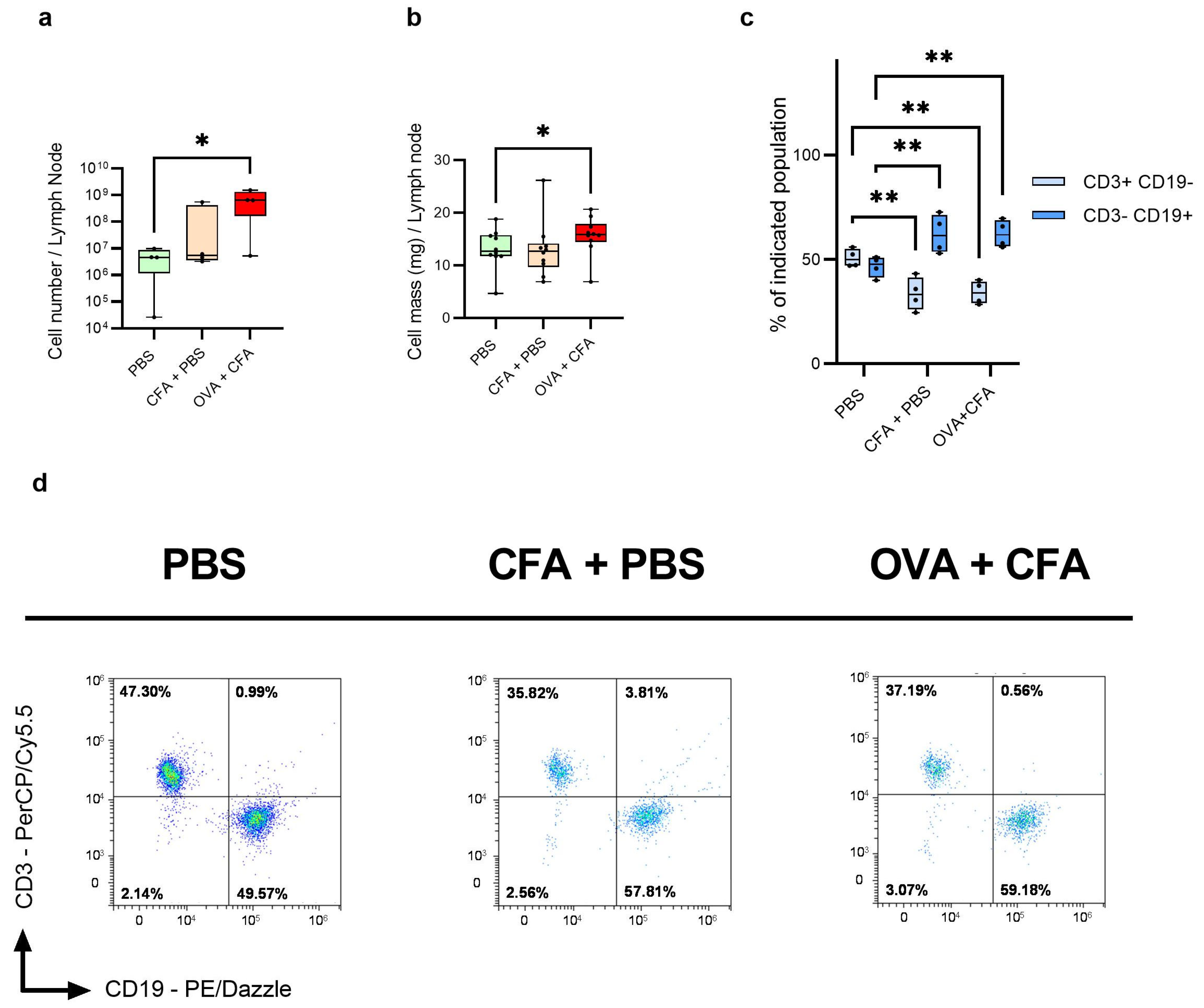
2.1. OVA + CFA Treatment Increases the Lymph Node Mass and Cell Number
2.2. Immunization Reduces the CD3+ CD19− T Cell Number and Increases the Number of CD3− CD19+ B Cells as Compared to Controls
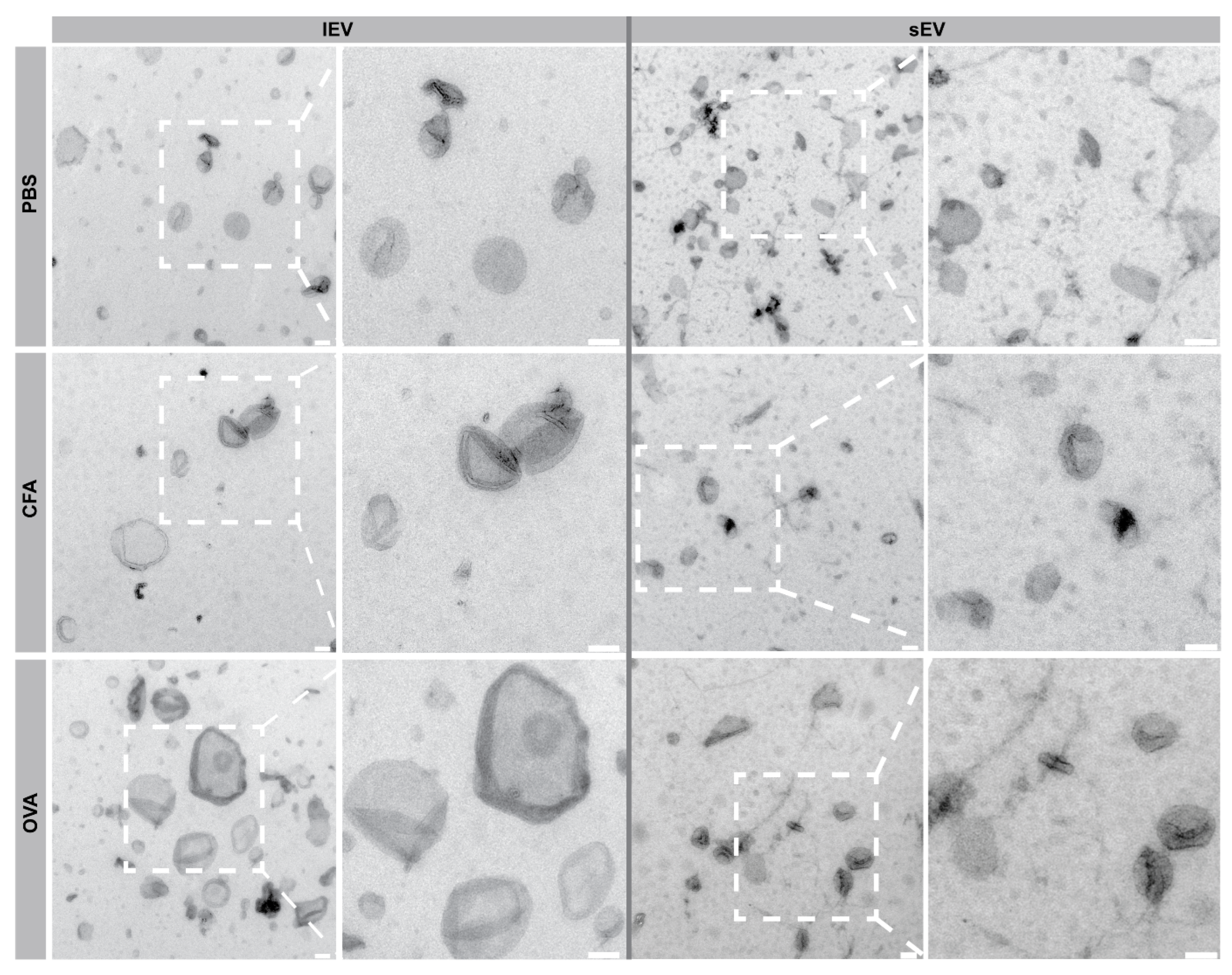
2.3. Transmission Electron Microscopy (TEM) Revealed lEV and sEV Morphology
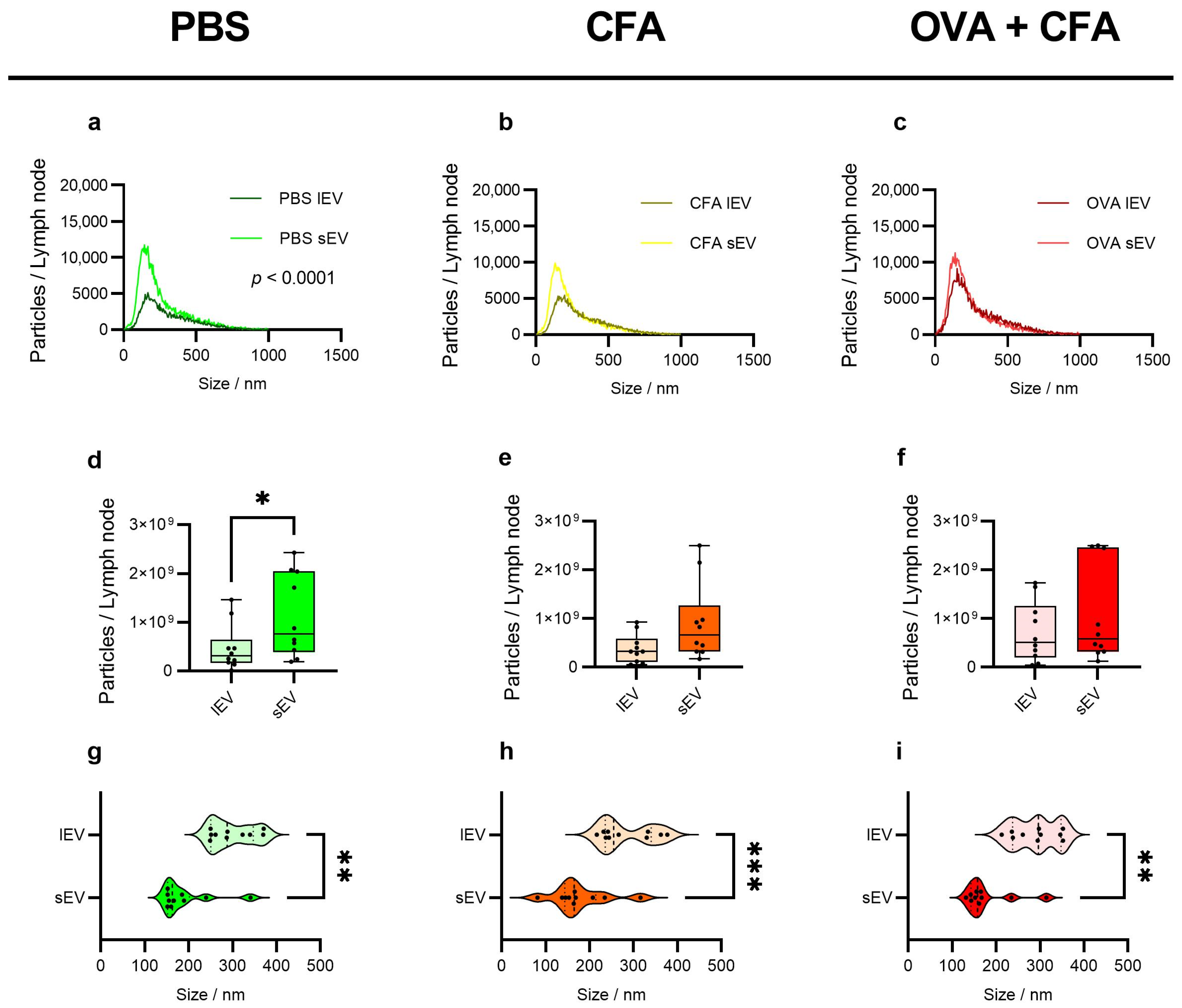
2.4. sEVs Show Higher Particle Counts than lEVs with Variable Trends Across Groups
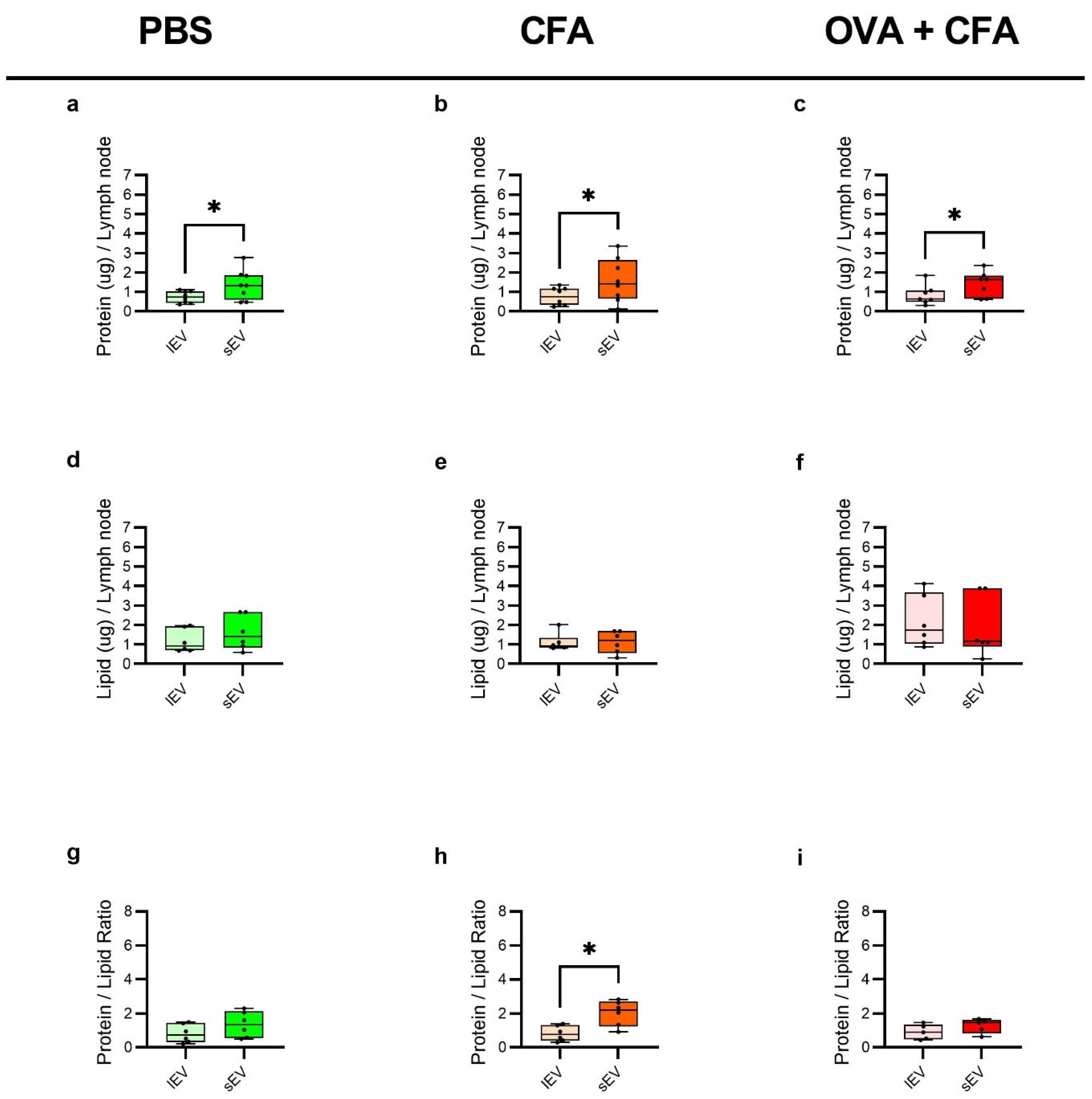
2.5. sEVs Show Higher Protein Content and Protein-to-Lipid Ratio than lEVs Across Groups, with Minimal Difference in Lipid Content
2.6. Surface Marker Profiling Reveals Distinct Immunological Signatures on lEVs and sEVs with Significant Shifts in CD146, MHC II, and CD45 upon OVA + CFA Treatment
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Immunization of the Mice
4.3. LN Isolation and Cell Extraction
4.4. Immunophenotyping of LN Cells
4.5. LN EVs Isolation by Differential Centrifugation Method
4.6. LN EVs Purification by SEC
4.7. TEM Analysis of LN EVs
4.8. Particle Size Distribution Analysis
4.9. Total Protein Content Quantification
4.10. Total Lipid Content Quantification
4.11. Calculation of the Protein per Particle, Lipid per Particle, Protein to Lipid Ratio, and EV Yield
4.12. Bead-Based Analysis of LN EVs
4.13. Western Blot Analysis
4.14. Software and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrugger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lan, M.; Chen, Y. Minimal Information for Studies of Extracellular Vesicles (MISEV): Ten-Year Evolution (2014–2023). Pharmaceutics 2024, 16, 1394. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2023, 23, 236–250. [Google Scholar] [CrossRef]
