
Pathogenic Crosstalk Between the Peripheral and Central Nervous System in Rheumatic Diseases: Emerging Evidence and Clinical Implications

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Neuroimmune Interface in Rheumatic Diseases
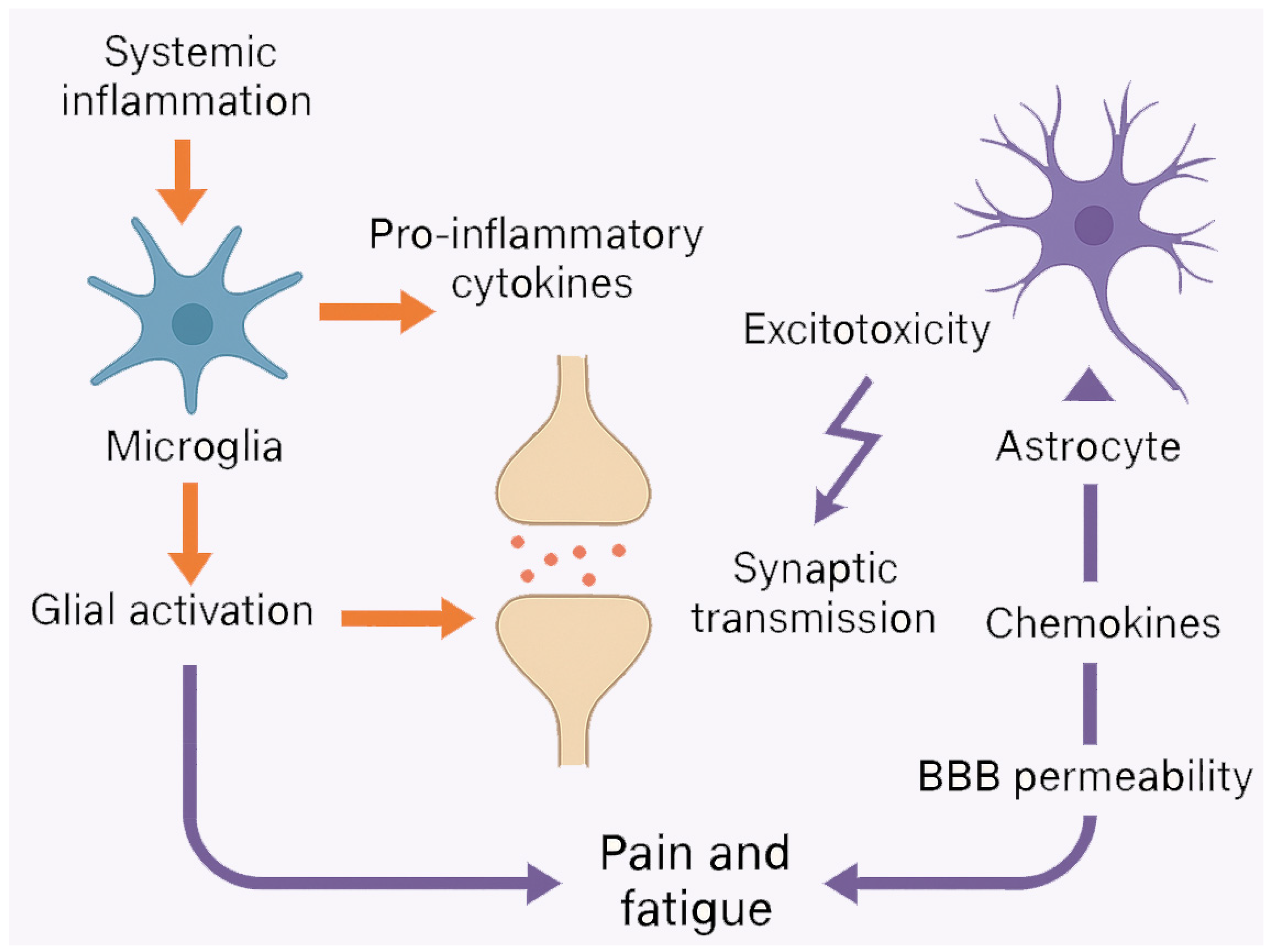
3. Neuroinflammation and Glial Activation
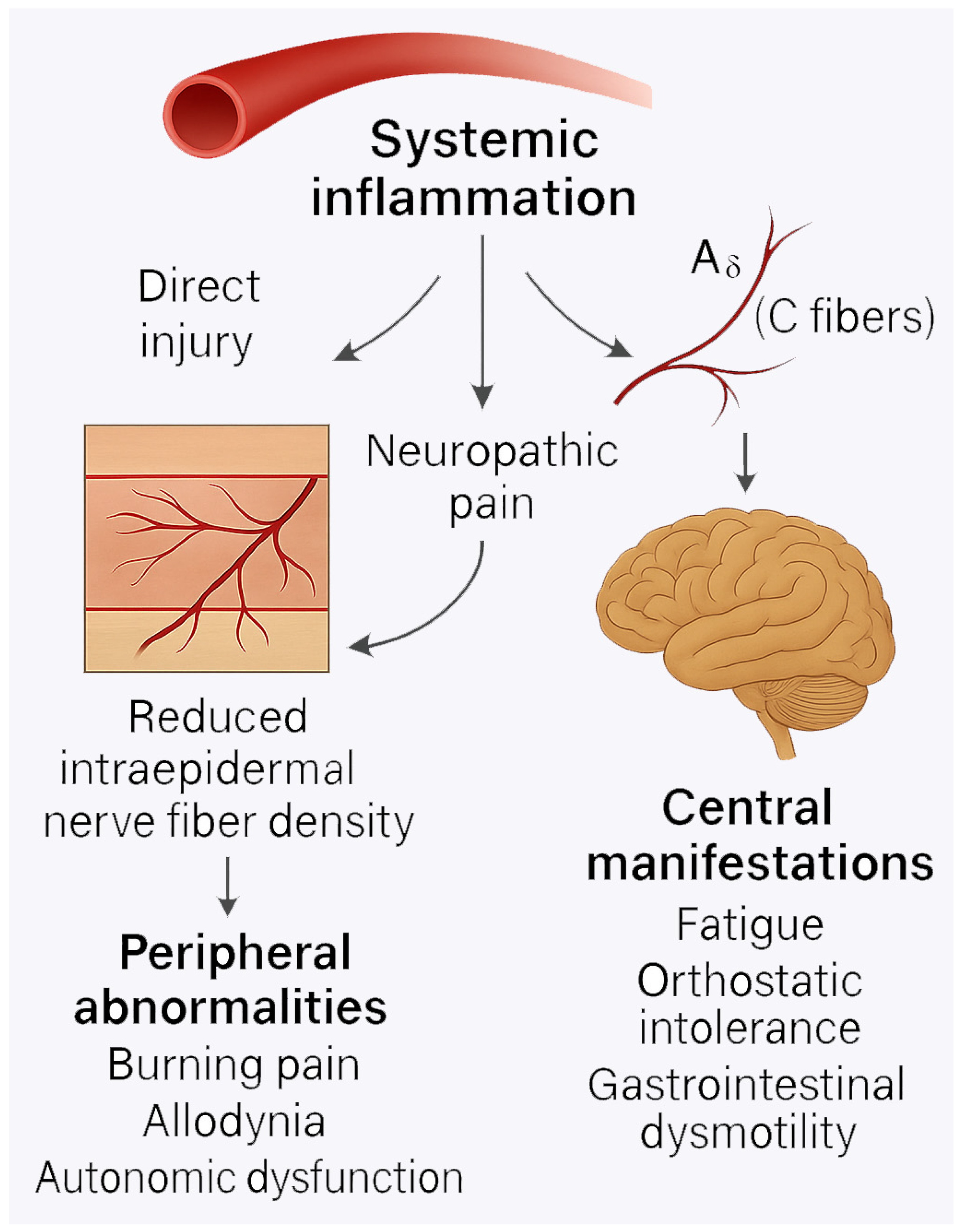
4. Small-Fiber Neuropathy and Peripheral Nerve Damage
5. Central Sensitization and Chronic Pain
6. Dysregulation of the Autonomic Nervous System
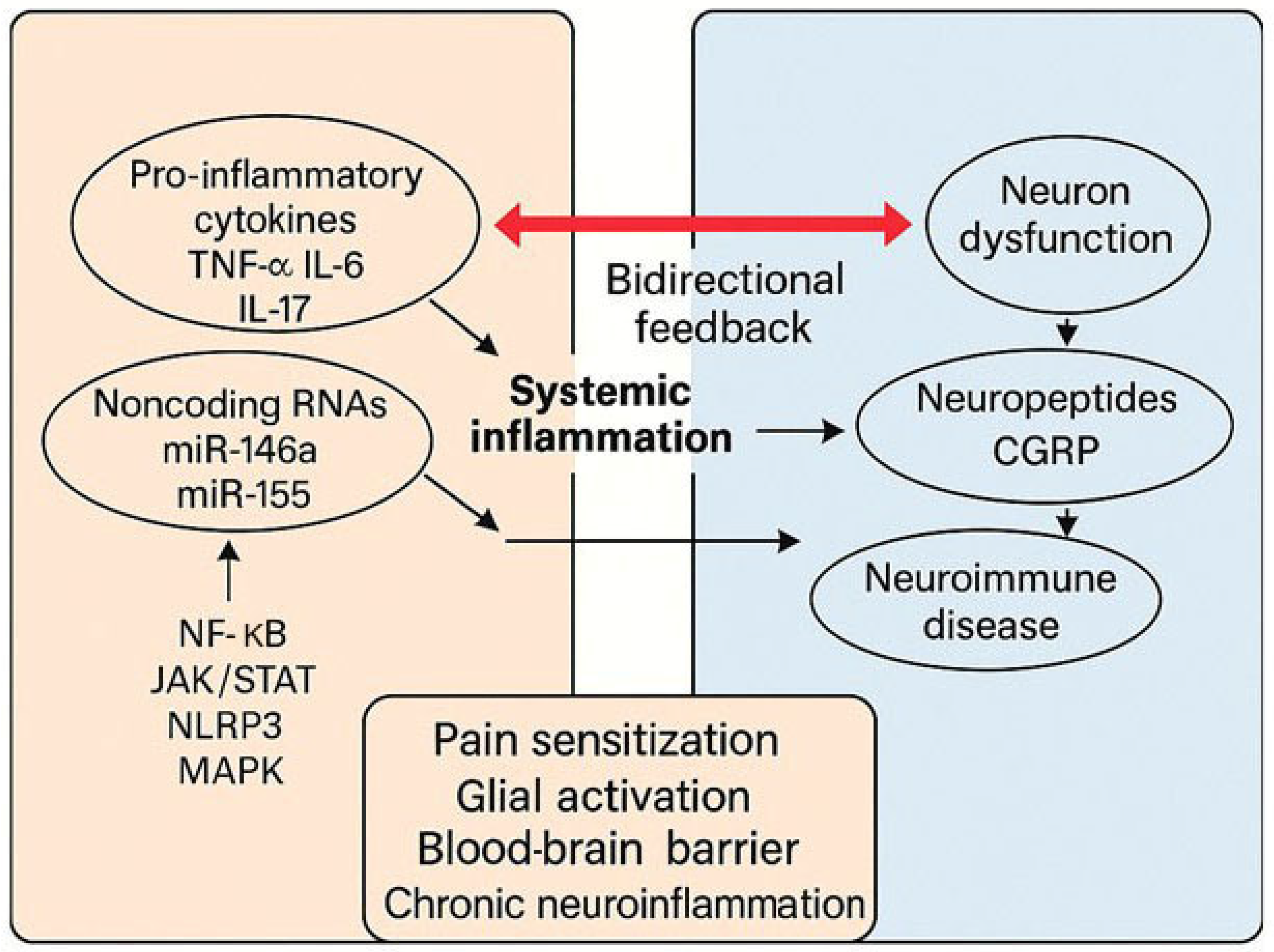
7. Emerging Molecular Mediators
8. Clinical Implications
9. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Mease, P.J. Navigating the complexity of pain in psoriatic arthritis and axial spondyloarthritis. Curr. Opin. Rheumatol. 2024, 36, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.E.; Minhas, D.; Clauw, D.J.; Lee, Y.C. Identification and management of nociplastic pain in individuals with rheumatic diseases: A Narrative Review. Arthritis Care Res. 2023, 75, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Lacagnina, M.J.; Heijnen, C.J.; Watkins, L.R.; Grace, P.M. Autoimmune regulation of chronic pain. PAIN Rep. 2021, 6, e905. [Google Scholar] [CrossRef]
- Davies, K.; Dures, E.; Ng, W.F. Fatigue in inflammatory rheumatic diseases: Current knowledge and areas for future research. Nat. Rev. Rheumatol. 2021, 17, 651–664. [Google Scholar] [CrossRef]
- Thombs, B.D.; Adams, C. Coping with fatigue in inflammatory rheumatic diseases. Lancet Rheumatol. 2022, 4, e526–e527. [Google Scholar] [CrossRef]
- Clauw, D.; Sarzi-Puttini, P.; Pellegrino, G.; Shoenfeld, Y. Is fibromyalgia an autoimmune disorder? Autoimmun. Rev. 2024, 23, 103424. [Google Scholar] [CrossRef]
- Myasoedova, E.; Sattui, S.E.; Lee, J.; O’Brien, J.T.; Makris, U.E. Cognitive impairment in individuals with rheumatic diseases: The role of systemic inflammation, immunomodulatory drugs, and comorbidities. Lancet Rheumatol. 2024, 6, e871–e880. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Song, G.; Li, J.; Song, Z.; Zhao, B.; Liang, L.; Li, W.; Hu, H.; Tu, H.; Li, S.; et al. Innervation of nociceptor neurons in the spleen promotes germinal center responses and humoral immunity. Cell 2024, 187, 2935–2951.e19. [Google Scholar] [CrossRef]
- Yu, J.; Xiao, K.; Chen, X.; Deng, L.; Zhang, L.; Li, Y.; Gao, A.; Gao, J.; Wu, C.; Yang, X.; et al. Neuron-derived neuropeptide Y regulates splenic immune responses. Neuron 2022, 110, 1326–1339. [Google Scholar] [CrossRef]
- Motyl, G.; Krupka, W.M.; Maslinska, M. The problem of residual pain in the evaluation of rheumatoid arthritis activity. Rheumatology 2024, 62, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Michaud, K.; Pope, J.; van de Laar, M.; Curtis, J.R.; Kannowski, C.; Mitchell, S.; Bell, J.; Workman, J.; Paik, J.; Cardoso, A.; et al. Systematic review of the literature on residual symptoms and unmet needs in patients with rheumatoid arthritis. Arthritis Care Res. 2021, 73, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of neuropathology-associated reactive astrocytes: A systematic review. Acta Neuropathol. Commun. 2023, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Amanollahi, M.; Jameie, M.; Heidari, A.; Rezaei, N. The dialogue between neuroinflammation and adult neurogenesis: Mechanisms involved and alterations in neurological diseases. Mol. Neurobiol. 2023, 60, 923–959. [Google Scholar] [CrossRef]
- Fisher, T.M.; Liddelow, S.A. Emerging roles of astrocytes as immune effectors in the central nervous system. Trends Immunol. 2024, 45, 824–836. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Drieu, A.; Mamuladze, T.; de Lima, K.A.; Dykstra, T.; Wall, M.; Papadopoulos, Z.; Kanamori, M.; Salvador, A.F.; Baker, W.; et al. Functional characterization of dural sinuses as a neuroimmune interface. Cell 2021, 184, 1000–1016.e27. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, L.; Chai, Y.; Zhang, J.; Deng, Q.; Chen, X. Reimagining the meninges from a neuroimmune perspective: A boundary, but not peripheral. J. Neuroinflammation 2024, 21, 299. [Google Scholar] [CrossRef]
- Seeliger, T.; Kramer, E.; Konen, F.F.; Zehrfeld, N.; Beider, S.; Prenzler, N.K.; Godecke, V.; Witte, T.; Skripuletz, T.; Ernst, D. Sjogren’s syndrome with and without neurological involvement. J. Neurol. 2023, 270, 2987–2996. [Google Scholar] [CrossRef]
- Brock, J.; Basu, N.; Schlachetzki, J.C.M.; Schett, G.; McInnes, I.B.; Cavanagh, J. Immune mechanisms of depression in rheumatoid arthritis. Nat. Rev. Rheumatol. 2023, 19, 790–804. [Google Scholar] [CrossRef]
- Carrion-Barbera, I.; Salman-Monte, T.C.; Vilchez-Oya, F.; Monfort, J. Neuropsychiatric involvement in systemic lupus erythematosus: A review. Autoimmun. Rev. 2021, 20, 102780. [Google Scholar] [CrossRef]
