
Immunopathology of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis



Abstract
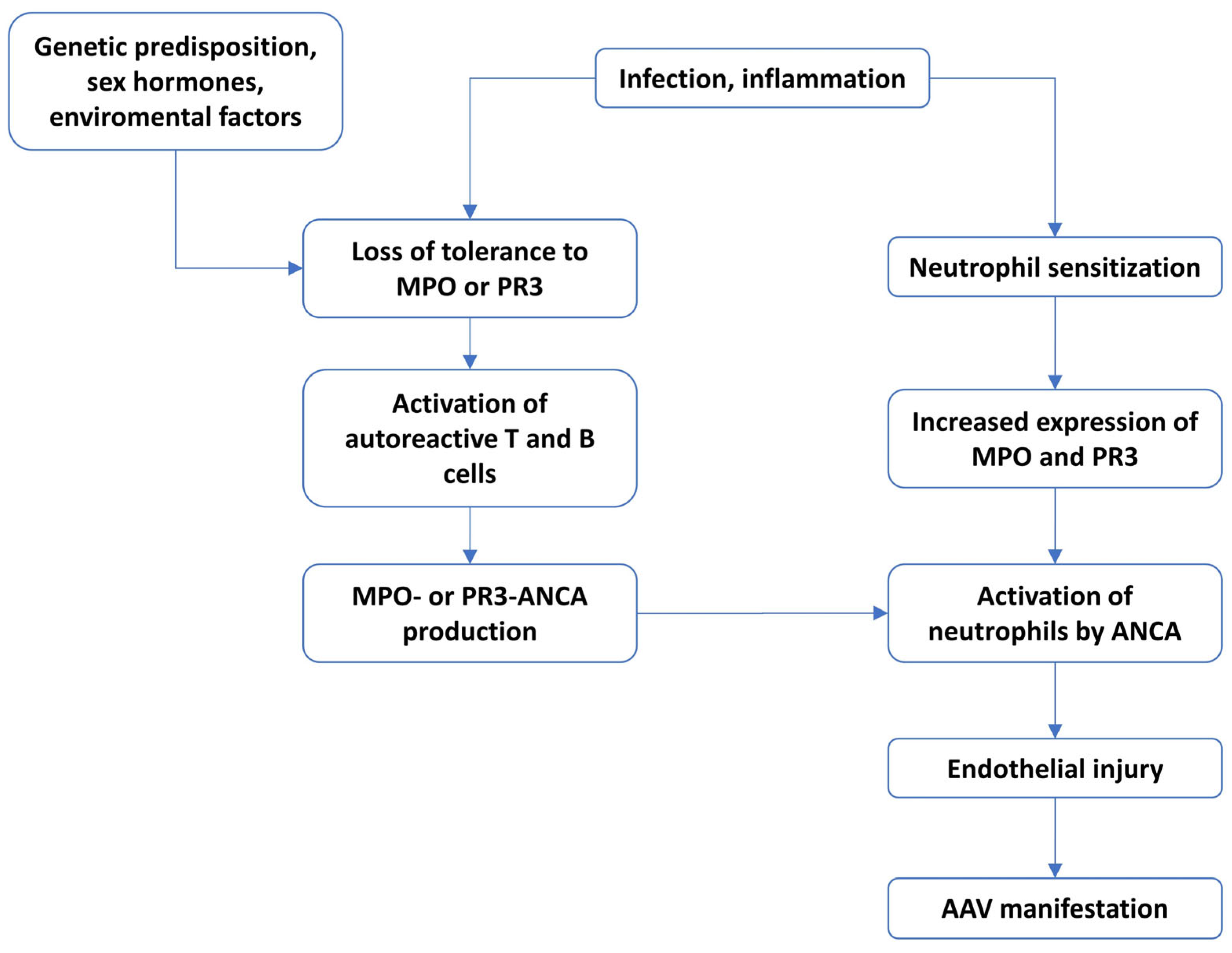
1. Introduction
2. Epidemiology
3. Clinical Manifestation
4. AAV Classification
5. Genetic Background of AAV
5.1. HLA Genes and Their Association with AAV
5.2. Role of Non-HLA Genes in AAV
5.3. Insights from Genome-Wide Association Studies (GWASs)
6. Immunopathomechanism in AAV
6.1. The Role of ANCA and Neutrophils
6.2. The Role of Monocytes in AAV
6.3. Eosinophils as Essential Cells in EGPA
6.4. Importance of DCs
6.5. Lymphocytes in AAV
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Cui, Z.; Long, J.; Huang, W.; Wang, J.; Wang, H.; Zhang, L.; Chen, M.; Zhao, M. The Frequency of ANCA-Associated Vasculitis in a National Database of Hospitalized Patients in China. Arthritis Res. Ther. 2018, 20, 226. [Google Scholar] [CrossRef] [PubMed]
- Berti, A.; Cornec, D.; Crowson, C.S.; Specks, U.; Matteson, E.L. The Epidemiology of Antineutrophil Cytoplasmic Autoantibody–Associated Vasculitis in Olmsted County, Minnesota: A Twenty-Year US Population–Based Study. Arthritis Rheumatol. 2017, 69, 2338–2350. [Google Scholar] [CrossRef]
- Coattrenec, Y.; Muller, Y.D.; Spoerl, D.; Lobrinus, J.A.; Seebach, J.D. Prevalence of large vessel vasculitis in ANCA-associated vasculitis: A retrospective cohort study. Rheumatol. Int. 2021, 41, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Kaymakci, M.S.; Elfishawi, M.M.; Langenfeld, H.E.; Hanson, A.C.; Crowson, C.S.; Bois, M.C.; Ghaffar, U.; Koster, M.J.; Specks, U.; Warrington, K.J. Large vessel involvement in antineutrophil cytoplasmic antibody-associated vasculitis. Rheumatology 2024, 63, 1682–1689. [Google Scholar] [CrossRef]
- Cartin-Ceba, R.; Peikert, T.; Specks, U. Pathogenesis of ANCA-Associated Vasculitis. Curr. Rheumatol. Rep. 2012, 14, 481–493. [Google Scholar] [CrossRef]
- Bonatti, F.; Reina, M.; Neri, T.M.; Martorana, D. Genetic Susceptibility to ANCA-Associated Vasculitis: State of the Art. Front. Immunol. 2014, 5, 577. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, P.A.; Lucas, R.M.; Engelsen, O.; Ponsonby, A.; Clements, M. Antineutrophil Cytoplasmic Antibody–Associated Vasculitides: Could Geographic Patterns Be Explained by Ambient Ultraviolet Radiation? Arthritis Rheum. 2009, 61, 1417–1424. [Google Scholar] [CrossRef]
- McDermott, G.; Fu, X.; Stone, J.H.; Wallwork, R.; Zhang, Y.; Choi, H.K.; Wallace, Z.S. Association of Cigarette Smoking With Antineutrophil Cytoplasmic Antibody–Associated Vasculitis. JAMA Intern. Med. 2020, 180, 870. [Google Scholar] [CrossRef]
- Lane, S.E.; Watts, R.A.; Bentham, G.; Innes, N.J.; Scott, D.G.I. Are Environmental Factors Important in Primary Systemic Vasculitis?: A Case–Control Study. Arthritis Rheum. 2003, 48, 814–823. [Google Scholar] [CrossRef]
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The Emerging Role of Epigenetics in Human Autoimmune Disorders. Clin. Epigenetics 2019, 11, 34. [Google Scholar] [CrossRef]
- Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis. Available online: https://www.orpha.net/en/disease/detail/156152 (accessed on 11 June 2025).