- Thery, C.; Duban, L.; Segura, E.; Veron, P.; Lantz, O.; Amigorena, S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Schneider, H.; Stumptner-Cuvelette, P.; Lankar, D.; Pain, S.; Raposo, G.; Benaroch, P.; Bonnerot, C. Exosomes bearing HLA-DR1 molecules need dendritic cells to efficiently stimulate specific T cells. Int. Immunol. 2002, 14, 713–722. [Google Scholar] [CrossRef]
- Kato, T.; Fahrmann, J.F.; Hanash, S.M.; Vykoukal, J. Extracellular Vesicles Mediate B Cell Immune Response and Are a Potential Target for Cancer Therapy. Cells 2020, 9, 1518. [Google Scholar] [CrossRef]
- Menay, F.; Cocozza, F.; Gravisaco, M.J.; Elisei, A.; Re, J.I.; Ferella, A.; Waldner, C.; Mongini, C. Extracellular vesicles derived from antigen-presenting cells pulsed with foot and mouth virus vaccine-antigens act as carriers of viral proteins and stimulate B cell response. Front. Immunol. 2024, 15, 1440667. [Google Scholar] [CrossRef]
- Nazimek, K.; Bryniarski, K. Approaches to inducing antigen-specific immune tolerance in allergy and autoimmunity: Focus on antigen-presenting cells and extracellular vesicles. Scand. J. Immunol. 2020, 91, e12881. [Google Scholar] [CrossRef]
- Safaei, S.; Alipour, S.; Bahojb Mahdavi, S.Z.; Shalmashi, H.; Shahgoli, V.K.; Shanehbandi, D.; Baradaran, B.; Kazemi, T. Triple-negative breast cancer-derived exosomes change the immunological features of human monocyte-derived dendritic cells and influence T-cell responses. Mol. Biol. Rep. 2024, 51, 1058. [Google Scholar] [CrossRef]