- Lu, Y.Z.; Nayer, B.; Singh, S.K.; Alshoubaki, Y.K.; Yuan, E.; Park, A.J.; Maruyama, K.; Akira, S.; Martino, M.M. CGRP sensory neurons promote tissue healing through neutrophils and macrophages. Nature 2024, 628, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Atta, A.A.; Ibrahim, W.W.; Mohamed, A.F.; Abdelkader, N.F. Polarization of microglia in nociplasmic pain: Mechanisms and perspectives. Inflammopharmacology 2023, 31, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Goebel, A.; Krock, E.; Gentry, C.; Israel, M.R.; Jurczak, A.; Urbina, C.M.; Sandor, K.; Vastani, N.; Maurer, M.; Cuhadar, U.; et al. Passive transfer of fibromyalgia symptoms from patients to mice. J. Clin. Investig. 2021, 131, e144201. [Google Scholar] [CrossRef] [PubMed]
- Midavaine, E.; Moraes, B.C.; Benitez, J.; Rodriguez, S.R.; Braz, J.M.; Kochhar, N.P.; Eckalbar, W.L.; Tian, L.; Domingos, A.I.; Pintar, J.E.; et al. Meningeal regulatory T cells inhibit nociception in female mice. Science 2025, 388, 96–104. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, S.; Lorenzo-Herrero, S.; Sordo-Bahamonde, C.; Hidalgo, A.; Gonzalez, S.; Menendez, L.; Baamonde, A. Involvement of CD4+ and CD8+ T lymphocytes in the modulation of CCL4-evoked nociceptive processing in mice. Life Sci. 2022, 291, 120302. [Google Scholar] [CrossRef]
- Taketani, Y.; Marmalidou, A.; Dohlman, T.H.; Singh, R.B.; Amouzegar, A.; Chauhan, S.K.; Chen, Y.; Dana, R. Restoration of Regulatory T-Cell Function in Dry Eye Disease by Antagonizing Substance P/Neurokinin-1 Receptor. Am. J. Pathol. 2020, 190, 1859–1866. [Google Scholar] [CrossRef]
- Furia, A.; Liguori, R.; Donadio, V. Small-Fiber Neuropathy: An Etiology-Oriented Review. Brain Sci. 2025, 15, 158. [Google Scholar] [CrossRef]
- Daifallah, O.; Farah, A.; Dawes, J.M. A role for pathogenic autoantibodies in small fiber neuropathy? Front. Mol. Neurosci. 2023, 16, 1254854. [Google Scholar] [CrossRef]
- Shinkarevsky Fleitman, I.; Nevo, Y.; Harel, L.; Amarilyo, G.; Dori, A.; Agmon-Levin, N.; Kachko, L.; Zaks Hoffer, G.; Dabby, R.; Rabie, M.; et al. Small fiber neuropathy associated with autoinflammatory syndromes in children and adolescents. Muscle Nerve 2020, 61, 791–796. [Google Scholar] [CrossRef]
- Gwathmey, K.G.; Satkowiak, K. Peripheral nervous system manifestations of rheumatologic diseases. J. Neurol. Sci. 2021, 424, 117421. [Google Scholar] [CrossRef]
- Zhao, Y.; Gan, L.; Ren, L.; Lin, Y.; Ma, C.; Lin, X. Factors influencing blood-brain barrier permeability. Brain Res. 2022, 1788, 147937. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Fitzcharles, M.A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Hauser, W. Nociplastic pain: Toward an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110. [Google Scholar] [CrossRef]
- Bjorklund, G.; Dadar, M.; Pivina, L.; Dosa, M.D.; Semenova, Y.; Maes, M. Environmental, Neuro-immune, and Neuro-oxidative Stress Interactions in Chronic Fatigue Syndrome. Mol. Neurobiol. 2020, 57, 4598–4607. [Google Scholar] [CrossRef]
- Jolly, A.A.; Brown, R.B.; Tozer, D.J.; Hong, Y.T.; Fryer, T.D.; Aigbirhio, F.I.; O’Brien, J.T.; Markus, H.S. Is central and systemic inflammation associated with fatigue in small vessel disease of the brain? Int. J. Stroke 2024, 19, 705–713. [Google Scholar] [CrossRef]
- Al-Hakeim, H.K.; Al-Rubaye, H.T.; Almulla, A.F.; Al-Hadrawi, D.S.; Maes, M. Chronic Fatigue, Depression and Anxiety Symptoms in Long COVID Are Strongly Predicted by Neuroimmune and Neuro-Oxidative Pathways Which Are Caused by the Inflammation during Acute Infection. J. Clin. Med. 2023, 12, 511. [Google Scholar] [CrossRef]
- Berntson, L.; Elfving, A.; Samuelsson, A.G.; Oman, A.; Mobarrez, F. Blood-brain barrier permeability and astrocyte-derived extracellular vesicles in children with juvenile idiopathic arthritis: A cross-sectional study. Pediatr. Rheumatol. 2024, 22, 47. [Google Scholar] [CrossRef]
- Matsushita, T.; Otani, K.; Yoshiga, M.; Hirano, M.; Noda, K.; Kurosaka, D. Inhibitory effect of baricitinib on microglia and STAT3 in a weak blood-brain barrier region in a mouse model of rheumatoid arthritis. Rheumatology 2023, 62, 2908–2917. [Google Scholar] [CrossRef]
- Chimenti, M.S.; Fonti, G.L.; Conigliaro, P.; Triggianese, P.; Bianciardi, E.; Coviello, M.; Lombardozzi, G.; Tarantino, G.; Niolu, C.; Siracusano, A.; et al. The burden of depressive disorders in musculoskeletal diseases: Is there an association between mood and inflammation? Ann. Gen. Psychiatry 2021, 20, 1. [Google Scholar] [CrossRef]
