
A Novel Pathogenic Variant Identified in HIKESHI-Related Hypomyelinating Leukodystrophy Disrupts Heat Shock Response in iPSCs

 , , , , , , , , , and
, , , , , , , , , and
Abstract

1. Introduction
2. Results
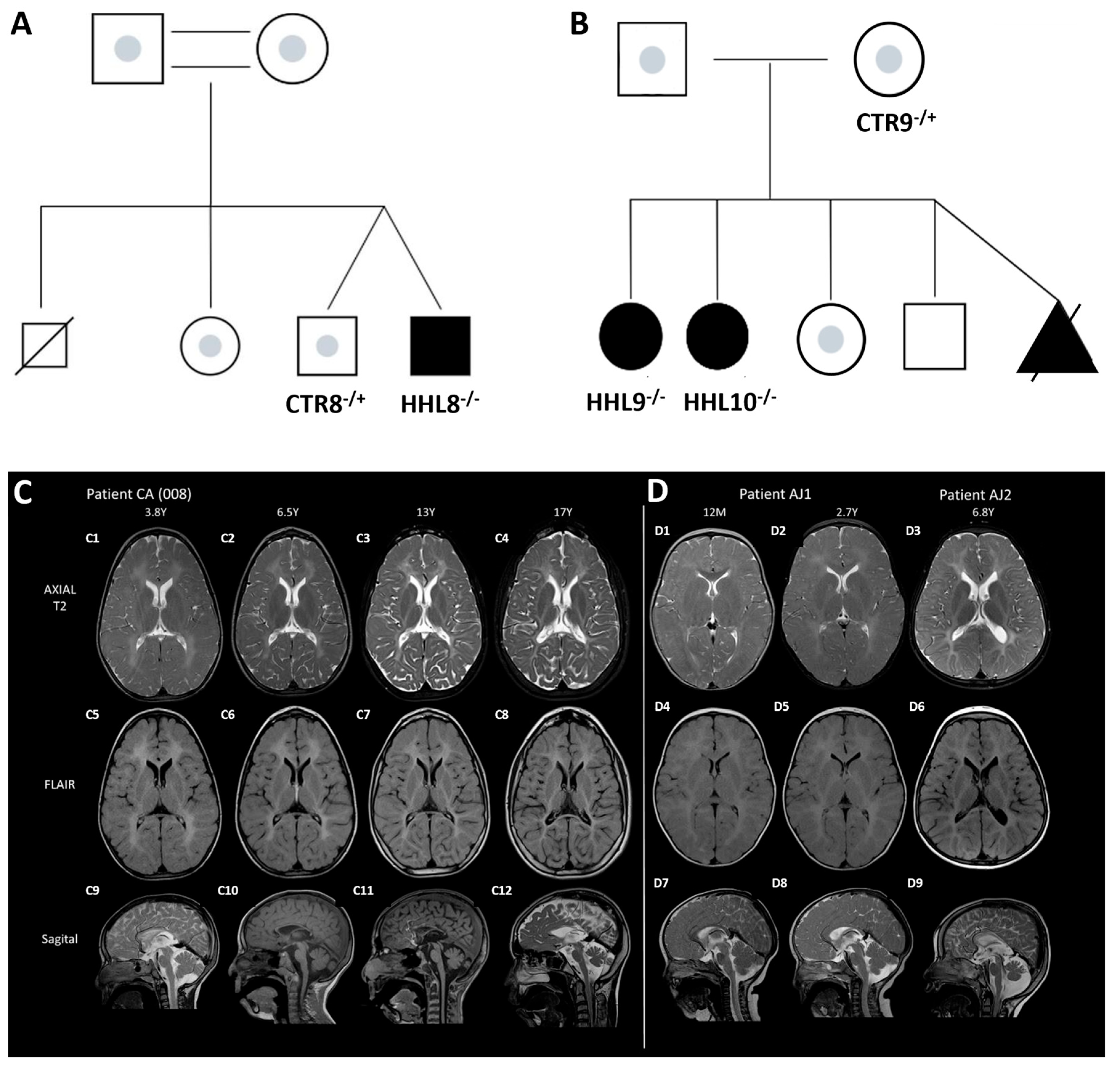
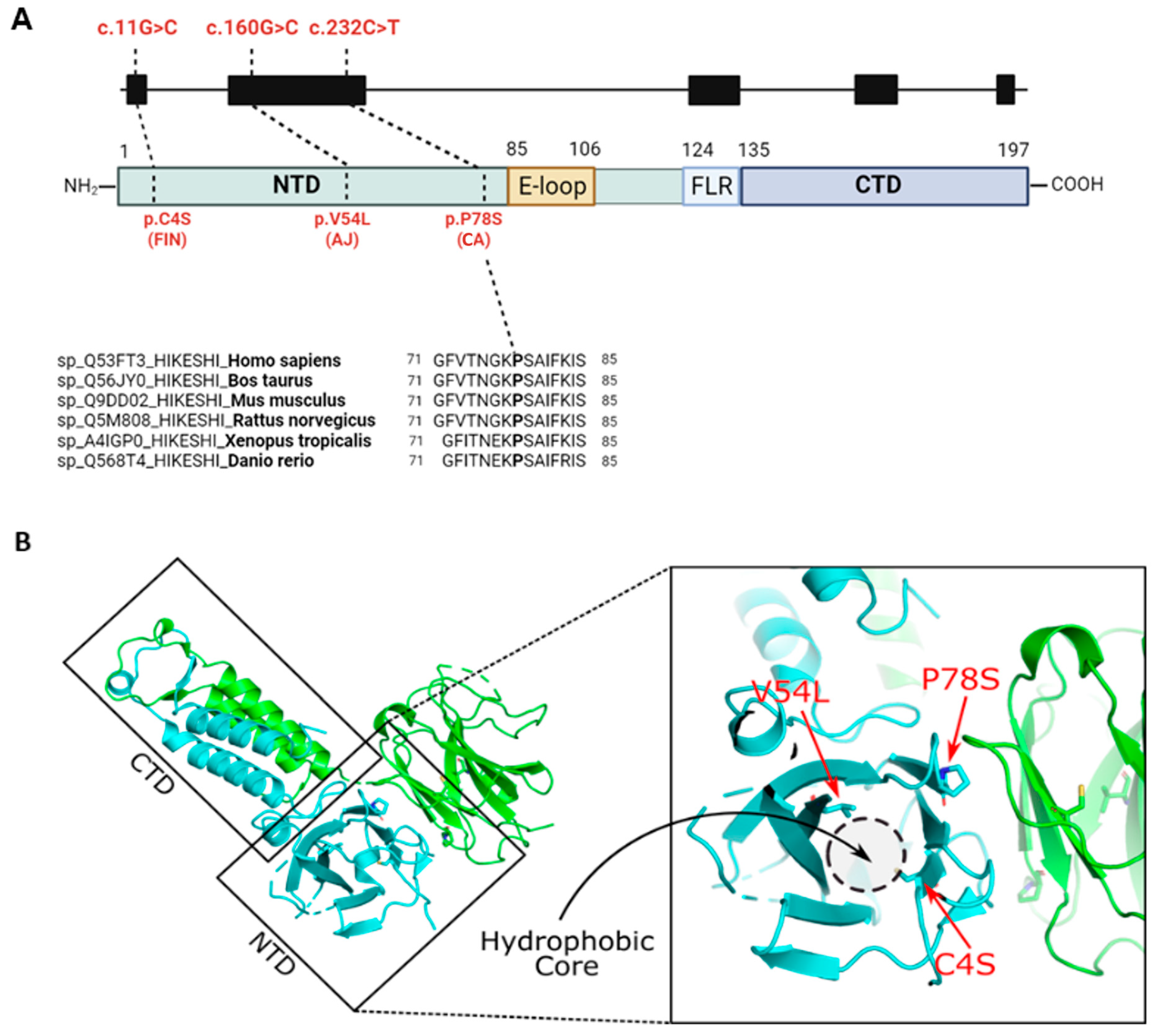
2.1. Identification of a Novel Homozygous Pathogenic Variant in an HHL Patient
2.2. In Silico Analysis of the HIKESHI Protein Reveals That the Pathogenic Variants Are Located Within a Hydrophobic Pocket
2.3. Generation of Patient-Specific iPSCs from HHL Patients and Healthy Controls
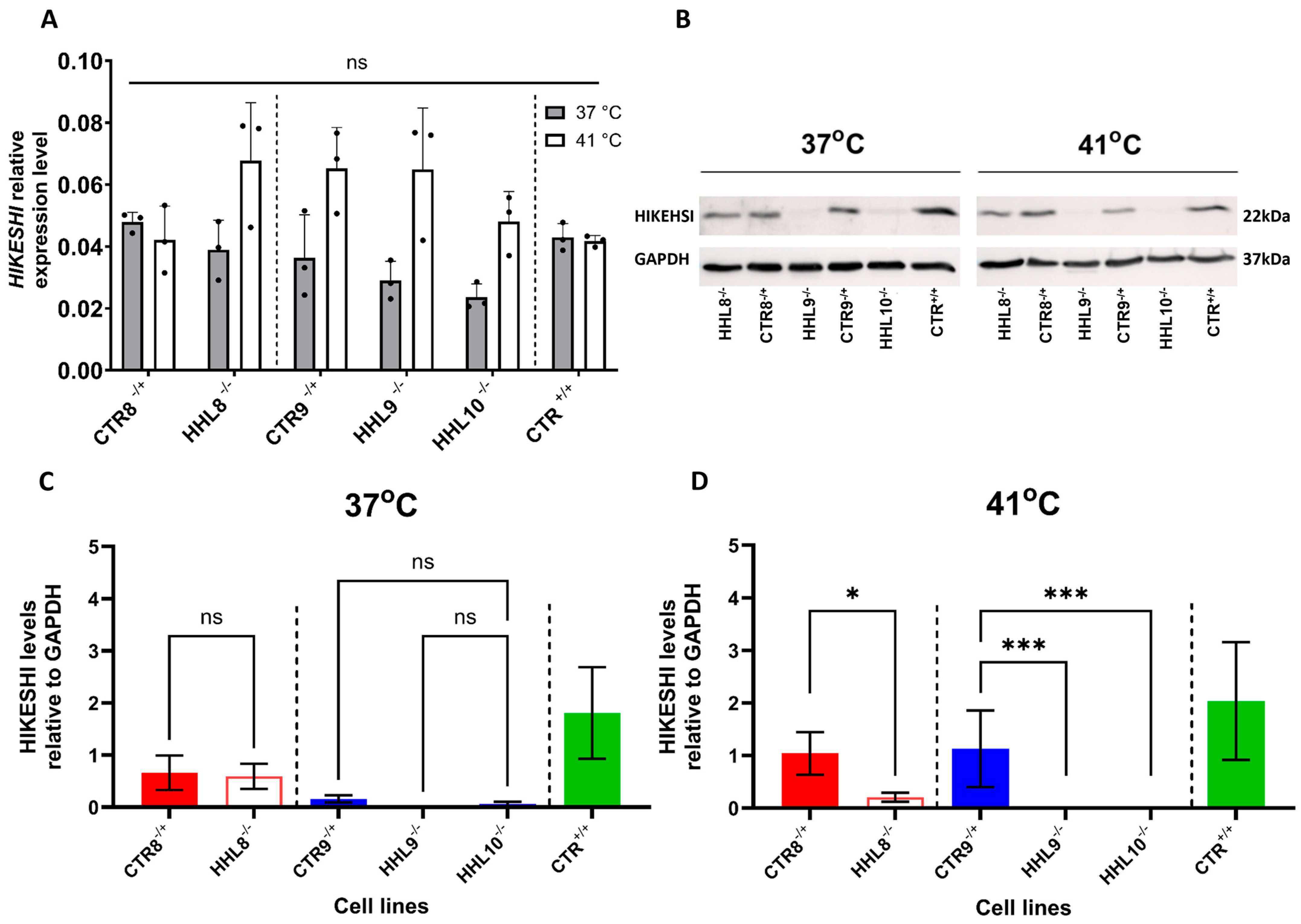
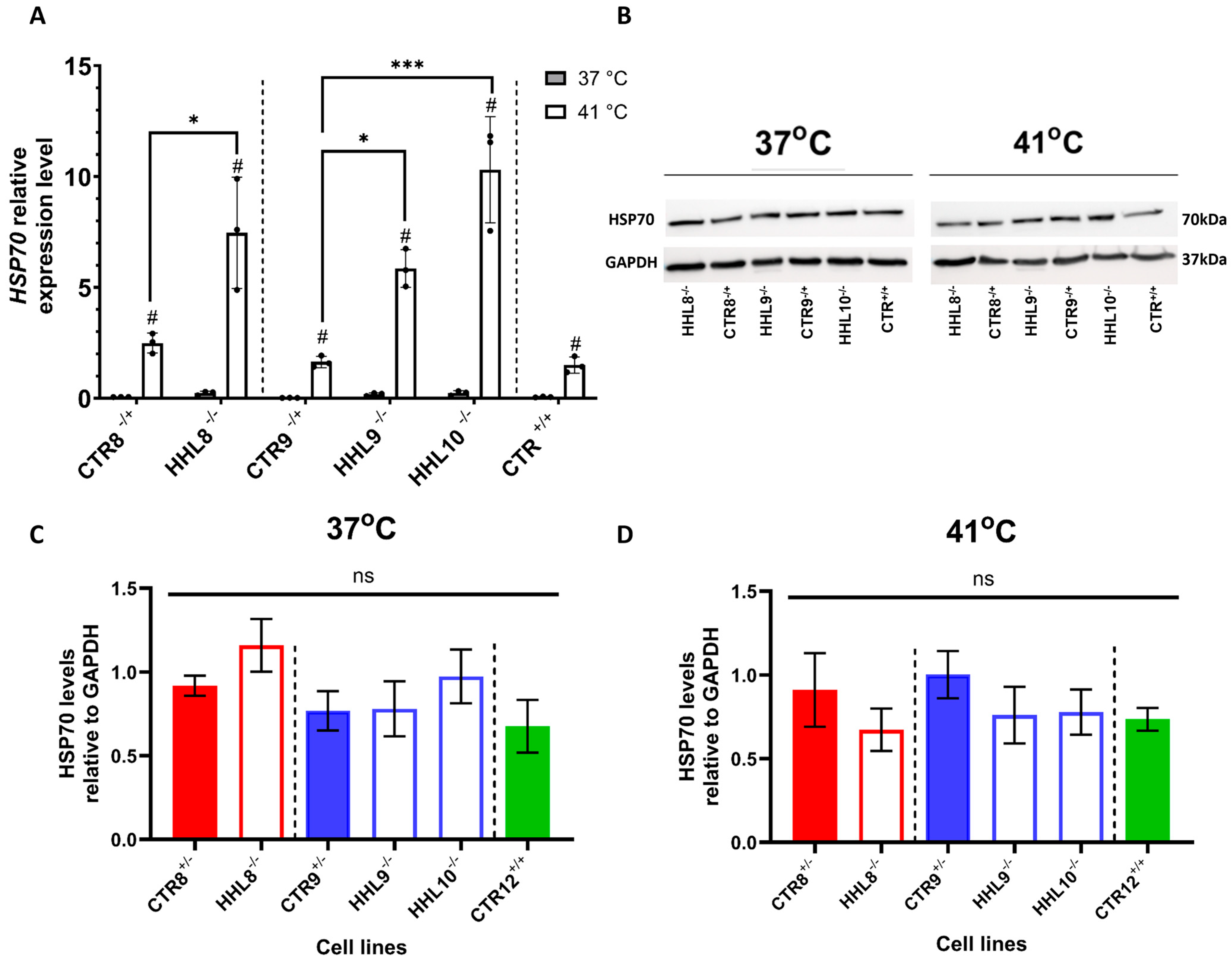
2.4. HIKESHI Levels Are Significantly Decreased in HHL iPSCs Following Heat Shock
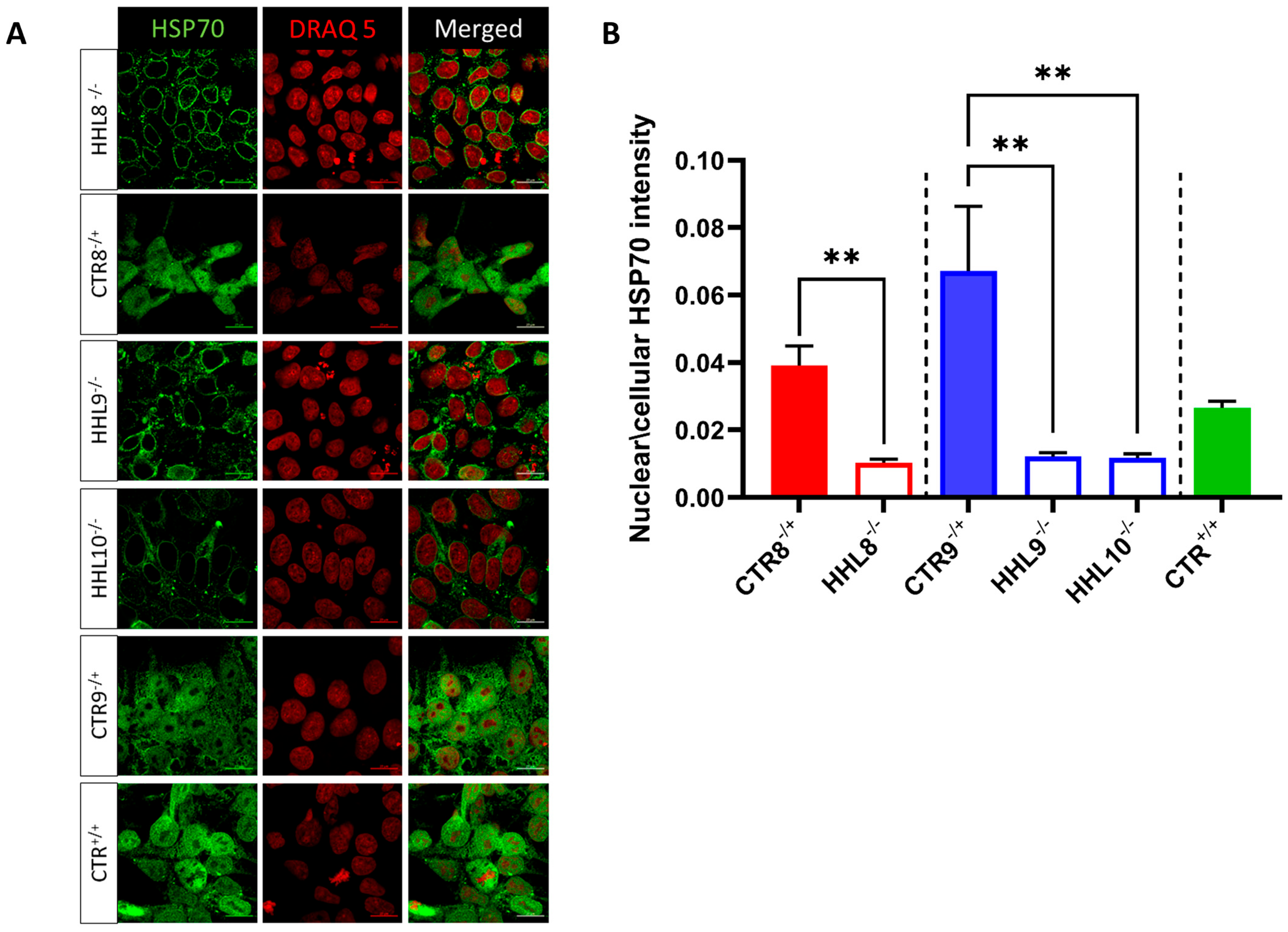
2.5. Impaired HSP70 Localization Following HS in HHL iPSCs
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Generation of Fibroblasts and Cell Reprogramming
4.3. iPSC Culture
4.4. Heat Shock Treatment
4.5. Immunocytochemistry Analysis
4.6. Genotyping