- Gyorgy, B.; Hung, M.E.; Breakefield, X.O.; Leonard, J.N. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439–464. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.A.; Lotvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef]
- Santos, P.; Almeida, F. Exosome-Based Vaccines: History, Current State, and Clinical Trials. Front. Immunol. 2021, 12, 711565. [Google Scholar] [CrossRef] [PubMed]
- Mustajab, T.; Kwamboka, M.S.; Choi, D.A.; Kang, D.W.; Kim, J.; Han, K.R.; Han, Y.; Lee, S.; Song, D.; Chwae, Y.J. Update on Extracellular Vesicle-Based Vaccines and Therapeutics to Combat COVID-19. Int. J. Mol. Sci. 2022, 23, 11247. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Chung, Y.; Tu, C.R.; Zhang, W.; Mu, X.; Wang, M.; Chan, G.C.; Leung, W.H.; Lau, Y.L.; et al. Tumor vaccine based on extracellular vesicles derived from gammadelta-T cells exerts dual antitumor activities. J. Extracell. Vesicles 2023, 12, e12360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Sim, W.K.; Shen, T.L.; Lim, S.K. Engineered EVs with pathogen proteins: Promising vaccine alternatives to LNP-mRNA vaccines. J. Biomed. Sci. 2024, 31, 9. [Google Scholar] [CrossRef]
- Phung, C.D.; Pham, T.T.; Nguyen, H.T.; Nguyen, T.T.; Ou, W.; Jeong, J.H.; Choi, H.G.; Ku, S.K.; Yong, C.S.; Kim, J.O. Anti-CTLA-4 antibody-functionalized dendritic cell-derived exosomes targeting tumor-draining lymph nodes for effective induction of antitumor T-cell responses. Acta Biomater. 2020, 115, 371–382. [Google Scholar] [CrossRef]
- Ke, Y.; Li, Y.; Kapp, J.A. Ovalbumin injected with complete Freund’s adjuvant stimulates cytolytic responses. Eur. J. Immunol. 1995, 25, 549–553. [Google Scholar] [CrossRef]
