Abstract
A series of novel podophyllotoxin derivatives containing benzothiazole scaffolds were synthesized and evaluated for their in vitro cytotoxic activity against five cancer cell lines (MCF-7, SKOV-3, B16F10, LOVO, and HeLa). Two compounds, 7 and 11, which are different only by the absence or presence of the ester group, showed the strongest cytotoxic effect towards all tested cancer cell lines with the IC50 0.68–2.88 µM. In addition, it was demonstrated that these compounds inhibit cancer cell proliferation by inducing G2/M phase arrest in HeLa cells. The structure–activity relationship was analyzed and it confirmed the importance of the core structural features like a dioxolane ring and free-rotating trimethoxyphenyl group for cytotoxicity. Moreover, the R configuration of the ester group at the C-8′ position proved to be substantial since its epimer was inactive. The molecular docking studies revealed that the most potent compounds have a different binding mode to β-tubulin than podophyllotoxin; however, the benzothiazole fragment docked in a similar location as the trimethoxyphenyl group of podophyllotoxin, exhibiting similar hydrophobic interactions. These findings clearly indicate that podophyllotoxin–benzothiazole derivatives could be addressed for further pharmacological studies in anticancer research.
1. Introduction
Podophyllotoxin 1 is an active compound from Podophyllum sp. that is well known for its broad spectrum of biological activities such as anticancer, antiviral, anti-inflammatory, and antibacterial properties [1,2,3,4]. Molecular mechanisms of podophyllotoxin 1 activity involve the inhibition of tubulin polymerization into microtubules and blocking the replication of cellular DNA; thus, it is considered an antitumor agent [4,5]. Unfortunately, despite such a promising bioactivity, podophyllotoxin 1 is highly toxic against normal cells; therefore, it is used only for the topical treatment of anogenital warts [6].
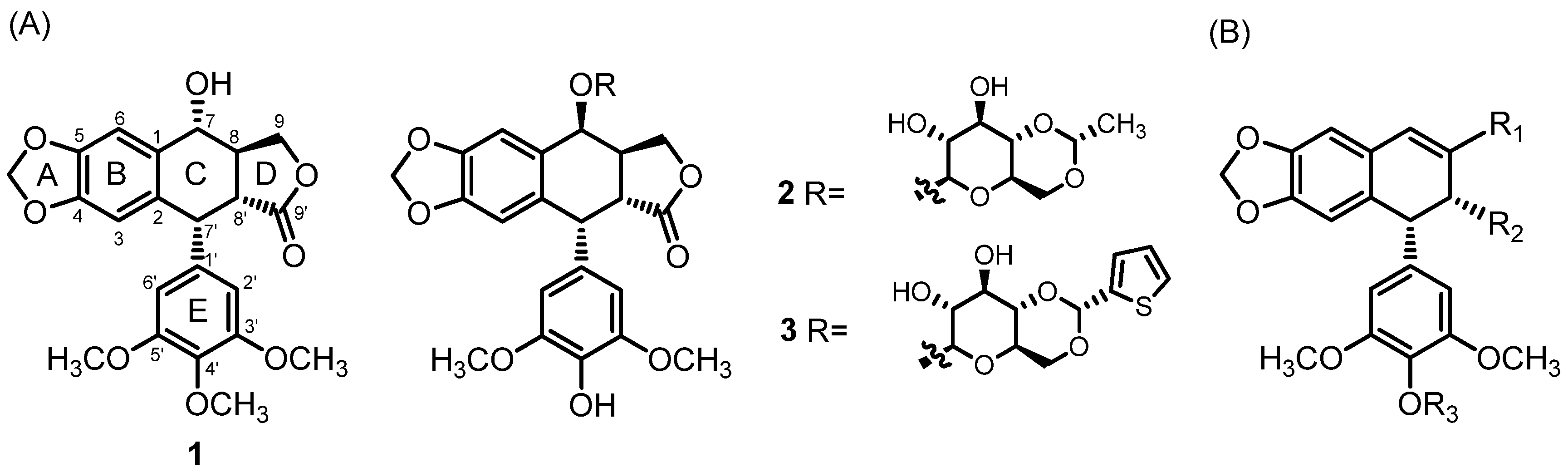
Cancer remains one of the leading causes of global mortality, with 20 million new cases and 9.7 million deaths reported in 2022 [7]. Breast and colorectal cancers are the second and third most prevalent malignancies, respectively, while cervical and ovarian cancers pose a significant concern due to high mortality rates among women [7,8]. Additionally, the incidence of melanoma continues to rise, correlating with increased exposure to ultraviolet (UV) radiation [9]. Numerous strategies have been developed to find novel drug candidates with high cytotoxicity to cancer cells, sufficient bioavailability, and acceptable toxicity to normal cells. Up to now, many podophyllotoxin 1 derivatives have been reported. The modifications in the C-ring of podophyllotoxin were predominantly explored. Among the most promising derivatives, etoposide 2 and teniposide 3 (Figure 1) were introduced in anticancer therapy [10]. Interestingly, these two derivatives have a different molecular mechanism of activity compared to the parent compound. While podophyllotoxin 1 inhibits the assembly of tubulin into microtubules, etoposide 2 and its analogs inhibit DNA topoisomerase, preventing relegation of the double-strand break [11]. The core structural features of podophyllotoxin 1, like trans-fused C-lactone, a fused dioxolane ring, and the almost-orthogonal free-rotating 3,4,5-trimethoxyphenyl fragment, are considered essential for the antitubulin activity of podophyllotoxin derivatives [12,13,14]. Although many of them have been synthesized so far, the most explored strategy is based on modifications at the C-7 position [10]. However, a literature survey revealed that podophyllic aldehyde 6 and its analogs lacking a trans-lactone D-ring exhibit significant cytotoxicity against various tumor cell lines [15,16]. Therefore, modification of the D-ring of podophyllotoxin 1 became of interest.