- Rathmann, J.; Segelmark, M.; Englund, M.; Mohammad, A.J. Stable Incidence but Increase in Prevalence of ANCA-Associated Vasculitis in Southern Sweden: A 23-Year Study. RMD Open 2023, 9, e002949. [Google Scholar] [CrossRef]
- Pearce, F.A.; Lanyon, P.C.; Grainge, M.J.; Shaunak, R.; Mahr, A.; Hubbard, R.B.; Watts, R.A. Incidence of ANCA-Associated Vasculitis in a UK Mixed Ethnicity Population. Rheumatology 2016, 55, 1656–1663. [Google Scholar] [CrossRef]
- Redondo-Rodriguez, R.; Mena-Vázquez, N.; Cabezas-Lucena, A.M.; Manrique-Arija, S.; Mucientes, A.; Fernández-Nebro, A. Systematic Review and Metaanalysis of Worldwide Incidence and Prevalence of Antineutrophil Cytoplasmic Antibody (ANCA) Associated Vasculitis. J. Clin. Med. 2022, 11, 2573. [Google Scholar] [CrossRef]
- Watts, R.A.; Mahr, A.; Mohammad, A.J.; Gatenby, P.; Basu, N.; Flores-Suárez, L.F. Classification, Epidemiology and Clinical Subgrouping of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. Nephrol. Dial. Transplant. 2015, 30, i14–i22. [Google Scholar] [CrossRef]
- Gatenby, P.A. Anti-neutrophil Cytoplasmic Antibody-associated Systemic Vasculitis: Nature or Nurture? Intern. Med. J. 2012, 42, 351–359. [Google Scholar] [CrossRef]
- Wójcik, K.; Wawrzycka-Adamczyk, K.; Włudarczyk, A.; Sznajd, J.; Zdrojewski, Z.; Masiak, A.; Czuszyńska, Z.; Majdan, M.; Jeleniewicz, R.; Klinger, M.; et al. Clinical Characteristics of Polish Patients with ANCA-Associated Vasculitides—Retrospective Analysis of POLVAS Registry. Clin. Rheumatol. 2019, 38, 2553–2563. [Google Scholar] [CrossRef]
- Almaani, S.; Fussner, L.A.; Brodsky, S.; Meara, A.S.; Jayne, D. ANCA-Associated Vasculitis: An Update. J. Clin. Med. 2021, 10, 1446. [Google Scholar] [CrossRef]
- Jayne, D. The diagnosis of vasculitis. Best Pract. Res. Clin. Rheumatol. 2009, 23, 445–453. [Google Scholar] [CrossRef]
- Solans-Laqué, R.; Fraile, G.; Rodriguez-Carballeira, M.; Caminal, L.; Castillo, M.J.; Martínez-Valle, F.; Sáez, L.; Rios, J.J.; Solanich, X.; Oristrell, J.; et al. Clinical characteristics and outcome of Spanish patients with ANCA-associated vasculitides: Impact of the vasculitis type, ANCA specificity, and treatment on mortality and morbidity. Medicine 2017, 96, e6083. [Google Scholar] [CrossRef]
- Puan, Y.; Ong, K.Y.; Tiew, P.Y.; Wen Chen, G.X.; Teo, N.W.Y.; Low, A.H.L.; Wechsler, M.E.; Koh, M.S. Characteristics of Severe Asthma Clinic Patients with Eosinophilic Granulomatosis with Polyangiitis. J. Allergy Clin. Immunol. Pract. 2025, 13, 361–368.e2. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef]
- Renauer, P.; Coit, P.; Sawalha, A.H. Epigenetics and Vasculitis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Brunini, F.; Page, T.H.; Gallieni, M.; Pusey, C.D. The Role of Monocytes in ANCA-Associated Vasculitides. Autoimmun. Rev. 2016, 15, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Robson, J.C.; Grayson, P.C.; Ponte, C.; Suppiah, R.; Craven, A.; Judge, A.; Khalid, S.; Hutchings, A.; Watts, R.A.; Merkel, P.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Granulomatosis with Polyangiitis. Ann. Rheum. Dis. 2022, 81, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, R.; Robson, J.C.; Grayson, P.C.; Ponte, C.; Craven, A.; Khalid, S.; Judge, A.; Hutchings, A.; Merkel, P.A.; Luqmani, R.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Microscopic Polyangiitis. Ann. Rheum. Dis. 2022, 81, 321–326. [Google Scholar] [CrossRef]
- Grayson, P.C.; Ponte, C.; Suppiah, R.; Robson, J.C.; Craven, A.; Judge, A.; Khalid, S.; Hutchings, A.; Luqmani, R.A.; Watts, R.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Ann. Rheum. Dis. 2022, 81, 309–314. [Google Scholar] [CrossRef]
- Geetha, D.; Jefferson, J.A. ANCA-Associated Vasculitis: Core Curriculum 2020. Am. J. Kidney Dis. 2020, 75, 124–137. [Google Scholar] [CrossRef]
- Cohen Tervaert, J.W.; Damoiseaux, J. Antineutrophil Cytoplasmic Autoantibodies: How Are They Detected and What Is Their Use for Diagnosis, Classification and Follow-Up? Clin. Rev. Allerg. Immunol. 2012, 43, 211–219. [Google Scholar] [CrossRef]
- Chejfec, G. Advanced Atlas of Autoantibody Patterns. Arch. Pathol. Lab. Med. 2000, 124, 472. [Google Scholar] [CrossRef]
- Radice, A.; Sinico, R.A. Antineutrophil Cytoplasmic Antibodies (ANCA). Autoimmunity 2005, 38, 93–103. [Google Scholar] [CrossRef]
- Elmér, E.; Smargianaki, S.; Pettersson, Å.; Skattum, L.; Ohlsson, S.; Hellmark, T.; Johansson, Å.C.M. Increased Frequencies of Switched Memory B Cells and Plasmablasts in Peripheral Blood from Patients with ANCA-Associated Vasculitis. J. Immunol. Res. 2020, 2020, 8209737. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Shen, C.; Meng, T.; Lin, W.; Hu, X.; Tang, R.; Xiong, Q.; Ooi, J.D.; Eggenhuizen, P.J.; Chen, J.; et al. Clinical Features and Prognosis of ANCA-Associated Vasculitis Patients Who Were Double-Seropositive for Myeloperoxidase-ANCA and Proteinase 3-ANCA. Clin. Exp. Med. 2024, 24, 66. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.J.; Hoffman, G.S. Controversies in Small Vessel Vasculitis—Comparing the Rheumatology and Nephrology Views. Curr. Opin. Rheumatol. 2007, 19, 1–9. [Google Scholar] [CrossRef]