4.7. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
4.8. Western Blot Analyses
4.9. Imaging
4.10. HSP70 Intensity Quantification of Confocal Microscopy Images
4.11. Statistical Analysis and Data Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HHL | Hikeshi-related hypomyelinating leukodystrophy |
| AJ | Ashkenazi Jewish |
| CA | Christian Arab |
| HS | Heat shock |
| iPSCs | Induced pluripotent stem cells |
| HSP70 | Heat Shock Protein 70 |
| CNS | Central nervous system |
| V54L | Val54Leu (mutation in HIKESHI gene) |
| P78S | Pro78Ser (mutation in HIKESHI gene) |
References
- Edvardson, S.; Kose, S.; Jalas, C.; Fattal-Valevski, A.; Watanabe, A.; Ogawa, Y.; Mamada, H.; Fedick, A.M.; Ben-Shachar, S.; Treff, N.R.; et al. Leukoencephalopathy and early death associated with an Ashkenazi-Jewish founder mutation in the Hikeshi gene. J. Med. Genet. 2016, 53, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Helman, G.; Zerem, A.; Almad, A.; Hacker, J.L.; Woidill, S.; Sase, S.; LeFevre, A.N.; Ekstein, J.; Johansson, M.M.; Stutterd, C.A.; et al. Further Delineation of the Clinical and Pathologic Features of HIKESHI-Related Hypomyelinating Leukodystrophy. Pediatr. Neurol. 