- Kwok, C.H.T.; Kohro, Y.; Mousseau, M.; O’Brien, M.S.; Matyas, J.R.; McDougall, J.J.; Trang, T. Role of Primary Afferents in Arthritis Induced Spinal Microglial Reactivity. Front. Immunol. 2021, 12, 626884. [Google Scholar] [CrossRef] [PubMed]
- Schaible, H.G.; Konig, C.; Ebersberger, A. Spinal pain processing in arthritis: The (inter)actions of neurons and glia. J. Neurochem. 2024, 168, 3644–3662. [Google Scholar] [CrossRef]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and nonimmune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Lee, J.H.; Flausino, L.E.; Quintana, F.J. Neuroinflammation: An astrocytic perspective. Sci. Transl. Med. 2023, 15, eadi7828. [Google Scholar] [CrossRef] [PubMed]
- Makabe, K.; Okada, H.; Tachibana, N.; Ishikura, H.; Ito, N.; Tanaka, M.; Chijimatsu, R.; Terashima, A.; Yano, F.; Asaka, M.; et al. Baricitinib ameliorates inflammatory and neuropathic pain in mice with collagen antibody-induced arthritis by modulating IL-6/JAK/STAT3 pathway and CSF-1 expression in dorsal root ganglion neurons. Arthritis Res. Ther. 2024, 26, 121. [Google Scholar] [CrossRef]
- Suss, P.; Rothe, T.; Hoffmann, A.; Schlachetzki, J.C.M.; Winkler, J. The Joint-Brain Axis: Insights From Rheumatoid Arthritis on the Crosstalk Between Chronic Peripheral Inflammation and the Brain. Front. Immunol. 2020, 11, 612104. [Google Scholar] [CrossRef]
- Nikolopoulos, D.; Manolakou, T.; Polissidis, A.; Filia, A.; Bertsias, G.; Koutmani, Y.; Boumpas, D.T. Activation of microglia in the presence of an intact blood-brain barrier and disruption of hippocampal neurogenesis by IL-6 and IL-18 mediate early diffuse neuropsychiatric lupus. Ann. Rheum. Dis. 2023, 82, 646–657. [Google Scholar] [CrossRef]
- Han, X.; Xu, T.; Ding, C.; Wang, D.; Yao, G.; Chen, H.; Fang, Q.; Hu, G.; Sun, L. Neuronal NR4A1 deficiency drives complement-coordinated synaptic stripping by microglia in a mouse model of lupus. Signal Transduct. Target. Ther. 2022, 7, 50. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, L.; Zheng, X.; Fang, Q.; Qian, Y.; Xu, T.; Liang, J.; Zhang, H.; Han, X.; Sun, L. Microglia orchestrate synaptic and neuronal stripping: Implication in neuropsychiatric lupus. J. Cell. Mol. Med. 2024, 28, e18190. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G. Special astrocytes release glutamate. Nat. Neurosci. 2023, 26, 1660. [Google Scholar] [CrossRef]
- Liu, D.; Liao, P.; Li, H.; Tong, S.; Wang, B.; Lu, Y.; Gao, Y.; Huang, Y.; Zhou, H.; Shi, L.; et al. Regulation of blood-brain barrier integrity by Dmp1-expressing astrocytes through mitochondrial transfer. Sci. Adv. 2024, 10, eadk2913. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; Bennett, R.E.; Hudry, E.; Hyman, B.T. Astrocyte-derived APOE4 leads to blood-brain barrier impairment. Brain 2022, 145, 3582–3593. [Google Scholar] [CrossRef] [PubMed]
- Hanani, M. Satellite glial cells in human diseases. Cells 2024, 13, 566. [Google Scholar] [CrossRef]
- Fang, S.; Wu, Z.; Guo, Y.; Zhu, W.; Wan, C.; Yuan, N.; Chen, J.; Hao, W.; Mo, X.; Guo, X.; et al. Roles of microglia in adult hippocampal neurogenesis in depression and their therapeutics. Front. Immunol. 2023, 14, 1193053. [Google Scholar] [CrossRef]
- Grubic Kezele, T.; Omrcen, H.; Baticic, L.; Sucurovic, S.; Zoricic Cvek, S. Joint Inflammation Correlates with Joint GPR30 Expression in Males and Hippocampal GPR30 Expression in Females in a Rat Model of Rheumatoid Arthritis. Int. J. Mol. Sci. 2024, 25, 7864. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Kim, H.K. Recent Developments in PET and SPECT Radiotracers as Radiopharmaceuticals for Hypoxia Tumors. Pharmaceutics 2023, 15, 1840. [Google Scholar] [CrossRef]
- Alshelh, Z.; Brusaferri, L.; Saha, A.; Morrissey, E.; Knight, P.; Kim, M.; Zhang, Y.; Hooker, J.M.; Albrecht, D.; Torrado-Carvajal, A.; et al. Neuroimmune signatures in chronic low back pain subtypes. Brain 2022, 145, 1098–1110. [Google Scholar] [CrossRef]
- Fukui, S.; Winkelmayer, W.C.; Tedeschi, S.K.; Marrugo, J.; Guan, H.; Harrold, L.; Litman, H.J.; Shinozaki, T.; Solomon, D.H. Disease activity of rheumatoid arthritis and kidney function decline: A large prospective registry study. Ann. Rheum. Dis. 2025, 84, 201–209. [Google Scholar] [CrossRef]
- Wang, Y.; Coughlin, J.M.; Ma, S.; Endres, C.J.; Kassiou, M.; Sawa, A.; Dannals, R.F.; Petri, M.; Pomper, M.G. Neuroimaging of translocator protein in patients with systemic lupus erythematosus: A pilot study using [11C]DPA-713 positron emission tomography. Lupus 2017, 26, 170–178. [Google Scholar] [CrossRef]
- Albrecht, D.S.; Forsberg, A.; Sandstrom, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Hoglund, C.O.; Catana, C.; et al. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef]