Figure 1.
(A) Podophyllotoxin 1 and its derivatives 2 (etoposide), 3 (teniposide); (B) novel lead structure with three modifiable positions, marked R1–R3.
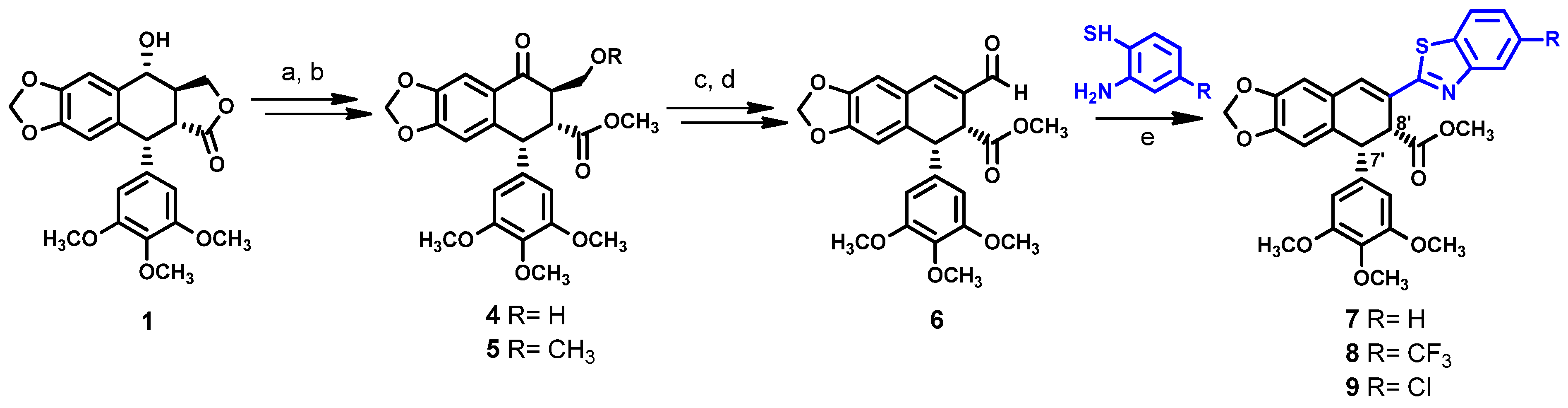
KL-3 7 is a derivative of podophyllotoxin 1 (Scheme 1) that was developed in our team [17,18]. It contains a benzothiazole group instead of the lactone ring. The synthesis of KL-3 7 can be effectively performed by the transformation of podophyllic aldehyde 6, the preparation of which was based on the photochemical cyclization [19]. In contrast to podophyllotoxin 1, which is highly toxic, KL-3 7 allows for the repair of normal cells and triggers mechanisms that restore non-cancer cell homeostasis [18]. This promising result inspired us to design and study new KL-3 7 analogs to obtain more potent drug candidates and evaluate their structure–activity relationships. We planned three modifications on 7, including adding substituents at the benzothiazole ring, modification of the ester part with hydrazines, and demethylation at the ring E. However, despite our efforts, the ester group modifications brought unexpected results. Nevertheless, we confirmed the leading structure for this class of podophyllotoxin–benzothiazole congeners with potent anticancer activity.
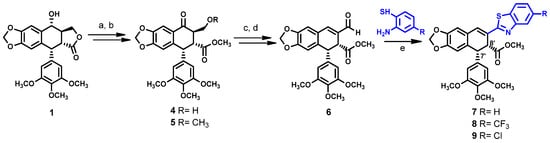
Scheme 1.
Reagents and conditions: a = PDC/CH2Cl2 rt, 2 h; b = H2SO4/MeOH reflux, 1 h; c = NaBH4, MeOH rt, 0.5 h; d = 1. (COCl)2, DMSO, −55 °C, 15 min, 2. Et3N, CH2Cl2, 0.5 h to 0 °C, 1 h; e = Na2S2O5, DMSO, 120 °C, 2 h.
2. Results and Discussion
2.1. Chemistry
Scheme 1 illustrates the synthetic pathway for compounds 7–9. The unsaturated aldehyde 6 was prepared according to the procedure previously described [15]. In short, the hydroxyl group in podophyllotoxin 1 was oxidized with pyridinium dichromate (PDC) and the resulting product was hydrolyzed in acidic conditions, using sulfuric acid in methanol to open the lactone ring. This strategy enabled us to maintain the desired cis-stereochemistry of substituents at C-7′ and C-8′ positions, while hydrolysis under alkaline conditions causes undesired epimerization at C-7′ position [12,20]. We observed that careful control of the reaction time enables us to obtain a good yield of compound 4, while extending this time beyond one hour resulted in the formation of compound 5. This result was confirmed with NMR by the appearance of additional methyl protons at 3.25 ppm and methoxy carbon at 70.2 ppm. The reduction of 4 with sodium borohydride afforded 1,3-diol, which was transformed into unsaturated aldehyde 6 using Swern oxidation. The 1H and 13C NMR of compounds 4 and 6 were identical, as reported in the literature [15]. Compounds 7–9 were synthesized by the condensation of aldehyde 6 with corresponding 2-aminothiophenol derivatives in the presence of sodium metabisulphite as an oxidizing agent [21].