2021, 121, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Vasilescu, C.; Isohanni, P.; Palomäki, M.; Pihko, H.; Suomalainen, A.; Carroll, C.J. Absence of Hikeshi, a nuclear transporter for heat-shock protein HSP70, causes infantile hypomyelinating leukoencephalopathy. Eur. J. Hum. Genet. 2017, 25, 366. [Google Scholar] [CrossRef]
- Mallack, E.J.; Wang, C.; Kim, J.S.; Ross, M.E. A novel missense variant in HIKESHI: Clinical phenotype, in vitro functional testing, and potential for gene therapy. Am. J. Med. Genet. Part A 2024, 194, e63790. [Google Scholar] [CrossRef]
- Kose, S.; Furuta, M.; Imamoto, N. Hikeshi, a nuclear import carrier for Hsp70s, protects cells from heat shock-induced nuclear damage. Cell 2012, 149, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Kose, S.; Imamoto, N. Nucleocytoplasmic transport under stress conditions and its role in HSP70 chaperone systems. Biochim. Biophys. Acta 2014, 1840, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Tago, K.; Memezawa, S.; Ochiai, A.; Sawaguchi, S.; Kato, Y.; Sato, T.; Tomizuka, K.; Ooizumi, H.; Ohbuchi, K.; et al. The Infantile Leukoencephalopathy-Associated Mutation of C11ORF73/HIKESHI Proteins Generates de novo Interactive Activity with Filamin A, Inhibiting Oligodendroglial Cell Morphological Differentiation. Medicines 2021, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hong, H.; Ko, J.; Park, E.; Park, S.; Son, S. Structure and nuclear transport mechanism of HSP70 nuclear transporter, Hikeshi. Biodesign 2015, 3, 117–122. [Google Scholar]
- Kose, S.; Imai, K.; Watanabe, A.; Nakai, A.; Suzuki, Y.; Imamoto, N. Lack of Hikeshi activates HSF1 activity under normal conditions and disturbs the heat-shock response. Life Sci. Alliance 2022, 5, e202101241. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kose, S.; Watanabe, A.; Son, S.Y.; Choi, S.; Hong, H.; Yamashita, E.; Park, I.Y.; Imamoto, N.; Lee, S.J. Structural and functional analysis of Hikeshi, a new nuclear transport receptor of Hsp70s. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, D.; Rabinski, T.; Ofir, R.; Hershkovitz, E.; Vatine, G.D. Generation of iPSC lines from two (BGUi002-A and BGUi003-A) homozygous p450 oxidoreductase-deficient patients and from one (BGUi001-A) heterozygous healthy family relative. Stem Cell Res. 2020, 48, 101975. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, D.; Rabinski, T.; Halfon, A.; Anzi, S.; Plaschkes, I.; Benyamini, H.; Nevo, Y.; Gershoni, O.Y.; Rosental, B.; Hershkovitz, E.; et al. P450 oxidoreductase regulates barrier maturation by mediating retinoic acid metabolism in a model of the human BBB. Stem Cell Rep. 2022, 17, 2050–2063. [Google Scholar] [CrossRef] [PubMed]
- Rabinski, T.; Sagiv, S.T.; Hausman-Kedem, M.; Fattal-Valevski, A.; Rubinstein, M.; Avraham, K.B.; Vatine, G.D. Reprogramming of two induced pluripotent stem cell lines from a heterozygous GRIN2D developmental and epileptic encephalopathy (DEE) patient (BGUi011-A) and from a healthy family relative (BGUi012-A). Stem Cell Res. 2021, 51, 102178. [Google Scholar] [CrossRef] [PubMed]
- Falik, D.; Rabinski, T.; Zlotnik, D.; Eshel, R.; Zorsky, M.; Garin-Shkolnik, T.; Ofir, R.; Adato, A.; Ashkenazi, A.; Vatine, G.D. Generation and characterization of iPSC lines (BGUi004-A, BGUi005-A) from two identical twins with polyalanine expansion in the paired-like homeobox 2B (PHOX2B) gene. Stem Cell Res. 2020, 48, 101955. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; ffrench-Constant, C.; van der Knaap, M.S. Hypomyelinating leukodystrophies—Unravelling myelin biology. Nat. Rev. Neurol. 2021, 17, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, N. Heat stress-induced nuclear transport mediated by Hikeshi confers nuclear function of Hsp70s. Curr. Opin. Cell Biol. 2018, 52, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Hikeshi, M.G.I. Mouse Gene Detail—MGI:96738—Heat Shock Protein Nuclear Import Factor. Available online: https://www.informatics.jax.org/marker/MGI:96738 (accessed on 24 October 2023).