- Rezende, R.M.; Lanser, A.J.; Rubino, S.; Kuhn, C.; Skillin, N.; Moreira, T.G.; Liu, S.; Gabriely, G.; David, B.A.; Menezes, G.B.; et al. γδ T cells control humoral immune response by inducing T follicular helper cell differentiation. Nat. Commun. 2018, 9, 3151. [Google Scholar] [CrossRef]
- Rivera, A.; Chen, C.C.; Ron, N.; Dougherty, J.P.; Ron, Y. Role of B cells as antigen-presenting cells in vivo revisited: Antigen-specific B cells are essential for T cell expansion in lymph nodes and for systemic T cell responses to low antigen concentrations. Int. Immunol. 2001, 13, 1583–1593. [Google Scholar] [CrossRef]
- Dotta, E.; Maciola, A.K.; Baccega, T.; Pasqual, G. Dendritic cells steering antigen and leukocyte traffic in lymph nodes. FEBS Lett. 2024. [Google Scholar] [CrossRef]
- Li, S.R.; Man, Q.W.; Gao, X.; Lin, H.; Wang, J.; Su, F.C.; Wang, H.Q.; Bu, L.L.; Liu, B.; Chen, G. Tissue-derived extracellular vesicles in cancers and non-cancer diseases: Present and future. J. Extracell. Vesicles 2021, 10, e12175. [Google Scholar] [CrossRef]
- Ye, Z.; Chen, W.; Li, G.; Huang, J.; Lei, J. Tissue-derived extracellular vesicles in cancer progression: Mechanisms, roles, and potential applications. Cancer Metastasis Rev. 2024, 43, 575–595. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.N.; Olcese, J.M.; Meckes, D.G., Jr. Extraction of Extracellular Vesicles from Whole Tissue. J. Vis. Exp. 2019, 144, e59143. [Google Scholar]
- Bordas, M.; Genard, G.; Ohl, S.; Nessling, M.; Richter, K.; Roider, T.; Dietrich, S.; Maass, K.K.; Seiffert, M. Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues. Int. J. Mol. Sci. 2020, 21, 5586. [Google Scholar] [CrossRef]