- Geladaris, A.; Torke, S.; Saberi, D.; Alankus, Y.B.; Streit, F.; Zechel, S.; Stadelmann-Nessler, C.; Fischer, A.; Boschert, U.; Hausler, D.; et al. BTK inhibition limits CNS inflammation perpetuated by microglia and promotes myelin repair. Acta Neuropathol. 2024, 147, 75. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Kang, X.; Zhang, H.; Xu, H.; Wu, X. Knockdown and inhibition of hippocampal GPR17 attenuate lipopolysaccharide-induced cognitive impairment in mice. J. Neuroinflammation 2023, 20, 271. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Nakamura, K.; Kohyama, K.; Harada, C.; Behanna, H.A.; Watterson, D.M.; Matsumoto, Y.; Harada, T. Inhibition of glial cell activation improves the severity of experimental autoimmune encephalomyelitis. Neurosci. Res. 2007, 59, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Devigili, G.; Cazzato, D.; Lauria, G. Clinical diagnosis and management of small fiber neuropathy: An update on best practices. Expert Rev. Neurother. 2020, 20, 967–980. [Google Scholar] [CrossRef]
- Kool, D.; Hoeijmakers, J.G.; Waxman, S.G.; Faber, C.G. Small fiber neuropathy. Int. Rev. Neurobiol. 2024, 179, 181–231. [Google Scholar] [CrossRef]
- Galosi, E.; Pirone, C.; Ceccarelli, F.; Esposito, N.; Falco, P.; Leopizzi, M.; Di Maio, V.; Tramontana, L.; De Stefano, G.; Di Pietro, G.; et al. Clinical, histologic, and immunologic signatures of Small Fiber Neuropathy in Systemic Lupus Erythematosus. J. Peripher. Nerv. Syst. 2024, 29, 315–328. [Google Scholar] [CrossRef]
- Seeliger, T.; Dreyer, H.N.; Siemer, J.M.; Bonig, L.; Gingele, S.; Dohrn, M.F.; Prenzler, N.; Ernst, D.; Witte, T.; Skripuletz, T. Clinical and paraclinical features of small-fiber neuropathy in Sjogren’s syndrome. J. Neurol. 2023, 270, 1004–1010. [Google Scholar] [CrossRef]
- Birnbaum, J. Small-fiber neuropathy presenting in the antecedent period of undifferentiated arthritis before rheumatoid arthritis. Neurol. Clin. Pract. 2017, 7, e47–e50. [Google Scholar] [CrossRef]
- Gavrilova, N.; Starshinova, A.; Zinchenko, Y.; Kudlay, D.; Shapkina, V.; Malkova, A.; Belyaeva, E.; Pavlova, M.; Yablonskiy, P.; Shoenfeld, Y. Small Fiber Neuropathy in Sarcoidosis. Pathophysiology 2021, 28, 544–550. [Google Scholar] [CrossRef]
- Saperstein, D.S. Small fiber neuropathy. Neurol. Clin. 2020, 38, 607–618. [Google Scholar] [CrossRef]
- Birnbaum, J.; Lalji, A.; Saed, A.; Baer, A.N. Biopsy-Proven Small-Fiber Neuropathy in Primary Sjogren’s Syndrome: Neuropathic pain characteristics, autoantibody findings, and histopathologic features. Arthritis Care Res. 2019, 71, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Liampas, A.; Parperis, K.; Erotocritou, M.F.; Nteveros, A.; Papadopoulou, M.; Moschovos, C.; Akil, M.; Coaccioli, S.; Hadjigeorgiou, G.M.; Hadjivassiliou, M.; et al. Primary peripheral neuropathy related to Sjogren’s syndrome: A systematic review and meta-analysis. Eur. J. Neurol. 2023, 30, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Zeidman, L.A.; Levine, T.; Cangelosi, J. Small-Vessel Vasculitis or Perifolliculitis in Small-Fiber Neuropathy with TS-HDS, FGFR-3, or Plexin D1 Antibodies. J. Clin. Neuromuscul. Dis. 2024, 26, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Kyle, K.; Hutto, S.K.; Reda, H.; Zonozi, R.; Farhad, K.; Jeyabalan, A.; Chwalisz, B.K. Small fiber neuropathy associated with ANCA positivity: A case series and brief literature review. Neurol. Sci. 2023, 44, 4473–4479. [Google Scholar] [CrossRef]
- Chan, A.C.Y.; Siah, K.T.H. Can Small Fiber Neuropathy Explain the Overlap Gastrointestinal and Non-gastrointestinal Symptoms in Some Irritable Bowel Syndrome Patients? J. Neurogastroenterol. Motil. 2024, 30, 116–118. [Google Scholar] [CrossRef]
- Azcue, N.; Del Pino, R.; Acera, M.; Fernandez-Valle, T.; Ayo-Mentxakatorre, N.; Perez-Concha, T.; Murueta-Goyena, A.; Lafuente, J.V.; Prada, A.; Lopez de Munain, A.; et al. Dysautonomia and small-fiber neuropathy in the post-COVID condition and chronic fatigue syndrome. J. Transl. Med. 2023, 21, 814. [Google Scholar] [CrossRef]
- Moak, J.P.; Ramwell, C.B.; Gordish-Dressman, H.; Sule, S.D.; Bettini, E. Small-fiber neuropathy in children, adolescents, and young adults with chronic orthostatic intolerance and tachycardia syndrome postural orthostatic tachycardia: A retrospective study. Auton. Neurosci. 2024, 253, 103163. [Google Scholar] [CrossRef]
- Dumolard, A.; Lefaucheur, J.P.; Hodaj, E.; Liateni, Z.; Payen, J.F.; Hodaj, H. Central Sensitization and Small-fiber Neuropathy Are Associated in Patients with Fibromyalgia. Clin. J. Pain 2023, 39, 8–14. [Google Scholar] [CrossRef]
- de Tommaso, M.; Vecchio, E.; Nolano, M. The fibromyalgia puzzle between central sensitization syndrome and small fiber neuropathy: A narrative review on neurophysiological and morphological evidence. Neurol. Sci. 2022, 43, 1667–1684. [Google Scholar] [CrossRef]