- Ohteru, Y.; Hamada, K.; Oishi, K.; Suizu, J.; Harada, M.; Murakawa, K.; Chikumoto, A.; Matsuda, K.; Uehara, S.; Ohata, S.; et al. Antineutrophil Cytoplasmic Antibody-Negative Granulomatosis with Polyangiitis Localized to the Lungs. Respir. Med. Case Rep. 2022, 36, 101600. [Google Scholar] [CrossRef]
- Eisenberger, U.; Fakhouri, F.; Vanhille, P.; Beaufils, H.; Mahr, A.; Guillevin, L.; Lesavre, P.; Noël, L.-H. ANCA-Negative Pauci-Immune Renal Vasculitis: Histology and Outcome. Nephrol. Dial. Transplant. 2005, 20, 1392–1399. [Google Scholar] [CrossRef]
- Falde, S.D.; Fussner, L.A.; Tazelaar, H.D.; O’Brien, E.K.; Lamprecht, P.; Konig, M.F.; Specks, U. Proteinase 3-Specific Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Lancet Rheumatol. 2024, 6, e314–e327. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wan, Q.; Liu, B. ANCA-Negative Eosinophilic Granulomatosis with Polyangiitis Complicated by Peripheral Nerve Damage: A Case Report. Medicine 2023, 102, e34450. [Google Scholar] [CrossRef]
- Tanna, A.; Salama, A.D.; Brookes, P.; Pusey, C.D. Familial Granulomatosis with Polyangiitis: Three Cases of This Rare Disorder in One Indoasian Family Carrying an Identical HLA DPB1 Allele. BMJ Case Rep. 2012, 2012, bcr0120125502. [Google Scholar] [CrossRef]
- Ueki, Y.; Oshikata, C.; Asai, Y.; Kaneko, T.; Tsurikisawa, N. Familial Eosinophilic Granulomatosis with Polyangiitis in a Sister and Brother. Intern. Med. 2020, 59, 991–995. [Google Scholar] [CrossRef]
- Quan, M.V.; Frankel, S.K.; Maleki-Fischbach, M.; Tan, L.D. A Rare Case Report of Polyangiitis Overlap Syndrome: Granulomatosis with Polyangiitis and Eosinophilic Granulomatosis with Polyangiitis. BMC Pulm. Med. 2018, 18, 181. [Google Scholar] [CrossRef]
- Kitching, A.R.; Anders, H.-J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-Associated Vasculitis. Nat. Rev. Dis. Primers 2020, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.; Sandin, S.; Askling, J. Risks and Relative Risks of Wegener’s Granulomatosis among Close Relatives of Patients with the Disease. Arthritis Rheum. 2008, 58, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Li, X.; Sundquist, J.; Sundquist, K. Familial Associations of Rheumatoid Arthritis with Autoimmune Diseases and Related Conditions. Arthritis Rheum. 2009, 60, 661–668. [Google Scholar] [CrossRef]
- Li, W.; Huang, H.; Cai, M.; Yuan, T.; Sheng, Y. Antineutrophil Cytoplasmic Antibody-Associated Vasculitis Update: Genetic Pathogenesis. Front. Immunol. 2021, 12, 624848. [Google Scholar] [CrossRef]
- Jagiello, P.; Gencik, M.; Arning, L.; Wieczorek, S.; Kunstmann, E.; Csernok, E.; Gross, W.L.; Epplen, J.T. New Genomic Region for Wegener?S Granulomatosis as Revealed by an Extended Association Screen with 202 Apoptosis-Related Genes. Hum. Genet. 2004, 114, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, M.; Holle, J.U.; Arning, L.; Knaup, S.; Hellmich, B.; Nothnagel, M.; Jagiello, P.; Gross, W.L.; Epplen, J.T.; Wieczorek, S. The Wegener’s Granulomatosis Quantitative Trait Locus on Chromosome 6p21.3 as Characterised by tagSNP Genotyping. Ann. Rheum. Dis. 2008, 67, 972–979. [Google Scholar] [CrossRef]
- Luo, H.; Chen, M.; Yang, R.; Xu, P.-C.; Zhao, M.-H. The Association of HLA-DRB1 Alleles with Antineutrophil Cytoplasmic Antibody-Associated Systemic Vasculitis in Chinese Patients. Hum. Immunol. 2011, 72, 422–425. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Kobayashi, S.; Hashimoto, H.; Ozaki, S.; Tokunaga, K. Association of HLA-DRB1*0901-DQB1*0303 Haplotype with Microscopic Polyangiitis in Japanese. Genes Immun. 2006, 7, 81–84. [Google Scholar] [CrossRef]
- Vaglio, A.; Martorana, D.; Maggiore, U.; Grasselli, C.; Zanetti, A.; Pesci, A.; Garini, G.; Manganelli, P.; Bottero, P.; Tumiati, B.; et al. HLA–DRB4 as a Genetic Risk Factor for Churg-Strauss Syndrome. Arthritis Rheum. 2007, 56, 3159–3166. [Google Scholar] [CrossRef]
- Wieczorek, S.; Hellmich, B.; Gross, W.L.; Epplen, J.T. Associations of Churg-Strauss Syndrome with the HLA–DRB1 Locus, and Relationship to the Genetics of Antineutrophil Cytoplasmic Antibody–Associated Vasculitides: Comment on the Article by Vaglio et Al. Arthritis Rheum. 2008, 58, 329–330. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.C.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; Van Der Merwe, P.A.; Davis, S.J. The Interaction Properties of Costimulatory Molecules Revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef]
- Carr, E.J.; Niederer, H.A.; Williams, J.; Harper, L.; Watts, R.A.; Lyons, P.A.; Smith, K.G. Confirmation of the Genetic Association of CTLA4 and PTPN22 with ANCA-Associated Vasculitis. BMC Med. Genet. 2009, 10, 121. [Google Scholar] [CrossRef]
- Kamesh, L.; Heward, J.M.; Williams, J.M.; Gough, S.C.L.; Chavele, K.-M.; Salama, A.; Pusey, C.; Savage, C.O.S.; Harper, L. CT60 and +49 Polymorphisms of CTLA 4 Are Associated with ANCA-Positive Small Vessel Vasculitis. Rheumatology 2009, 48, 1502–1505. [Google Scholar] [CrossRef]