- Crescitelli, R.; Lasser, C.; Lotvall, J. Isolation and characterization of extracellular vesicle subpopulations from tissues. Nat. Protoc. 2021, 16, 1548–1580. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.J.; Chau, Z.L.; Chen, S.Y.; Hill, J.J.; Korpany, K.V.; Liang, N.W.; Lin, L.H.; Lin, Y.H.; Liu, J.K.; Liu, Y.C.; et al. Exosome Processing and Characterization Approaches for Research and Technology Development. Adv. Sci. 2022, 9, e2103222. [Google Scholar] [CrossRef]
- Crescitelli, R.; Lasser, C.; Jang, S.C.; Cvjetkovic, A.; Malmhall, C.; Karimi, N.; Hoog, J.L.; Johansson, I.; Fuchs, J.; Thorsell, A.; et al. Subpopulations of extracellular vesicles from human metastatic melanoma tissue identified by quantitative proteomics after optimized isolation. J. Extracell. Vesicles 2020, 9, 1722433. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Wang, J.; Liu, T.; Tian, T.; Li, T.; Ding, L.; Chen, W.; Wang, H.; Zhu, J.; et al. Optimizing of a suitable protocol for isolating tissue-derived extracellular vesicles and profiling small RNA patterns in hepatocellular carcinoma. Liver Int. 2024, 44, 2672–2686. [Google Scholar] [CrossRef]
- Webster, B.; Ekland, E.H.; Agle, L.M.; Chyou, S.; Ruggieri, R.; Lu, T.T. Regulation of lymph node vascular growth by dendritic cells. J. Exp. Med. 2006, 203, 1903–1913. [Google Scholar] [CrossRef]
- Chyou, S.; Benahmed, F.; Chen, J.; Kumar, V.; Tian, S.; Lipp, M.; Lu, T.T. Coordinated regulation of lymph node vascular-stromal growth first by CD11c+ cells and then by T and B cells. J. Immunol. 2011, 187, 5558–5567. [Google Scholar] [CrossRef]