- Blake, J.A.; Baldarelli, R.; Kadin, J.A.; Richardson, J.E.; Smith, C.L.; Bult, C.J. Mouse Genome Database (MGD): Knowledgebase for mouse-human comparative biology. Nucleic Acids Res. 2021, 49, D981–D9817. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, D.; Sun, D.; Zhang, X.; Lei, W.; Wu, M.; Huang, Q.; Nian, X.; Dai, W.; Lu, X.; et al. Nuclear import carrier Hikeshi cooperates with HSP70 to promote murine oligodendrocyte differentiation and CNS myelination. Dev. Cell 2023, 58, 2275–2291.e6. [Google Scholar] [CrossRef] [PubMed]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A generalist algorithm for cellular segmentation. Nat. Methods 2020, 18, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Direction | Primer Sequence 5′-3′ |
|---|---|---|
| HIKESHI Intron 1 | Forward | GGCCTCCCACAAAGTGTTGG |
| HIKESHI Intron 2 | Reverse | ATTTTTTTTGGCCGGGCGC |
| HIKESHI Intron 1 | Forward | CAGAAACTCATAGAACCATTGAACTG |
| HIKESHI Intron 2 | Reverse | TATGACTTTTCCCCCTTTAGATTGTG |
| Gene Name | Primer Sequence 5′-3′ | Assay ID |
|---|---|---|
| HIKESHI | GTCTTAAATCTGGAGAAGGAAGCCA | Hs01581971_g1 |
| HSP70 | TTTTCCGGTTTCTACATGCAGAGAT | Hs00359163_s1 |
| GAPDH | CGCTGCCAAGGCTGTGGGCAAGGTC | Hs02786624_g1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, M.A.; Boichuck, M.; Ottolenghi, A.; Rabinski, T.; Goldenthal, O.; Sanchez, D.S.; Fattal-Valevski, A.; Heimer, G.; Ben-Shachar, S.; Libzon, S.; et al. A Novel Pathogenic Variant Identified in HIKESHI-Related Hypomyelinating Leukodystrophy Disrupts Heat Shock Response in iPSCs. Int. J. Mol. Sci. 2025, 26, 6037. https://doi.org/10.3390/ijms26136037
Saleh MA, Boichuck M, Ottolenghi A, Rabinski T, Goldenthal O, Sanchez DS, Fattal-Valevski A, Heimer G, Ben-Shachar S, Libzon S, et al. A Novel Pathogenic Variant Identified in HIKESHI-Related Hypomyelinating Leukodystrophy Disrupts Heat Shock Response in iPSCs. International Journal of Molecular Sciences. 2025; 26(13):6037. https://doi.org/10.3390/ijms26136037
Chicago/Turabian StyleSaleh, Mahmood Ali, Maria Boichuck, Aner Ottolenghi, Tatiana Rabinski, Omri Goldenthal, Daniel Sevilla Sanchez, Aviva Fattal-Valevski, Gali Heimer, Shay Ben-Shachar, Stephanie Libzon, and et al. 2025. "A Novel Pathogenic Variant Identified in HIKESHI-Related Hypomyelinating Leukodystrophy Disrupts Heat Shock Response in iPSCs" International Journal of Molecular Sciences 26, no. 13: 6037. https://doi.org/10.3390/ijms26136037
APA StyleSaleh, M. A., Boichuck, M., Ottolenghi, A., Rabinski, T., Goldenthal, O., Sanchez, D. S., Fattal-Valevski, A., Heimer, G., Ben-Shachar, S., Libzon, S., Gershoni-Yahalom, O., Ben-Zvi, A., Zarivach, R., Zerem, A., Rosental, B., & Vatine, G. D. (2025). A Novel Pathogenic Variant Identified in HIKESHI-Related Hypomyelinating Leukodystrophy Disrupts Heat Shock Response in iPSCs. International Journal of Molecular Sciences, 26(13), 6037. https://doi.org/10.3390/ijms26136037