- Husmann, C.A.; Holle, J.U.; Moosig, F.; Mueller, S.; Wilde, B.; Cohen Tervaert, J.W.; Harper, L.; Assmann, G.; Gross, W.L.; Epplen, J.T.; et al. Genetics of Toll like Receptor 9 in ANCA Associated Vasculitides. Ann. Rheum. Dis. 2014, 73, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Jagiello, P.; Aries, P.; Arning, L.; Wagenleiter, S.E.N.; Csernok, E.; Hellmich, B.; Gross, W.L.; Epplen, J.T. The PTPN22 620W Allele Is a Risk Factor for Wegener’s Granulomatosis. Arthritis Rheum. 2005, 52, 4039–4043. [Google Scholar] [CrossRef] [PubMed]
- Martorana, D.; Maritati, F.; Malerba, G.; Bonatti, F.; Alberici, F.; Oliva, E.; Sebastio, P.; Manenti, L.; Brugnano, R.; Catanoso, M.G.; et al. PTPN22 R620W Polymorphism in the ANCA-Associated Vasculitides. Rheumatology 2012, 51, 805–812. [Google Scholar] [CrossRef]
- Gencik, M.; Meller, S.; Borgmann, S.; Fricke, H. Proteinase 3 Gene Polymorphisms and Wegener’s Granulomatosis. Kidney Int. 2000, 58, 2473–2477. [Google Scholar] [CrossRef]
- Merkel, P.A.; Xie, G.; Monach, P.A.; Ji, X.; Ciavatta, D.J.; Byun, J.; Pinder, B.D.; Zhao, A.; Zhang, J.; Tadesse, Y.; et al. Identification of Functional and Expression Polymorphisms Associated with Risk for Antineutrophil Cytoplasmic Autoantibody–Associated Vasculitis. Arthritis Rheumatol. 2017, 69, 1054–1066. [Google Scholar] [CrossRef]
- Elzouki, A.-N.Y.; Segelmark, M.; Wieslander, J.; Eriksson, S. Strong Link between the Alpha1 -antitrypsin PiZ Allele and Wegener’s Granulomatosis. J. Intern. Med. 1994, 236, 543–548. [Google Scholar] [CrossRef]
- Borgmann, S.; Endisch, G.; Urban, S.; Sitter, T.; Fricke, H. A Linkage Disequilibrium between Genes at the Serine Protease Inhibitor Gene Cluster on Chromosome 14q32.1 Is Associated with Wegener’s Granulomatosis. Clin. Immunol. 2001, 98, 244–248. [Google Scholar] [CrossRef]
- Lyons, P.A.; Peters, J.E.; Alberici, F.; Liley, J.; Coulson, R.M.R.; Astle, W.; Baldini, C.; Bonatti, F.; Cid, M.C.; Elding, H.; et al. Genome-Wide Association Study of Eosinophilic Granulomatosis with Polyangiitis Reveals Genomic Loci Stratified by ANCA Status. Nat. Commun. 2019, 10, 5120. [Google Scholar] [CrossRef] [PubMed]
- Lyons, P.A.; Rayner, T.F.; Trivedi, S.; Holle, J.U.; Watts, R.A.; Jayne, D.R.W.; Baslund, B.; Brenchley, P.; Bruchfeld, A.; Chaudhry, A.N.; et al. Genetically Distinct Subsets within ANCA-Associated Vasculitis. N. Engl. J. Med. 2012, 367, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Roshandel, D.; Sherva, R.; Monach, P.A.; Lu, E.Y.; Kung, T.; Carrington, K.; Zhang, S.S.; Pulit, S.L.; Ripke, S.; et al. Association of Granulomatosis with Polyangiitis (Wegener’s) With HLA–DPB1*04 and SEMA6A Gene Variants: Evidence From Genome-Wide Analysis. Arthritis Rheum. 2013, 65, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, A.; Motevalizadeh Ardekani, A. Role of Epigenetics in Biology and Human Diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Wardowska, A. The Epigenetic Face of Lupus: Focus on Antigen-Presenting Cells. Int. Immunopharmacol. 2020, 81, 106262. [Google Scholar] [CrossRef]
- Karagianni, P.; Tzioufas, A.G. Epigenetic Perspectives on Systemic Autoimmune Disease. J. Autoimmun. 2019, 104, 102315. [Google Scholar] [CrossRef]
- Jones, B.E.; Yang, J.; Muthigi, A.; Hogan, S.L.; Hu, Y.; Starmer, J.; Henderson, C.D.; Poulton, C.J.; Brant, E.J.; Pendergraft, W.F.; et al. Gene-Specific DNA Methylation Changes Predict Remission in Patients with ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 1175–1187. [Google Scholar] [CrossRef]
- Ciavatta, D.J.; Yang, J.; Preston, G.A.; Badhwar, A.K.; Xiao, H.; Hewins, P.; Nester, C.M.; Pendergraft, W.F.; Magnuson, T.R.; Jennette, J.C.; et al. Epigenetic Basis for Aberrant Upregulation of Autoantigen Genes in Humans with ANCA Vasculitis. J. Clin. Investig. 2010, 120, 3209–3219. [Google Scholar] [CrossRef]
- Yang, J.; Ge, H.; Poulton, C.J.; Hogan, S.L.; Hu, Y.; Jones, B.E.; Henderson, C.D.; McInnis, E.A.; Pendergraft, W.F.; Jennette, J.C.; et al. Histone Modification Signature at Myeloperoxidase and Proteinase 3 in Patients with Anti-Neutrophil Cytoplasmic Autoantibody-Associated Vasculitis. Clin. Epigenetics 2016, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-K.; Dong, J.; Lam, C.W.-K. Molecular Mechanisms Regulating the Synergism between IL-32γ and NOD for the Activation of Eosinophils. J. Leukoc. Biol. 2013, 95, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.L.; Little, C. Purification and Some Properties of Myeloperoxidase and Eosinophil Peroxidase from Human Blood. Biochem. J. 1983, 209, 781–787. [Google Scholar] [CrossRef]
- Shao, B.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Myeloperoxidase: An Oxidative Pathway for Generating Dysfunctional High-Density Lipoprotein. Chem. Res. Toxicol. 2010, 23, 447–454. [Google Scholar] [CrossRef]
- Sugawara, S. Immune Functions of Proteinase 3. Crit. Rev. Immunol. 2005, 25, 343–360. [Google Scholar] [CrossRef]
- Schreiber, A.; Luft, F.C.; Kettritz, R. Membrane Proteinase 3 Expression and ANCA-Induced Neutrophil Activation. Kidney Int. 2004, 65, 2172–2183. [Google Scholar] [CrossRef]
- van der Geld, Y.M.; Limburg, P.C.; Kallenberg, C.G. Proteinase 3, Wegener’s Autoantigen: From Gene to Antigen. J. Leukoc. Biol. 2001, 69, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Silvescu, C.I.; Sackstein, R. G-CSF Induces Membrane Expression of a Myeloperoxidase Glycovariant That Operates as an E-Selectin Ligand on Human Myeloid Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10696–10701. [Google Scholar] [CrossRef]