- Abe, J.; Ueha, S.; Yoneyama, H.; Shono, Y.; Kurachi, M.; Goto, A.; Fukayama, M.; Tomura, M.; Kakimi, K.; Matsushima, K. B cells regulate antibody responses through the medullary remodeling of inflamed lymph nodes. Int. Immunol. 2012, 24, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Ray, R.M.; Scott, T.A. Prospects and challenges of tissue-derived extracellular vesicles. Mol. Ther. 2024, 32, 2950–2978. [Google Scholar] [CrossRef] [PubMed]
- Courtney, A.H.; Shvets, A.A.; Lu, W.; Griffante, G.; Mollenauer, M.; Horkova, V.; Lo, W.L.; Yu, S.; Stepanek, O.; Chakraborty, A.K.; et al. CD45 functions as a signaling gatekeeper in T cells. Sci. Signal. 2019, 12, eaaw8151. [Google Scholar] [CrossRef] [PubMed]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Van Niel, G.; Mallegol, J.; Bevilacqua, C.; Candalh, C.; Brugiere, S.; Tomaskovic-Crook, E.; Heath, J.K.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial exosomes carry MHC class II/peptides able to inform the immune system in mice. Gut 2003, 52, 1690–1697. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Denzer, K.; van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; de Groot, C.; Geuze, H.J. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef]
- Buschow, S.I.; Nolte-‘t Hoen, E.N.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Buschow, S.I.; van Balkom, B.W.; Aalberts, M.; Heck, A.J.; Wauben, M.; Stoorvogel, W. MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol. Cell Biol. 2010, 88, 851–856. [Google Scholar] [CrossRef]