- Volcheck, M.M.; Graham, S.M.; Fleming, K.C.; Mohabbat, A.B.; Luedtke, C.A. Central sensitization, chronic pain, and other symptoms: Better understanding, better management. Clevel. Clin. J. Med. 2023, 90, 245–254. [Google Scholar] [CrossRef]
- Lepri, B.; Romani, D.; Storari, L.; Barbari, V. Effectiveness of pain neuroscience training in patients with chronic musculoskeletal pain and central sensitization: A Systematic Review. Int. J. Environ. Res. Public Health 2023, 20, 4098. [Google Scholar] [CrossRef] [PubMed]
- Trouvin, A.P.; Attal, N.; Perrot, S. Evaluation of central sensitization with quantitative sensory testing in inflammatory rheumatic diseases: A systematic review. Jt. Bone Spine 2022, 89, 105399. [Google Scholar] [CrossRef] [PubMed]
- Dougados, M.; Perrot, S. Fibromyalgia and central sensitization in chronic inflammatory joint disease. Jt. Bone Spine 2017, 84, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Guler, M.A.; Celik, O.F.; Ayhan, F.F. The important role of central sensitization in chronic musculoskeletal pain observed in various rheumatic diseases. Clin. Rheumatol. 2020, 39, 269–274. [Google Scholar] [CrossRef]
- Kosek, E.; Cohen, M.; Baron, R.; Gebhart, G.F.; Mico, J.A.; Rice, A.S.C.; Rief, W.; Sluka, A.K. Do we need a third mechanistic descriptor for chronic pain states? Pain 2016, 157, 1382–1386. [Google Scholar] [CrossRef]
- Jensen, T.S.; Finnerup, N.B. Allodynia and hyperalgesia in neuropathic pain: Clinical manifestations and mechanisms. Lancet Neurol. 2014, 13, 924–935. [Google Scholar] [CrossRef]
- Melvin, B.; Wright, R.; McNally, A.; Elmofty, D. Allodynia: A Review Article. Curr. Pain Headache Rep. 2025, 29, 49. [Google Scholar] [CrossRef]
- Sarzi-Puttini, P.; Giorgi, V.; Marotto, D.; Atzeni, F. Fibromyalgia: An update on clinical features, etiopathogenesis, and treatment. Nat. Rev. Rheumatol. 2020, 16, 645–660. [Google Scholar] [CrossRef]
- Fitzcharles, M.A.; Perrot, S.; Hauser, W. Comorbid fibromyalgia: A qualitative review of prevalence and significance. Eur. J. Pain 2018, 22, 1565–1576. [Google Scholar] [CrossRef]
- Zhao, S.S.; Duffield, S.J.; Goodson, N.J. The prevalence and impact of comorbid fibromyalgia in inflammatory arthritis. Best Pract. Res. Clin. Rheumatol. 2019, 33, 101423. [Google Scholar] [CrossRef]
- Coskun Benlidayi, I. Fibromyalgia interferes with disease activity and response to biological therapy in inflammatory rheumatic diseases. Rheumatol. Int. 2020, 40, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.T.; Mallawaarachchi, B.; Shim, J.; Lock, J.; Macfarlane, G.J. The prevalence of fibromyalgia in axial spondyloarthritis. Rheumatol. Int. 2020, 40, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zeng, X.; Qin, G.; Zhang, D.; Zhou, J.; Chen, L. Downregulation of metabotropic glutamate 5 receptor alleviates central sensitization by activating autophagy through inhibition of mTOR pathway in a rat model of chronic migraine. Neurosci. Lett. 2021, 743, 135552. [Google Scholar] [CrossRef]
- Merighi, A. Brain-Derived Neurotrophic Factor, Nociception, and Pain. Biomolecules 2024, 14, 539. [Google Scholar] [CrossRef]
- Nugraha, B.; Karst, M.; Engeli, S.; Gutenbrunner, C. Brain-derived neurotrophic factor and exercise in fibromyalgia syndrome patients: A mini review. Rheumatol. Int. 2012, 32, 2593–2599. [Google Scholar] [CrossRef]
- Souza Monteiro de Araujo, D.; Nassini, R.; Geppetti, P.; De Logu, F. TRPA1 as a therapeutic target for nociceptive pain. Expert Opin. Ther. Targets 2020, 24, 997–1008. [Google Scholar] [CrossRef]
- De Preter, C.C.; Heinricher, M.M. The ‘in’s and out’s’ of descending pain modulation from the rostral ventromedial medulla. Trends Neurosci. 2024, 47, 447–460. [Google Scholar] [CrossRef]
- Gao, Z.R.; Chen, W.Z.; Liu, M.Z.; Chen, X.J.; Wan, L.; Zhang, X.Y.; Yuan, L.; Lin, J.K.; Wang, M.; Zhou, L.; et al. Tac1-expressing neurons in the periaqueductal gray facilitate the pruritus-gratate cycle through down-regulation. Neuron 2019, 101, 45–59.e9. [Google Scholar] [CrossRef]
- Pinto-Ribeiro, F.; Amorim, D.; David-Pereira, A.; Monteiro, A.M.; Costa, P.; Pertovaara, A.; Almeida, A. Pronoception from the dorsomedial nucleus of the hypothalamus is mediated by the rostral ventromedial medulla in healthy controls but is absent in arthritic animals. Brain Res. Bull. 2013, 99, 100–108. [Google Scholar] [CrossRef]
- Kandemirli, S.G.; Bathla, G. Neuroimaging findings in rheumatologic disorders. J. Neurol. Sci. 2021, 427, 117531. [Google Scholar] [CrossRef]