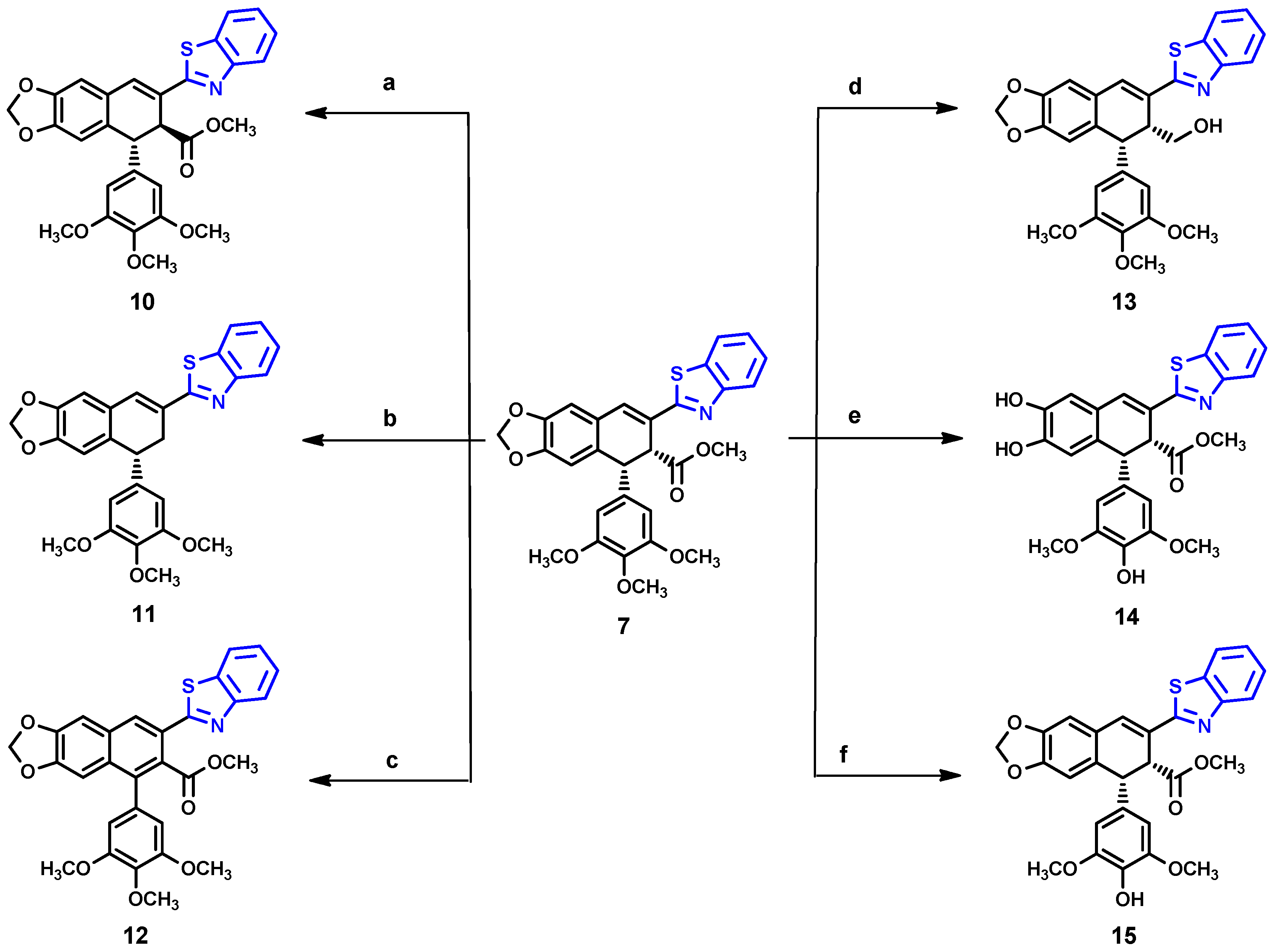
As shown in Scheme 2, we further modified compound 7, obtaining six derivatives (10–15). Attempted hydrazinolysis of compound 7 gave rise to compound 10 as a result of epimerization of the ester group, as confirmed by the 1H NMR spectrum, in which the methyl ester protons were shifted to 3.63 ppm. Alternatively, compound 11 was obtained by treatment of 7 with potassium hydroxide in methanol, followed by neutralization with hydrochloric acid. The absence of a 1H NMR signal of methyl protons from the ester group, and the appearance of two new methylene protons at 3.38 and 3.18 ppm, confirmed the unexpected removal of the ester group. On the other hand, the one-pot reaction of 7 with potassium hydroxide solution followed by the reaction with oxalyl chloride and hydrazine treatment led to aromatization of ring C 12, which was identified from the disappearance of two methine protons in the range 4.0–5.0 ppm and the appearance of two quaternary carbons at 129.2 ppm and 132.9 ppm. The reaction of 7 with DIBAL-H resulted in the formation of a complex mixture of products difficult to identify, from which only compound 13 was isolated. Considering the fact that many podophyllotoxin derivatives with a free 4′-hydroxy group, such as etoposide 2, have potential as anticancer agents, especially targeting topoisomerase II [22,23,24], we considered performing a demethylation of 7. Thus, the reaction of 7 with methionine in the presence of methanesulfonic acid was performed according to the procedure previously reported [23]; however, we observed that not only did demethylation occurred, but the dioxolane ring was also opened to form compound 14. Fortunately, the reaction of compound 7 with trimethylsilyl iodide (TMSI) [25] successfully produced compound 15.
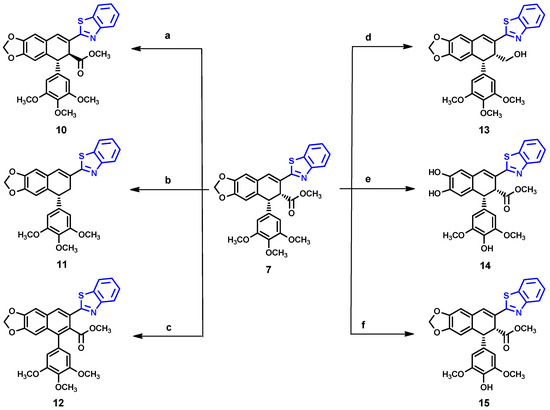
Scheme 2.