- Testa, J.S.; Apcher, G.S.; Comber, J.D.; Eisenlohr, L.C. Exosome-driven antigen transfer for MHC class II presentation facilitated by the receptor binding activity of influenza hemagglutinin. J. Immunol. 2010, 185, 6608–6616. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, H.O.; Ju, Y.J.; Kye, Y.C.; Lee, G.W.; Lee, S.W.; Yun, C.H.; Bottini, N.; Webster, K.; Goodnow, C.C.; et al. CD45-mediated control of TCR tuning in naive and memory CD8+ T cells. Nat. Commun. 2016, 7, 13373. [Google Scholar] [CrossRef]
- Dube, J.Y.; McIntosh, F.; Zarruk, J.G.; David, S.; Nigou, J.; Behr, M.A. Synthetic mycobacterial molecular patterns partially complete Freund’s adjuvant. Sci. Rep. 2020, 10, 5874. [Google Scholar] [CrossRef]
- Osteikoetxea, X.; Silva, A.; Lazaro-Ibanez, E.; Salmond, N.; Shatnyeva, O.; Stein, J.; Schick, J.; Wren, S.; Lindgren, J.; Firth, M.; et al. Engineered Cas9 extracellular vesicles as a novel gene editing tool. J. Extracell. Vesicles 2022, 11, e12225. [Google Scholar] [CrossRef]
- Osteikoetxea, X.; Balogh, A.; Szabó-Taylor, K.; Németh, A.; Szabó, T.G.; Pálóczi, K.; Sódar, B.; Kittel, Á.; György, B.; Pállinger, É.; et al. Improved Characterization of EV Preparations Based on Protein to Lipid Ratio and Lipid Properties. PLoS ONE 2015, 10, e0121184. [Google Scholar] [CrossRef]
- Visnovitz, T.; Osteikoetxea, X.; Sodar, B.W.; Mihaly, J.; Lorincz, P.; Vukman, K.V.; Toth, E.A.; Koncz, A.; Szekacs, I.; Horvath, R.; et al. An improved 96 well plate format lipid quantification assay for standardisation of experiments with extracellular vesicles. J. Extracell. Vesicles 2019, 8, 1565263. [Google Scholar] [CrossRef]