- Tan, D.S.; Gan, P.Y.; O’Sullivan, K.M.; Hammett, M.V.; Summers, S.A.; Ooi, J.D.; Lundgren, B.A.; Boyd, R.L.; Scott, H.S.; Kitching, A.R.; et al. Thymic deletion and regulatory T cells prevent antimyeloperoxidase GN. J. Am. Soc. Nephrol. 2013, 24, 573–585. [Google Scholar] [CrossRef]
- Gan, P.Y.; Holdsworth, S.R.; Kitching, A.R.; Ooi, J.D. Myeloperoxidase (MPO)-specific CD4+ T cells contribute to MPO-anti-neutrophil cytoplasmic antibody (ANCA) associated glomerulonephritis. Cell. Immunol. 2013, 282, 21–27. [Google Scholar] [CrossRef]
- Primo, V.C.; Marusic, S.; Franklin, C.C.; Goldmann, W.H.; Achaval, C.G.; Smith, R.N.; Arnaout, M.A.; Nikolic, B. Anti-PR3 immune responses induce segmental and necrotizing glomerulonephritis. Clin. Exp. Immunol. 2010, 159, 327–337. [Google Scholar] [CrossRef]
- Sharma, R.K.; Yoosuf, N.; Afonso, M.; Scheffschick, A.; Avik, A.; Bartoletti, A.; Horuluoglu, B.; Diaz Boada, J.S.; Boddul, S.K.; Jonasdottir, A.D.; et al. Identification of proteinase 3 autoreactive CD4+T cells and their T-cell receptor repertoires in antineutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2023, 103, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Porges, A.J.; Redecha, P.B.; Kimberly, W.T.; Csernok, E.; Gross, W.L.; Kimberly, R.P. Anti-Neutrophil Cytoplasmic Antibodies Engage and Activate Human Neutrophils via Fc Gamma RIIa. J. Immunol. 1994, 153, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Ben-Smith, A.; Hewins, P.; Dove, S.K.; Hughes, P.; McEwan, R.; Wakelam, M.J.O.; Savage, C.O.S. Activation of the Gi Heterotrimeric G Protein by ANCA IgG F(Ab′)2 Fragments Is Necessary but Not Sufficient to Stimulate the Recruitment of Those Downstream Mediators Used by Intact ANCA IgG. J. Am. Soc. Nephrol. 2003, 14, 661–669. [Google Scholar] [CrossRef]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the Immune System and The Altered Glycan Theory of Autoimmunity: A Critical Review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.J.; Terrell, R.S.; Charles, L.A.; Jennette, J.C. Anti-Neutrophil Cytoplasmic Autoantibodies Induce Neutrophils to Degranulate and Produce Oxygen Radicals in Vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4115–4119. [Google Scholar] [CrossRef]
- Thomas, C.J.; Schroder, K. Pattern recognition receptor function in neutrophils. Trends Immunol. 2013, 34, 317–328. [Google Scholar] [CrossRef]
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; Silva, L.S.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; De Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020, 16, e1008230. [Google Scholar] [CrossRef]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef]
- Schreiber, A.; Rousselle, A.; Becker, J.U.; von Mässenhausen, A.; Linkermann, A.; Kettritz, R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E9618–E9625. [Google Scholar] [CrossRef]
- Sangaletti, S.; Tripodo, C.; Chiodoni, C.; Guarnotta, C.; Cappetti, B.; Casalini, P.; Piconese, S.; Parenza, M.; Guiducci, C.; Vitali, C.; et al. Neutrophil Extracellular Traps Mediate Transfer of Cytoplasmic Neutrophil Antigens to Myeloid Dendritic Cells toward ANCA Induction and Associated Autoimmunity. Blood 2012, 120, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting Neutrophils in Autoimmune Small-Vessel Vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Hamayasu, H.; Seki, A.; Nonaka, K.; Wang, T.; Matsumoto, T.; Hamano, Y.; Sumikura, H.; Kumasaka, T.; Murayama, S.; et al. Presence of Citrullinated Histone H3-Positive Neutrophils in Microscopic Polyangiitis from the Early Phase: An Autopsy Proven Case. Pathol. Int. 2016, 66, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Kawasaki, T.; Shigematsu, K.; Kawamura, K.; Oka, N. Neutrophil extracellular traps in neuropathy with anti-neutrophil cytoplasmic autoantibody-associated microscopic polyangiitis. Clin. Rheumatol. 2017, 36, 913–917. [Google Scholar] [CrossRef]
- Meng, W.; Paunel-Görgülü, A.; Flohé, S.; Witte, I.; Schädel-Höpfner, M.; Windolf, J.; Lögters, T.T. Deoxyribonuclease is a potential counter regulator of aberrant neutrophil extracellular traps formation after major trauma. Mediat. Inflamm. 2012, 2012, 149560. [Google Scholar] [CrossRef]
- Söderberg, D.; Kurz, T.; Motamedi, A.; Hellmark, T.; Eriksson, P.; Segelmark, M. Increased levels of neutrophil extracellular trap remnants in the circulation of patients with small vessel vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatology 2015, 54, 2085–2094. [Google Scholar] [CrossRef]
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulou, O.D.; Goules, A.V.; Boutzios, G.; Tsirogianni, A.; Sfontouris, C.; Manoussakis, M.N.; Vlachoyiannopoulos, P.G.; Tzioufas, A.G.; Kapsogeorgou, E.K. Occurrence and Antigenic Specificity of Perinuclear Anti-Neutrophil Cytoplasmic Antibodies (P-ANCA) in Systemic Autoimmune Diseases. Cells 2021, 10, 2128. [Google Scholar] [CrossRef]