Reagents and conditions: a = Hydrazine, MeOH 50 °C, 2 h; b = 5% KOH/MeOH, 3 d, neut. 2N HCl; c = 1. KOH/MeOH, rt, 1 h, 2. (COCl)2/DMF-DCM 0–10 °C, 3. Hydrazine-Acetic Acid/THF, reflux 2.5 h; d = DIBAL-H/DCM, −78 °C, 15 min; e = Methionine, MsOH, 40 °C, 45 min; f = TMSI, 0°, 24 h).
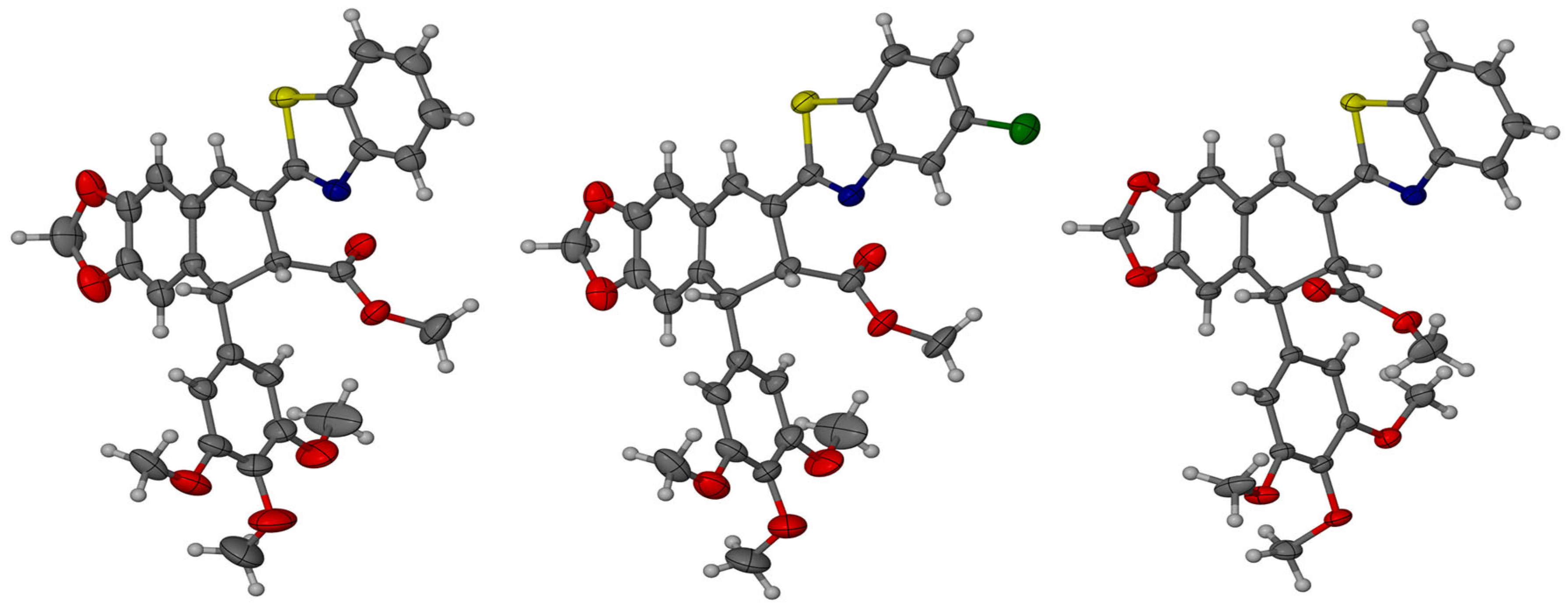
The absolute configurations of compounds 7, 9, and 10 were assigned by X-ray diffraction analysis. Compound 7 and its derivative 9 crystallize in the orthorhombic P212121 space group with Z = 8 molecules in the unit cell, whereas crystals of 10 crystallize in the monoclinic P21 space group with Z = 4. Although crystals are built of the single diastereoisomer, which is consistent with the non-centrosymmetric space group, the independent part of the unit cell is composed of two molecules (Z’ = 2) in each crystal of the compounds 7, 9, 10. Two molecules differ in geometrical parameters: bond lengths and angles. The most spectacular difference is, however, the difference in the conformation of the methoxy group of the phenyl ring. In general, molecules in the crystal structure are connected by a series of weak CH···O and CH···N hydrogen bonds, which are probably responsible for the conformational richness of the molecules in the solid state.
In all cases, the data were collected with full Friedel pair coverage, ensuring sufficient sensitivity to anomalous scattering effects to allow reliable determination of the absolute configuration. The resulting Flack parameter values, which are close to zero, confirm the accurate assignment of the enantiomeric form in each case and the high reliability of the absolute structure determination. The compound configuration was determined as (7′R,8′R) for compounds 7 and 9, and (7′R,8′S) for compound 10, as shown in Figure 2.
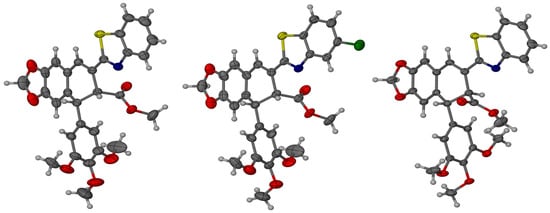
Figure 2.