- Kestecher, B.M.; Nemeth, K.; Ghosal, S.; Sayour, N.V.; Gergely, T.G.; Bodnar, B.R.; Forsonits, A.I.; Sodar, B.W.; Oesterreicher, J.; Holnthoner, W.; et al. Reduced circulating CD63+ extracellular vesicle levels associate with atherosclerosis in hypercholesterolaemic mice and humans. Cardiovasc. Diabetol. 2024, 23, 368. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodnár, B.R.; Ghosal, S.; Kestecher, B.M.; Királyhidi, P.; Försönits, A.; Fekete, N.; Bugyik, E.; Komlósi, Z.I.; Pállinger, É.; Nagy, G.; et al. Isolation and Characterization of Tissue-Derived Extracellular Vesicles from Mouse Lymph Nodes. Int. J. Mol. Sci. 2025, 26, 6092. https://doi.org/10.3390/ijms26136092
Bodnár BR, Ghosal S, Kestecher BM, Királyhidi P, Försönits A, Fekete N, Bugyik E, Komlósi ZI, Pállinger É, Nagy G, et al. Isolation and Characterization of Tissue-Derived Extracellular Vesicles from Mouse Lymph Nodes. International Journal of Molecular Sciences. 2025; 26(13):6092. https://doi.org/10.3390/ijms26136092
Chicago/Turabian StyleBodnár, Bernadett R., Sayam Ghosal, Brachyahu M. Kestecher, Panna Királyhidi, András Försönits, Nóra Fekete, Edina Bugyik, Zsolt I. Komlósi, Éva Pállinger, György Nagy, and et al. 2025. "Isolation and Characterization of Tissue-Derived Extracellular Vesicles from Mouse Lymph Nodes" International Journal of Molecular Sciences 26, no. 13: 6092. https://doi.org/10.3390/ijms26136092
APA StyleBodnár, B. R., Ghosal, S., Kestecher, B. M., Királyhidi, P., Försönits, A., Fekete, N., Bugyik, E., Komlósi, Z. I., Pállinger, É., Nagy, G., Buzás, E. I., & Osteikoetxea, X. (2025). Isolation and Characterization of Tissue-Derived Extracellular Vesicles from Mouse Lymph Nodes. International Journal of Molecular Sciences, 26(13), 6092. https://doi.org/10.3390/ijms26136092