- Novikov, P.; Smitienko, I.; Bulanov, N.; Zykova, A.; Moiseev, S. Testing for antineutrophil cytoplasmic antibodies (ANCAs) in patients with systemic vasculitides and other diseases. Ann. Rheum. Dis. 2017, 76, e23. [Google Scholar] [CrossRef]
- Kagiyama, N.; Takayanagi, N.; Kanauchi, T.; Ishiguro, T.; Yanagisawa, T.; Sugita, Y. Antineutrophil cytoplasmic antibody-positive conversion and microscopic polyangiitis development in patients with idiopathic pulmonary fibrosis. BMJ Open Respir. Res. 2015, 2, e000058. [Google Scholar] [CrossRef]
- Johansson, Å.C.; Ohlsson, S.; Pettersson, Å.; Bengtsson, A.A.; Selga, D.; Hansson, M.; Hellmark, T. Impaired Phagocytosis and Reactive Oxygen Species Production in Phagocytes Is Associated with Systemic Vasculitis. Arthritis Res. Ther. 2016, 18, 92. [Google Scholar] [CrossRef]
- Mishra, R.; Behera, L.M.; Rana, S. Binding of raloxifene to human complement fragment 5a (hC5a): A perspective on cytokine storm and COVID19. J. Biomol. Struct. Dyn. 2022, 40, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Xiao, H.; Jennette, J.C.; Schneider, W.; Luft, F.C.; Kettritz, R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Rzeniewicz, K.; Newe, A.; Rey Gallardo, A.; Davies, J.; Holt, M.R.; Patel, A.; Charras, G.T.; Stramer, B.; Molenaar, C.; Tedder, T.F.; et al. L-selectin shedding is activated specifically within transmigrating pseudopods of monocytes to regulate cell polarity in vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E1461–E1470. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Løbner, M.; Cédile, O.; Owens, T. Comparison of microglia and infiltrating CD11c⁺ cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflammation 2014, 11, 57. [Google Scholar] [CrossRef]
- Hattar, K.; Bickenbach, A.; Csernok, E.; Rosseau, S.; Grandel, U.; Seeger, W.; Grimminger, F.; Sibelius, U. Wegener’s Granulomatosis: Antiproteinase 3 Antibodies Induce Monocyte Cytokine and Prostanoid Release—Role of Autocrine Cell Activation. J. Leukoc. Biol. 2002, 71, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Weidner, S.; Neupert, W.; Goppelt-Struebe, M.; Rupprecht, H.D. Antineutrophil Cytoplasmic Antibodies Induce Human Monocytes to Produce Oxygen Radicals in Vitro. Arthritis Rheum. 2001, 44, 1698–1706. [Google Scholar] [CrossRef]
- Tarzi, R.M.; Liu, J.; Schneiter, S.; Hill, N.R.; Page, T.H.; Cook, H.T.; Pusey, C.D.; Woollard, K.J. CD14 Expression Is Increased on Monocytes in Patients with Anti-Neutrophil Cytoplasm Antibody (ANCA)-Associated Vasculitis and Correlates with the Expression of ANCA Autoantigens. Clin. Exp. Immunol. 2015, 181, 65–75. [Google Scholar] [CrossRef]
- Matsumoto, T.; Matsui, T.; Hirano, F.; Tohma, S.; Mori, M. Disease Activity, Treatment and Long-Term Prognosis of Adult Juvenile Idiopathic Arthritis Patients Compared with Rheumatoid Arthritis Patients. Mod. Rheumatol. 2020, 30, 78–84. [Google Scholar] [CrossRef]
- Kobold, A.C.M.; Kallenberg, C.G.M.; Tervaert, J.W.C. Monocyte Activation in Patients with Wegener’s Granulomatosis. Ann. Rheum. Dis. 1999, 58, 237–245. [Google Scholar] [CrossRef]
- Haller, H.; Eichhorn, J.; Pieper, K.; Göbel, U.; Luft, F.C. Circulating Leukocyte Integrin Expression in Wegener’s Granulomatosis. J. Am. Soc. Nephrol. 1996, 7, 40–48. [Google Scholar] [CrossRef]
- Emmi, G.; Bettiol, A.; Gelain, E.; Bajema, I.M.; Berti, A.; Burns, S.; Cid, M.C.; Cohen Tervaert, J.W.; Cottin, V.; Durante, E.; et al. Evidence-Based Guideline for the Diagnosis and Management of Eosinophilic Granulomatosis with Polyangiitis. Nat. Rev. Rheumatol. 2023, 19, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Hashimoto, M. Eosinophilic Granulomatosis with Polyangiitis: Latest Findings and Updated Treatment Recommendations. J. Clin. Med. 2023, 12, 5996. [Google Scholar] [CrossRef]
- Kotas, M.E.; Dion, J.; Van Dyken, S.; Ricardo-Gonzalez, R.R.; Danel, C.J.; Taillé, C.; Mouthon, L.; Locksley, R.M.; Terrier, B. A Role for IL-33–Activated ILC2s in Eosinophilic Vasculitis. JCI Insight 2021, 6, e143366. [Google Scholar] [CrossRef] [PubMed]
- Fulkerson, P.C.; Schollaert, K.L.; Bouffi, C.; Rothenberg, M.E. IL-5 Triggers a Cooperative Cytokine Network That Promotes Eosinophil Precursor Maturation. J. Immunol. 2014, 193, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E.; Akuthota, P.; Jayne, D.; Khoury, P.; Klion, A.; Langford, C.A.; Merkel, P.A.; Moosig, F.; Specks, U.; Cid, M.C.; et al. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N. Engl. J. Med. 2017, 376, 1921–1932. [Google Scholar] [CrossRef]
- Fukuchi, M.; Kamide, Y.; Ueki, S.; Miyabe, Y.; Konno, Y.; Oka, N.; Takeuchi, H.; Koyota, S.; Hirokawa, M.; Yamada, T.; et al. Eosinophil ETosis–Mediated Release of Galectin-10 in Eosinophilic Granulomatosis with Polyangiitis. Arthritis Rheumatol. 2021, 73, 1683–1693. [Google Scholar] [CrossRef]
- Rodríguez-Alcázar, J.F.; Ataide, M.A.; Engels, G.; Schmitt-Mabmunyo, C.; Garbi, N.; Kastenmüller, W.; Latz, E.; Franklin, B.S. Charcot–Leyden Crystals Activate the NLRP3 Inflammasome and Cause IL-1β Inflammation in Human Macrophages. J. Immunol. 2019, 202, 550–558. [Google Scholar] [CrossRef]