Molecular models with thermal ellipsoids showing the absolute stereochemistry of compounds 7, 9, and 10. The ellipsoids are drawn at the 30% probability level.
2.2. Biological Evaluation
2.2.1. The In Vitro Cytotoxicity Assay
The cytotoxicity properties of semi-synthesized compounds 7–15 was evaluated against five types of cancer cell lines including, human: HeLa (cervix cancer), LoVo (colorectal cancer), MCF-7 (breast cancer), SKOV-3 (ovarian cancer), and murine: B16F10 (malignant melanoma). Thus, various human tumor cell lines representing major types of malignant cancers were included in the study protocol. Non-tumorigenic HaCaT cells were used as a representative model of normal (non-cancerous) cells. The crystal violet assay and PrestoBlue assay were used to evaluate the cytotoxic/cytostatic effect. All compounds showed diverse cytotoxicity activity against all the tested cell lines, as shown in Table 1. We also included the cytotoxicity data of podophyllotoxin and etoposide as reference values. As expected, compound 7 showed a significant growth inhibition to all tested cancer cell lines; however, it was also significantly toxic to HaCaT cells. Compound 8 with an additional CF3 group on the benzothiazole ring was inactive on all tested cell lines, while compound 9 with a Cl-substituent was selective to HeLa and MCF-7 with IC50 1.81–2.02 µM, and had moderate activity on SKOV-3 and LOVO. In addition, compound 9 showed higher inhibition to cell lines derived from cancer than to the non-tumorigenic one. In contrast, compound 10 showed no cytotoxicity on all tested cell lines, which is consistent with the literature data on similar compounds that were found to be inactive on HepG2 (human liver cancer cell) [26], A549 (lung cancer), HT-29 (colorectal cancer), and Mel-28 (melanoma) [27]. Interestingly, compound 11 revealed a strong cytotoxic/cytostatic effect against all tested cell lines with IC50 1.54–2.88 µM, despite the absence of the ester group. Moreover, the IC50 value of this compound showed no significant difference with the parent compound 7. On the other hand, compounds 12 and 13 showed no effect on cancer cells nor non-cancer cells, indicating that the presence of the methyl ester group and the free-rotating three-methoxy phenyl ring are important. Furthermore, compound 14, lacking the dioxolane ring, showed low activity on LOVO, MCF-7, SKOV-3, and HaCaT cells. Lastly, compound 15 was found to exhibit moderate cytotoxicity on all the cell lines tested, except for LoVo cells with an IC50 in range 2.49–3.76 µM, and lower activity to HaCaT. Despite this, the demethylation product 15 displayed inferior activity compared to its parent compound, but still shows potential for future studies.

Table 1.
The in vitro cytotoxicity assay of compounds 7–15.