- Vassilaki, M.; Crowson, C.S.; Davis Iii, J.M.; Duong, S.Q.; Jones, D.T.; Nguyen, A.; Mielke, M.M.; Vemuri, P.; Myasoedova, E. Rheumatoid Arthritis, Cognitive Impairment, and Neuroimaging Biomarkers: Results from the Mayo Clinic Study of Aging. J. Alzheimer’s Dis. 2022, 89, 943–954. [Google Scholar] [CrossRef]
- Taylor, P.C. Joint pain and beyond: The challenge of rheumatoid arthritis. Lancet Rheumatol. 2023, 5, e351–e360. [Google Scholar] [CrossRef] [PubMed]
- de la Coba, P.; Montoro, C.I.; Reyes Del Paso, G.A.; Galvez-Sanchez, C.M. Algometry for the assessment of central pain sensitization in fibromyalgia patients: A systematic review. Ann. Med. 2022, 54, 1403–1422. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, A.; Ashrafi, M.; Deodhar, A. Difficult-to-treat psoriatic arthritis: How to manage it? Clin. Rheumatol. 2023, 42, 2251–2265. [Google Scholar] [CrossRef]
- Currado, D.; Saracino, F.; Ruscitti, P.; Marino, A.; Pantano, I.; Vomero, M.; Berardicurti, O.; Pavlych, V.; Di Vico, C.; Caso, F.; et al. Pain catastrophizing negatively affects drug retention rate in patients with psoriatic arthritis and axial spondyloarthritis: Results of a 2-year multicenter GIRRCS (Italian Research Group in Clinical Rheumatology) study. Arthritis Res. Ther. 2024, 26, 162. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Herrem, C.; Ndlovu, N.; O’Brien, C.; Yazici, Y. A somatization comorbidity phenotype affects response to therapy in rheumatoid arthritis: Post-hoc results from the phase 4 PREDICT study of certolizumab pegol. Arthritis Res. Ther. 2017, 19, 215. [Google Scholar] [CrossRef]
- Cooper, T.E.; Derry, S.; Wiffen, P.J.; Moore, R.A. Gabapentin for fibromyalgia pain in adults. Cochrane Database Syst. Rev. 2017, 1, CD012188. [Google Scholar] [CrossRef]
- VanderWeide, L.A.; Smith, S.M.; Trinkley, K.E. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J. Clin. Pharm. Ther. 2015, 40, 1–6. [Google Scholar] [CrossRef]
- Macfarlane, G.J.; Kronisch, C.; Dean, L.E.; Atzeni, F.; Hauser, W.; Fluss, E.; Choy, E.; Kosek, E.; Amris, K.; Branco, J.; et al. EULAR revised recommendations for the management of fibromyalgia. Ann. Rheum. Dis. 2017, 76, 318–328. [Google Scholar] [CrossRef]
- Serrat, M.; Sanabria-Mazo, J.P.; Almirall, M.; Muste, M.; Feliu-Soler, A.; Mendez-Ulrich, J.L.; Sanz, A.; Luciano, J.V. Effectiveness of a Multicomponent Treatment Based on Pain Neuroscience Education, Therapeutic Exercise, Cognitive Behavioral Therapy, and Mindfulness in Patients with Fibromyalgia (FIBROWALK Study): A Randomized Controlled Trial. Phys. Ther. 2021, 101, pzab200. [Google Scholar] [CrossRef]
- Ambrose, K.R.; Golightly, Y.M. Exercise as a nonpharmacologic treatment of chronic pain: Why and when. Best Pract. Res. Clin. Rheumatol. 2015, 29, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Wehrwein, E.A.; Orer, H.S.; Barman, S.M. Overview of the Anatomy, Physiology, and Pharmacology of the Autonomic Nervous System. Compr. Physiol. 2016, 6, 1239–1278. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.; Ng, W.F. Autonomic Nervous System Dysfunction in Primary Sjogren’s Syndrome. Front. Immunol. 2021, 12, 702505. [Google Scholar] [CrossRef]
- Bortoluzzi, A.; Silvagni, E.; Furini, F.; Piga, M.; Govoni, M. Peripheral nervous system involvement in systemic lupus erythematosus: A review of the evidence. Clin. Exp. Rheumatol. 2019, 37, 146–155. [Google Scholar]
- Bellocchi, C.; Carandina, A.; Montinaro, B.; Targetti, E.; Furlan, L.; Rodrigues, G.D.; Tobaldini, E.; Montano, N. The Interplay between Autonomic Nervous System and Inflammation across Systemic Autoimmune Diseases. Int. J. Mol. Sci. 2022, 23, 2449. [Google Scholar] [CrossRef]
- Tumiati, B.; Perazzoli, F.; Negro, A.; Pantaleoni, M.; Regolisti, G. Heart rate variability in patients with Sjogren’s syndrome. Clin. Rheumatol. 2000, 19, 477–480. [Google Scholar] [CrossRef]
- Erelund, S.; Sodergren, A.; Wiklund, U.; Sundstrom, N. Heart rate variability and cardiovascular risk factors in patients with rheumatoid arthritis: A longitudinal study. Auton. Neurosci. 2023, 249, 103119. [Google Scholar] [CrossRef]
- Rovsing, C.; Rovsing, H.; Liboriussen, C.H.; Jensen, M.K.; Andersen, S.S.; Andersen, S.S.; Kristensen, S.; Jochumsen, M. Deep breathing increases heart rate variability in patients with rheumatoid arthritis and systemic lupus erythematosus. J. Clin. Rheumatol. 2021, 27, 261–266. [Google Scholar] [CrossRef]
- Stojanovich, L. Autonomic dysfunction in autoimmune rheumatic disease. Autoimmun. Rev. 2009, 8, 569–572. [Google Scholar] [CrossRef]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef]