- Hilhorst, M.; Shirai, T.; Berry, G.; Goronzy, J.J.; Weyand, C.M. T Cell-acrophage Interactions and Granuloma Formation in Vasculitis. Front. Immunol. 2014, 5, 432. [Google Scholar] [CrossRef]
- Su, B.; Mao, X.; Yin, B.; Chen, C.; Zhang, M.; Cui, T.; Hao, Y. TIM-3 Regulates the NETs-Mediated Dendritic Cell Activation in Myeloperoxidase-ANCA-Associated Vasculitis. Clin. Exp. Rheumatol. 2021, 39, 13–20. [Google Scholar] [CrossRef]
- Uno, K.; Muso, E.; Ito-Ihara, T.; Endo, T.; Yasuda, Y.; Yagi, K.; Suzuki, K. Impaired HVJ-Stimulated Interferon Producing Capacity in MPO-ANCA-Associated Vasculitis with Rapidly Progressive Glomerulonephritis Lead to Susceptibility to Infection. Cytokine 2020, 136, 155221. [Google Scholar] [CrossRef] [PubMed]
- Odobasic, D.; Holdsworth, S.R. Emerging Cellular Therapies for Anti-Myeloperoxidase Vasculitis and Other Autoimmune Diseases. Front. Immunol. 2021, 12, 642127. [Google Scholar] [CrossRef]
- Braudeau, C.; Néel, A.; Amouriaux, K.; Martin, J.C.; Rimbert, M.; Besançon, A.; Giraudet, S.; Terrien, C.; Aliaga, M.; Salabert-Le Guen, N.; et al. Dysregulated Responsiveness of Circulating Dendritic Cells to Toll-Like Receptors in ANCA-Associated Vasculitis. Front. Immunol. 2017, 8, 102. [Google Scholar] [CrossRef]
- Wilde, B.; Van Paassen, P.; Damoiseaux, J.; Heerings-Rewinkel, P.; Van Rie, H.; Witzke, O.; Tervaert, J.W.C. Dendritic Cells in Renal Biopsies of Patients with ANCA-Associated Vasculitis. Nephrol. Dial. Transplant. 2009, 24, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Tsurikisawa, N.; Saito, H.; Oshikata, C.; Tsuburai, T.; Ishiyama, M.; Mitomi, H.; Akiyama, K. An Increase of CD83+ Dendritic Cells Ex Vivo Correlates with Increased Regulatory T Cells in Patients with Active Eosinophilic Granulomatosis and Polyangiitis. BMC Immunol. 2014, 15, 32. [Google Scholar] [CrossRef]
- Fijolek, J.; Radzikowska, E. Eosinophilic granulomatosis with polyangiitis—Advances in pathogenesis, diagnosis, and treatment. Front. Med. 2023, 10, 1145257. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Nagao, T.; Nagi-Miura, N.; Ohno, N.; Yasuhara, M.; Yamamoto, K.; Nakayama, T.; Suzuki, K. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J. Autoimmun. 2008, 31, 79–89. [Google Scholar] [CrossRef]
- Mulder, A.H.; Stegeman, C.A.; Kallenberg, C.G. Activation of granulocytes by anti-neutrophil cytoplasmic antibodies (ANCA) in Wegener’s granulomatosis: A predominant role for the IgG3 subclass of ANCA. Clin. Exp. Immunol. 1995, 101, 227–232. [Google Scholar] [CrossRef]
- Nozaki, Y. New Insights into Novel Therapeutic Targets in ANCA-Associated Vasculitis. Front. Immunol. 2021, 12, 631055. [Google Scholar] [CrossRef]
- Martinez Valenzuela, L.; Bordignon Draibe, J.; Fulladosa Oliveras, X.; Bestard Matamoros, O.; Cruzado Garrit, J.M.; Torras Ambrós, J. T-Lymphocyte in ANCA-Associated Vasculitis: What Do We Know? A Pathophysiological and Therapeutic Approach. Clin. Kidney J. 2019, 12, 503–511. [Google Scholar] [CrossRef]
- Balding, C.E.J.; Howie, A.J.; Drake-Lee, A.B.; Savage, C.O.S. Th2 Dominance in Nasal Mucosa in Patients with Wegener’s Granulomatosis. Clin. Exp. Immunol. 2001, 125, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Abdulahad, W.H.; Van Der Geld, Y.M.; Stegeman, C.A.; Kallenberg, C.G.M. Persistent Expansion of CD4+ Effector Memory T Cells in Wegener’s Granulomatosis. Kidney Int. 2006, 70, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, W.; Jakieła, B.; Wawrzycka-Adamczyk, K.; Sanak, M.; Hubalewska-Mazgaj, M.; Padjas, A.; Surmiak, M.; Szczeklik, K.; Sznajd, J.; Musiał, J. Skewing toward Treg and Th2 Responses Is a Characteristic Feature of Sustained Remission in ANCA-positive Granulomatosis with Polyangiitis. Eur. J. Immunol. 2017, 47, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.; Thewissen, M.; Damoiseaux, J.; Van Paassen, P.; Witzke, O.; Tervaert, J.W. T Cells in ANCA-Associated Vasculitis: What Can We Learn from Lesional versus Circulating T Cells? Arthritis Res. Ther. 2010, 12, 204. [Google Scholar] [CrossRef]
- Marinaki, S.; Kälsch, A.-I.; Grimminger, P.; Breedijk, A.; Birck, R.; Schmitt, W.H.; Weiss, C.; Van Der Woude, F.J.; Yard, B.A. Persistent T-Cell Activation and Clinical Correlations in Patients with ANCA-Associated Systemic Vasculitis. Nephrol. Dial. Transplant. 2006, 21, 1825–1832. [Google Scholar] [CrossRef]
- Marinaki, S.; Neumann, I.; Kälsch, A.-I.; Grimminger, P.; Breedijk, A.; Birck, R.; Schmitt, W.; Waldherr, R.; Yard, B.A.; Van Der Woude, F.J. Abnormalities of CD4+ T Cell Subpopulations in ANCA-Associated Vasculitis. Clin. Exp. Immunol. 2005, 140, 181–191. [Google Scholar] [CrossRef]
- Moosig, F.; Csernok, E.; Wang, G.; Gross, W.L. Costimulatory Molecules in Wegener’s Granulomatosis (WG): Lack of Expression of CD28 and Preferential up-Regulation of Its Ligands B7-1 (CD80) and B7-2 (CD86) on T Cells. Clin. Exp. Immunol. 2001, 114, 113–118. [Google Scholar] [CrossRef]
- Komocsi, A.; Lamprecht, P.; Csernok, E.; Mueller, A.; Holl-Ulrich, K.; Seitzer, U.; Moosig, F.; Schnabel, A.; Gross, W.L. Peripheral Blood and Granuloma CD4+CD28− T Cells Are a Major Source of Interferon-γ and Tumor Necrosis Factor-α in Wegener’s Granulomatosis. Am. J. Pathol. 2002, 160, 1717–1724. [Google Scholar] [CrossRef]