Additionally, the selectivity index (SI) was calculated as the ratio of the IC50 values for HaCaT and HeLa cells (SI = IC50HaCaT/IC50HeLa), where a higher SI value indicates the greater selectivity of the compound towards cancer cells. In general, all active compounds showed selectivity against HeLa cells, except for compound 12. Compounds 9 and 15 demonstrated the highest selectivity, with SI values of 7.31 and 3.05, respectively. Although compounds 5 and 11 showed lower selectivity, their SI values were still above 1.00, and they are therefore still considered to be potent anticancer agents [28].
2.2.2. Cell Cycle Analysis
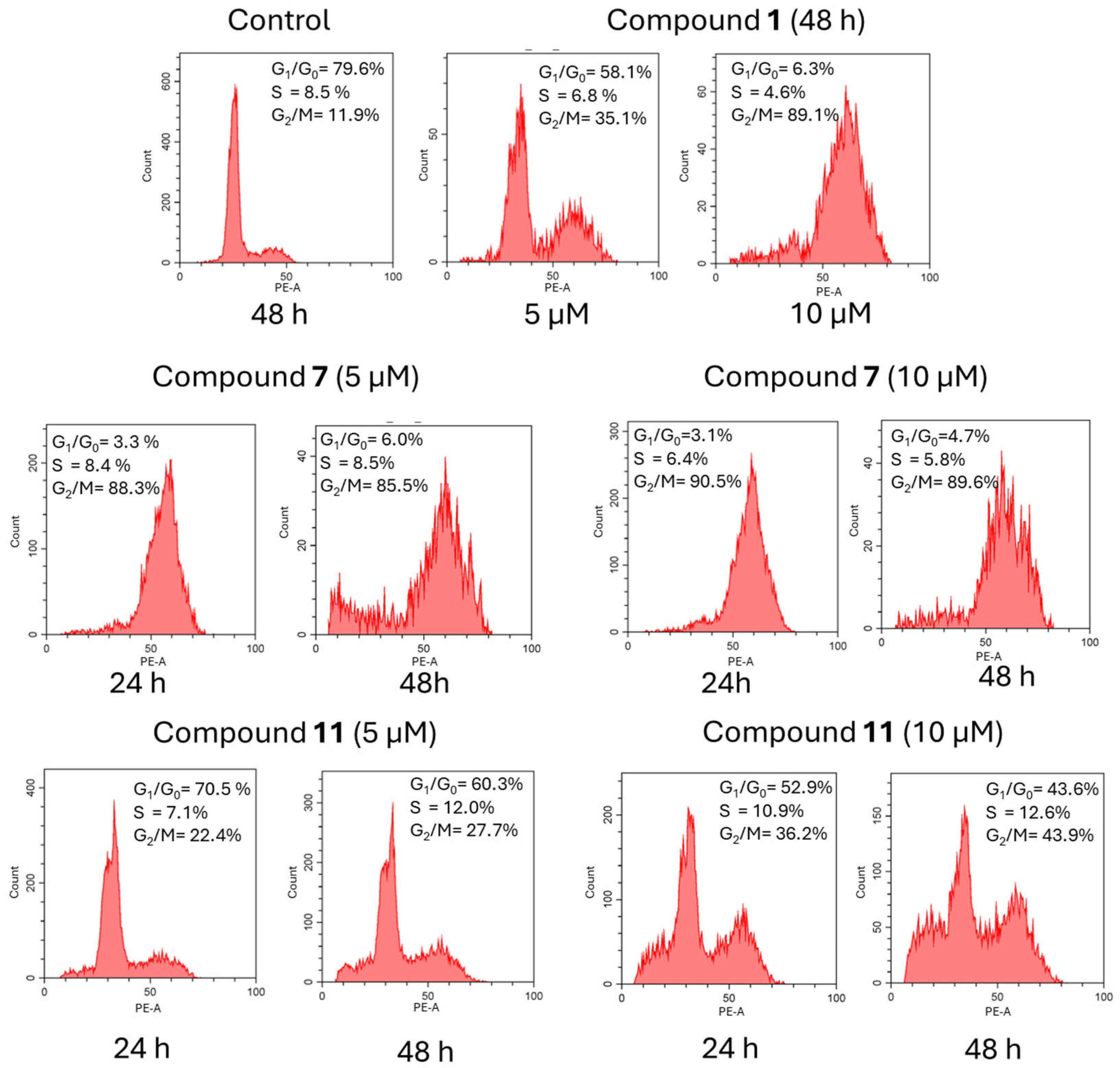
To determine the possible mechanisms of compounds 7 and 11 in inhibiting the proliferation of cancer cells, cell cycle analysis was performed with the flow cytometry method, as shown in Figure 3. Compound 7 was previously reported to inhibit proliferation of HaCaT cells via cell cycle arrest in G2/M [18]. As a reference, HeLa cells were treated with the indicated concentrations of compounds 7 and 11 for 24 h and 48 h, and the cell cycle phases distribution of treated cells was determined with the propidium iodide (PI) method, as previously described [18]. The effects of novel podophyllotoxin derivatives on cell cycle phase distribution were evaluated in cells exposed to these compounds for 24 h or 48 h. Three compounds were compared: podophyllotoxin 1, KL-3 derivative 7, and its analog lacking the ester group 11. Each compound induced cell cycle arrest in the G2/M phase. Compounds 7 and 1 were the most potent, with approximately 90% of the cells arrested in the G2/M phase. In contrast, compound 11, at a low concentration of 5 µM, induced only a minor block in the G2/M phase. However, at higher concentrations and after 48 h of exposure, the percentage of cells arrested in the G2/M phase increased to approximately 43%. These findings suggest that the structure and concentration of podophyllotoxin derivatives significantly influence their ability to block the G2/M transition in the cell cycle.
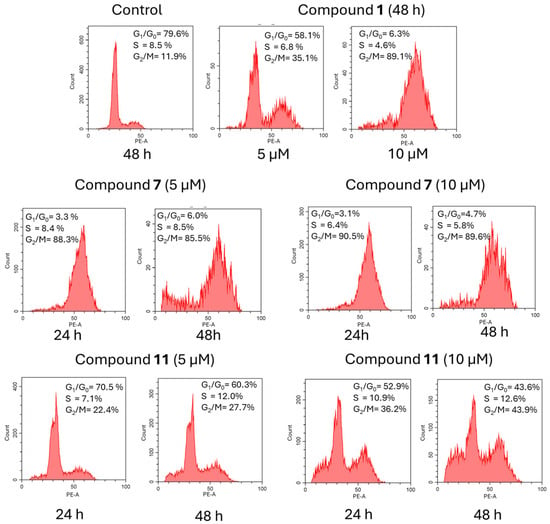
Figure 3.
Cell cycle distribution for compounds 1, 7, and 11.
2.3. Molecular Docking Studies
Podophyllotoxin is known as a tubulin polymerization inhibitor for disrupting microtubule formation, which leads to cell cycle arrest in mitosis [29,30]. While podophyllotoxin binds to tubulin at the same site as colchicine, the trimethoxyphenyl group binds to the active site of tubulin via hydrophobic interactions. Therefore, to predict the binding interaction between the synthetized compounds 7, 11, and 15 with β-tubulin (PDB: 1SA1), we performed a molecular docking analysis via SwissDock with AutoDock Vina 1.2.5 algorithm. SwissDock is a freely accessible web tool for small molecules, allowing docking with automatized ligand preparation and targeting. Moreover, this program exhibits a good accuracy of docking predictions with rapid execution times [31,32].