- Occhinegro, A.; McAllen, R.M.; McKinley, M.J.; Martelli, D. Acute inhibition of sympathetic nerve-mediated inflammation: The inflammatory reflex. Neuroimmunomodulation 2023, 30, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Occhinegro, A.; Wong, C.Y.; Chua, B.Y.; Jackson, D.C.; McKinley, M.J.; McAllen, R.M.; Martelli, D. Endogenous inflammatory reflex inhibits the inflammatory response to different immune challenges in mice. Brain Behav. Immun. 2021, 97, 371–375. [Google Scholar] [CrossRef]
- Yang, H.; George, S.J.; Thompson, D.A.; Silverman, H.A.; Tsaava, T.; Tynan, A.; Pavlov, V.A.; Chang, E.H.; Andersson, U.; Brines, M.; et al. Famotidine activates the vagus nerve inflammatory reflex to attenuate cytokine storm. Mol. Med. 2022, 28, 57. [Google Scholar] [CrossRef] [PubMed]
- Oyama, J.; Node, K. Sympathetic nerve activity and endothelial function. Hypertens. Res. 2014, 37, 1035–1036. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.E.; Pfeiffer-Jensen, M.; Drewes, A.M.; Farmer, A.D.; Deleuran, B.W.; Stengaard-Pedersen, K.; Brock, B.; Brock, C. Vagal influences in rheumatoid arthritis. Scand. J. Rheumatol. 2018, 47, 1–11. [Google Scholar] [CrossRef]
- Tyagi, S.; Higerd-Rusli, G.P.; Ghovanloo, M.R.; Dib-Hajj, F.; Zhao, P.; Liu, S.; Kim, D.H.; Shim, J.S.; Park, K.S.; Waxman, S.G.; et al. Compartment-specific regulation of Na(V)1.7 in sensory neurons after acute exposure to TNF-alpha. Cell Rep. 2024, 43, 113685. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Heffner, D.L.; Kim, S.; Espy, S.M.; Spano, A.J.; Cleland, C.L.; Deppmann, C.D. TNF-alpha/TNFR1 signaling is required for primary nociceptor development and function. Neuron 2014, 82, 587–602. [Google Scholar] [CrossRef]
- Zhou, Y.Q.; Liu, Z.; Liu, Z.H.; Chen, S.P.; Li, M.; Shahveranov, A.; Ye, D.W.; Tian, Y.K. Interleukin-6: An emerging regulator of pathological pain. J. Neuroinflammation 2016, 13, 141. [Google Scholar] [CrossRef]
- Choy, E.H.S.; Calabrese, L.H. Neuroendocrine and neurophysiologic effects of interleukin-6 in rheumatoid arthritis. Rheumatology 2018, 57, 1885–1895. [Google Scholar] [CrossRef]
- Wanigatunga, A.A.; Varadhan, R.; Simonsick, E.M.; Carlson, O.D.; Studenski, S.; Ferrucci, L.; Schrack, J.A. Longitudinal Relationship Between Interleukin-6 and Perceived Fatigability Among Well-Functioning Adults in Mid-to-Late Life. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 720–725. [Google Scholar] [CrossRef]
- Wang, J.; Wu, T.; Zhao, Y.; Mao, L.; Ding, J.; Wang, X. IL-17A exacerbates blood-brain barrier disruption through activation of Src signaling in epileptic mice. Neurobiol. 2024, 61, 11012–11025. [Google Scholar] [CrossRef]
- Khan, A.W.; Farooq, M.; Hwang, M.J.; Haseeb, M.; Choi, S. Autoimmune neuroinflammatory diseases: Role of interleukins. Int. J. Mol. Sci. 2023, 24, 7960. [Google Scholar] [CrossRef] [PubMed]
- Brigas, H.C.; Ribeiro, M.; Coelho, J.E.; Gomes, R.; Gomez-Murcia, V.; Carvalho, K.; Faivre, E.; Costa-Pereira, S.; Darrigues, J.; de Almeida, A.A.; et al. IL-17 triggers the onset of cognitive and synaptic deficits in the early stages of Alzheimer’s disease. Cell Rep. 2021, 36, 109574. [Google Scholar] [CrossRef] [PubMed]
- Schou, W.S.; Ashina, S.; Amin, F.M.; Goadsby, P.J.; Ashina, M. Calcitonin gene-related peptide and pain: A systematic review. J. Headache Pain 2017, 18, 34. [Google Scholar] [CrossRef]
- Iyengar, S.; Ossipov, M.H.; Johnson, K.W. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 2017, 158, 543–559. [Google Scholar] [CrossRef]
- Ding, W.; Stohl, L.L.; Xu, L.; Zhou, X.K.; Manni, M.; Wagner, J.A.; Granstein, R.D. Calcitonin Gene-Related Peptide-Exposed Endothelial Cells Bias Antigen Presentation to CD4+ T Cells toward a Th17 Response. J. Immunol. 2016, 196, 2181–2194. [Google Scholar] [CrossRef]
- Mikami, N.; Sueda, K.; Ogitani, Y.; Otani, I.; Takatsuji, M.; Wada, Y.; Watanabe, K.; Yoshikawa, R.; Nishioka, S.; Hashimoto, N.; et al. Calcitonin gene-related peptide regulates type IV hypersensitivity through dendritic cell functions. PLoS ONE 2014, 9, e86367. [Google Scholar] [CrossRef]
- Schank, J.R.; Heilig, M. Substance P and the neurokinin-1 receptor: The novel CRF. Int. Rev. Neurobiol. 2017, 136, 151–175. [Google Scholar] [CrossRef]
- Puts, S.; Liberman, K.; Leysen, L.; Forti, L.; Muyldermans, E.; Vaes, P.; Nijs, J.; Beckwee, D.; Bautmans, I. Effects of exercise on inflammatory markers and brain-derived neurotrophic factor in patients with knee osteoarthritis. A systematic review with meta-analysis. Exerc. Immunol. Rev. 2023, 29, 22–53. [Google Scholar]
- Tanaka, T.; Okuda, H.; Isonishi, A.; Terada, Y.; Kitabatake, M.; Shinjo, T.; Nishimura, K.; Takemura, S.; Furue, H.; Ito, T.; et al. Cutaneous macrophages regulate pain sensitivity by modulating the amount of tissue NGF through an SNX25-Nrf2 pathway. Nat. Immunol. 2023, 24, 439–451. [Google Scholar] [CrossRef]