- Capraru, D.; Müller, A.; Csernok, E.; Gross, W.L.; Holl-Ulrich, K.; Northfield, J.; Klenerman, P.; Herlyn, K.; Holle, J.; Gottschlich, S.; et al. Expansion of Circulating NKG2D+ Effector Memory T-Cells and Expression of NKG2D-Ligand MIC in Granulomaous Lesions in Wegener’s Granulomatosis. Clin. Immunol. 2008, 127, 144–150. [Google Scholar] [CrossRef]
- Żabińska, M.; Kościelska-Kasprzak, K.; Krajewska, J.; Bartoszek, D.; Augustyniak-Bartosik, H.; Krajewska, M. Immune Cells Profiling in ANCA-Associated Vasculitis Patients—Relation to Disease Activity. Cells 2021, 10, 1773. [Google Scholar] [CrossRef]
- Evans, H.G.; Gullick, N.J.; Kelly, S.; Pitzalis, C.; Lord, G.M.; Kirkham, B.W.; Taams, L.S. In Vivo Activated Monocytes from the Site of Inflammation in Humans Specifically Promote Th17 Responses. Proc. Natl. Acad. Sci. USA 2009, 106, 6232–6237. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Tsurikisawa, N.; Tsuburai, T.; Oshikata, C.; Akiyama, K. Cytokine Production Profile of CD4+ T Cells from Patients with Active Churg-Strauss Syndrome Tends Toward Th17. Int. Arch. Allergy Immunol. 2009, 149, 61–65. [Google Scholar] [CrossRef]
- Nogueira, E.; Hamour, S.; Sawant, D.; Henderson, S.; Mansfield, N.; Chavele, K.-M.; Pusey, C.D.; Salama, A.D. Serum IL-17 and IL-23 Levels and Autoantigen-Specific Th17 Cells Are Elevated in Patients with ANCA-Associated Vasculitis. Nephrol. Dial. Transplant. 2010, 25, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Scheffschick, A.; Gunnarsson, I.; Brauner, H. Natural Killer Cells in Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis—A Review of the Literature. Front. Immunol. 2022, 12, 796640. [Google Scholar] [CrossRef]
- Tognarelli, S.; Gayet, J.; Lambert, M.; Dupuy, S.; Karras, A.; Cohen, P.; Guillevin, L.; De Menthon, M.; Caillat-Zucman, S. Tissue-Specific Microvascular Endothelial Cells Show Distinct Capacity to Activate NK Cells: Implications for the Pathophysiology of Granulomatosis with Polyangiitis. J. Immunol. 2014, 192, 3399–3408. [Google Scholar] [CrossRef] [PubMed]
- Oleinika, K.; Mauri, C.; Salama, A.D. Effector and Regulatory B Cells in Immune-Mediated Kidney Disease. Nat. Rev. Nephrol. 2019, 15, 11–26. [Google Scholar] [CrossRef]
- Rincón-Arévalo, H.; Sanchez-Parra, C.C.; Castaño, D.; Yassin, L.; Vásquez, G. Regulatory B Cells and Mechanisms. Int. Rev. Immunol. 2015, 35, 156–176. [Google Scholar] [CrossRef]
- Lepse, N.; Abdulahad, W.H.; Kallenberg, C.G.M.; Heeringa, P. Immune Regulatory Mechanisms in ANCA-Associated Vasculitides. Autoimmun. Rev. 2011, 11, 77–83. [Google Scholar] [CrossRef]
- Jones, R.B.; Cohen Tervaert, J.W.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; Van Paassen, P.; et al. Rituximab versus Cyclophosphamide in ANCA-Associated Renal Vasculitis. N. Engl. J. Med. 2010, 363, 211–220. [Google Scholar] [CrossRef]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; St. Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef]
- Alberici, F.; Smith, R.M.; Jones, R.B.; Roberts, D.M.; Willcocks, L.C.; Chaudhry, A.; Smith, K.G.C.; Jayne, D.R.W. Long-Term Follow-up of Patients Who Received Repeat-Dose Rituximab as Maintenance Therapy for ANCA-Associated Vasculitis. Rheumatology 2015, 54, 1153–1160. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| p-ANCA | c-ANCA | |
|---|---|---|
| Autoantigens | MPO, cathepsin G, elastase, lactoferrin, and lysozyme | proteinase 3 |
| Staining pattern | perinuclear | cytoplasmic |
| AAV | MPA, EGPA | GPA |
| GPA | MPA | EGPA | |
|---|---|---|---|
| Clinical presentation | Respiratory tract involvement, and ocular symptoms (uveitis, ocular ulcers, episcleritis, and scleritis) | Glomerulonephritis, pulmonary capillaritis, skin, nerves, and gastrointestinal tract involvement | Respiratory tract involvement (asthma as a typical early feature), and neurological symptoms |
| Granulomas | Present | Not present | Present |
| The main type of ANCA | PR3-ANCA | MPO-ANCA | MPO-ANCA |
| Percentage of ANCA(−) patients | 10% | 10% | 50% |
| Main effector cells | Neutrophils | Neutrophils | Neutrophils in ANCA(+), and eosinophils in ANCA(−) |
| T-cell polarization | Th1 (localized disease), and Th2 (generalized disease) | Th1 | Th1, Th17 in ANCA(+), and Th2 in ANCA(−) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisowska, K.A.; Wardowska, A. Immunopathology of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. Int. J. Mol. Sci. 2025, 26, 6065. https://doi.org/10.3390/ijms26136065
Lisowska KA, Wardowska A. Immunopathology of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. International Journal of Molecular Sciences. 2025; 26(13):6065. https://doi.org/10.3390/ijms26136065
Chicago/Turabian StyleLisowska, Katarzyna Aleksandra, and Anna Wardowska. 2025. "Immunopathology of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis" International Journal of Molecular Sciences 26, no. 13: 6065. https://doi.org/10.3390/ijms26136065
APA StyleLisowska, K. A., & Wardowska, A. (2025). Immunopathology of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. International Journal of Molecular Sciences, 26(13), 6065. https://doi.org/10.3390/ijms26136065