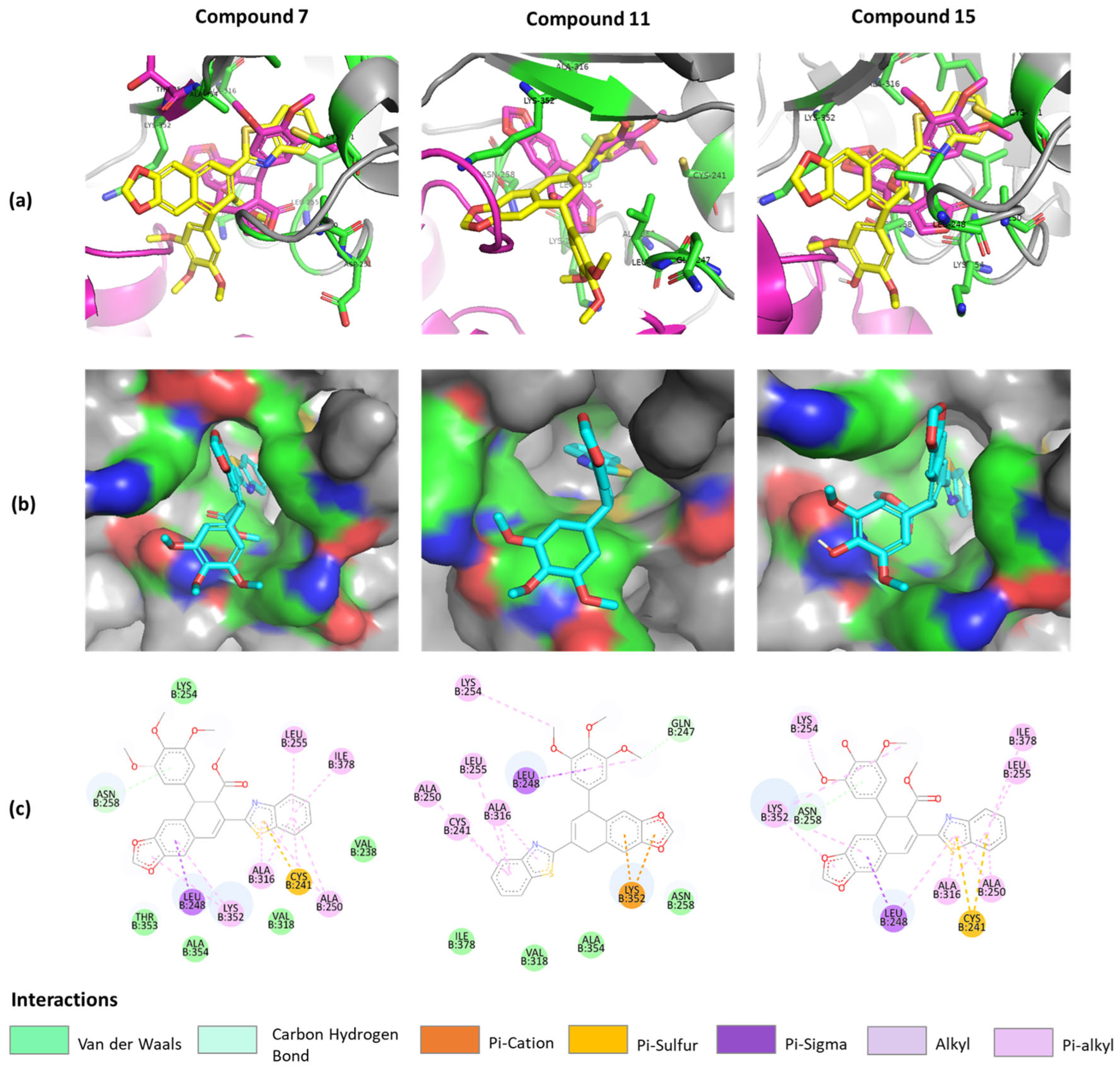
As a result of using this program, we found that compounds 7, 11, and 15 are subject to conformation change at the podophyllotoxin binding site, with a binding affinity −7.795 kcal/mol, −7.227 kcal/mol, and −7.965 kcal/mol, respectively, compared to the podophyllotoxin binding affinity equal to −7.682 kcal/mol. Interestingly, all docked benzothiazole groups (Figure 4a) were overlapping with the 3′,4′,5′-trimethoxyphenyl fragment of podophyllotoxin, having similar hydrophobic interactions with ALA316, CYS241, LEU255, and ALA250 [14]. The surface mode (Figure 4b) displayed that benzothiazole groups were located inside the binding pocket of β-tubulin, proving the crucial role of this structure motif responsible for potent inhibition [33,34].
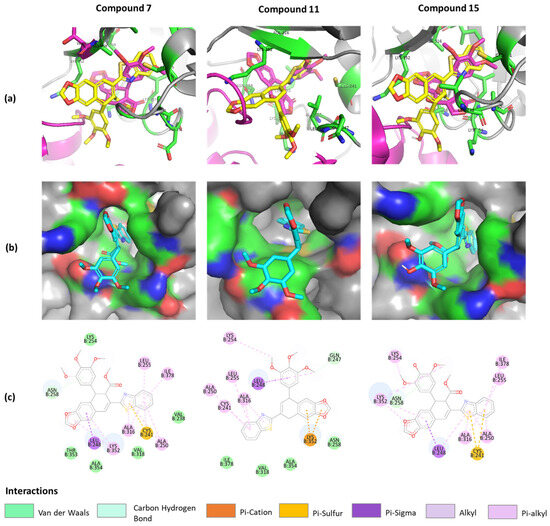
Figure 4.
Predicted binding model of compounds 7, 11, and 15 in tubulin (PDB ID: 1SA1). (a) Merged compounds (yellow) and podophyllotoxin (magenta) models at the binding site. (b) Surface version of compounds. (c) The 2D interactions of compounds with protein residues.
Despite the prediction that the unsubstituted benzothiazole moiety plays an important role in inhibiting cancer cells, other structural elements such as a dioxolane ring, free-rotating 3′,4′,5′-trimethoxyphenyl group, and cis-methyl ester group also contribute to the binding mode. Therefore, careful and thorough analysis of the molecular mechanism of these potent compounds (7, 11, and 15) should be conducted.