- Zaninelli, T.H.; Factors, V.; Heintz, O.K.; Wright, K.R.; Bennallack, P.R.; Sim, D.; Bukhari, H.; Terry, K.L.; Vitonis, A.F.; Missmer, S.A.; et al. Targeting NGF but not VEGFR1 or BDNF signaling reduces endometriosis-associated pain in mice. J. Adv. Res. 2024, 73, 593–605. [Google Scholar] [CrossRef]
- Patel, F.; Hess, D.K.; Maher, D.P. Antibodies against nerve growth factor for the treatment of low back pain. Expert Rev. Clin. Pharmacol. 2020, 13, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ma, M.; Yu, H.; Yu, H. Inhibition of specific lncRNA X-inactivated transcript ameliorates inflammatory pain by suppressing satellite glial cell activation and inflammation by acting as a sponge of miR-146a to inhibit Na(v)1,7. J. Cell. Biochem. 2018, 119, 9888–9898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Q.; Nai, Y.; Cao, C. Suppression of miR-155 attenuates neuropathic pain by inducing a switch from M1 to M2 in microglia. Folia Neuropathol. 2020, 58, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, H.L.; An, L.J.; Li, L.; Wei, M.; Ge, D.J.; Su, Z. miR-124-3p attenuates neuropathic pain induced by chronic sciatic nerve injury in rats through targeting EZH2. J. Cell. Biochem. 2019, 120, 5747–5755. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Rodriguez-Palma, E.J.; Huerta de la Cruz, S.; Islas-Espinoza, A.M.; Castaneda-Corral, G.; Granados-Soto, V.; Khanna, R. Nociplastic pain mechanisms and toll-like receptors as promising targets for its management. Pain 2024, 165, 2150–2164. [Google Scholar] [CrossRef]
- Hu, S.Q.; Hu, J.L.; Zou, F.L.; Liu, J.P.; Luo, H.L.; Hu, D.X.; Wu, L.D.; Zhang, W.J. P2X7 receptor in inflammation and pain. Brain Res. Bull. 2022, 187, 199–209. [Google Scholar] [CrossRef]
- Aminin, D.; Illes, P. Purinergic Signaling in Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 12895. [Google Scholar] [CrossRef]
- Fadda, G.; Flanagan, E.P.; Cacciaguerra, L.; Jitprapaikulsan, J.; Solla, P.; Zara, P.; Sechi, E. Myelitis features and outcomes in CNS demyelinating disorders: Comparison between multiple sclerosis, MOGAD, and AQP4-IgG-positive NMOSD. Front. Neurol. 2022, 13, 1011579. [Google Scholar] [CrossRef]
- Liu, Y.; Tu, Z.; Zhang, X.; Du, K.; Xie, Z.; Lin, Z. Pathogenesis and treatment of neuropsychiatric systemic lupus erythematosus: A review. Front. Cell Dev. Biol. 2022, 10, 998328. [Google Scholar] [CrossRef] [PubMed]
- Sletten, D.M.; Suarez, G.A.; Low, P.A.; Mandrekar, J.; Singer, W. COMPASS 31: A refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin. Proc. 2012, 87, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K.; Lee, G.S. Clinical assessment of autonomic function in fibromyalgia using the Refined and Abbreviated Composite Autonomic Symptom Score (COMPASS 31): A case-control study. Rev. Recent Clin. Trials 2022, 17, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Pindi Sala, T.; Villedieu, M.; Damian, L.; Crave, J.C.; Pautot, V.; Stojanovich, L.; Tervaert, J.W.C.; Cherin, P.; Belizna, C. Long-term efficacy of immunoglobulins in Sjogren’s syndrome-related small fiber neuropathy. J. Neurol. 2020, 267, 3499–3507. [Google Scholar] [CrossRef]
- Giordano, F.; Zicca, A.; Barba, C.; Guerrini, R.; Parent, L. Vagus nerve stimulation: Surgical technique of implantation and revision and related morbidity. Epilepsia 2017, 58 (Suppl. S1), 85–90. [Google Scholar] [CrossRef]
- Kudrina, I.; Shir, Y.; Fitzcharles, M.A. Multidisciplinary treatment of rheumatic pain. Best Pract. Res. Clin. Rheumatol. 2015, 29, 156–163. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paroli, M.; Sirinian, M.I. Pathogenic Crosstalk Between the Peripheral and Central Nervous System in Rheumatic Diseases: Emerging Evidence and Clinical Implications. Int. J. Mol. Sci. 2025, 26, 6036. https://doi.org/10.3390/ijms26136036
Paroli M, Sirinian MI. Pathogenic Crosstalk Between the Peripheral and Central Nervous System in Rheumatic Diseases: Emerging Evidence and Clinical Implications. International Journal of Molecular Sciences. 2025; 26(13):6036. https://doi.org/10.3390/ijms26136036
Chicago/Turabian StyleParoli, Marino, and Maria Isabella Sirinian. 2025. "Pathogenic Crosstalk Between the Peripheral and Central Nervous System in Rheumatic Diseases: Emerging Evidence and Clinical Implications" International Journal of Molecular Sciences 26, no. 13: 6036. https://doi.org/10.3390/ijms26136036
APA StyleParoli, M., & Sirinian, M. I. (2025). Pathogenic Crosstalk Between the Peripheral and Central Nervous System in Rheumatic Diseases: Emerging Evidence and Clinical Implications. International Journal of Molecular Sciences, 26(13), 6036. https://doi.org/10.3390/ijms26136036