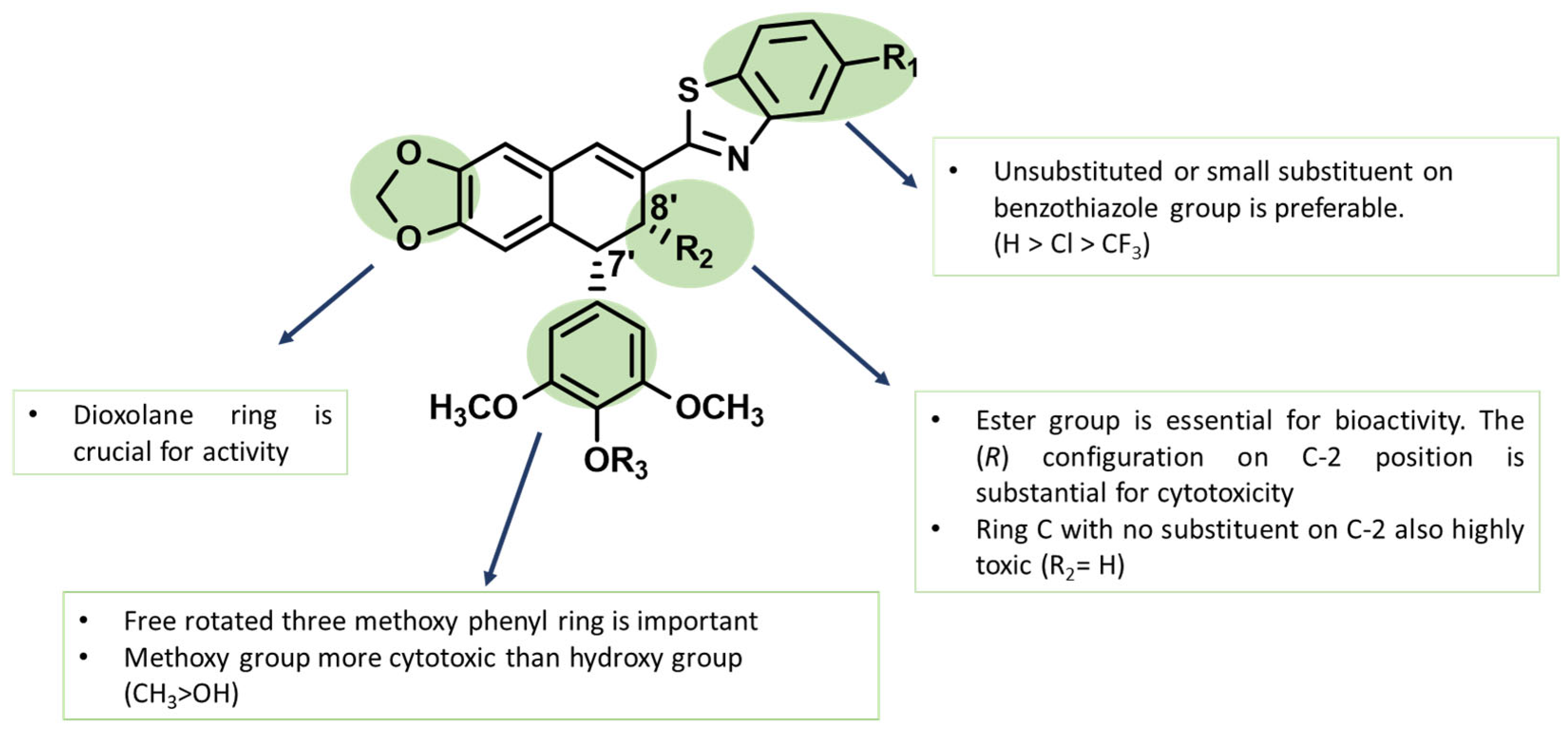
2.4. The Structure–Activity Relationships
Using compound 7, we analyzed the influence of substituents at the benzothiazole ring, the modification in ring A and ring E, and the presence of different functional groups at the C-8′ position. The structure–activity relationships (SARs) are presented in Figure 5. The results reveal that the presence of larger substituents at the benzothiazole ring leads to lower cytotoxicity (–H > –Cl > –CF3); therefore, it is clear that the unsubstituted benzothiazole ring is preferred for the desired bioactivity. In addition, the stereochemistry of the ester group has a crucial effect on its ability to inhibit cancer cells proliferation as compound 10 with S-configuration at C-8′ atom showed no effect on any of the tested cell lines. Compound 11 presents an interesting case because despite lacking cis-stereochemistry at the C ring, it still demonstrates significant anticancer activity. The fact that compound 13 shows no activity against cancer cells suggests that the hydroxymethyl group likely has insufficient interaction with the binding pocket. The aromatization of ring C in compound 12 resulted in the loss the cytotoxic activity to all tested cells. Finally, compound 15 better and broader antiproliferation activity than 14, which proves that the dioxolane ring is important for anticancer activity.
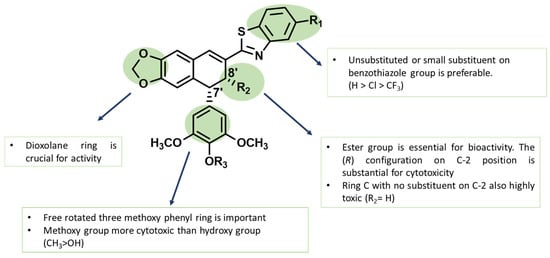
Figure 5.
The structure–activity relationships (SARs) of novel podophyllotoxin–benzothiazole derivatives.
2.5. Prediction of ADMET Parameter with SwissADME
To predict the pharmacokinetic properties and drug-likeness of the novel compounds, the ADME parameter (for absorption, distribution, metabolism, and excretion) was analyzed via the SwissADME web tool (http://www.swissadme.ch/, accessed on 9 April 2025) [35]. The physicochemical properties of prospective compounds 7, 11, and 15 (Table 2) suggest their promising drug-likeness, particularly for compound 11 with zero violations of Lipinski’s rule. In addition, compound 11 is predicted to have high GI absorption, indicating a high probability of passive absorption through the gastrointestinal tract [35,36]. However, experimental studies are required to confirm these predictions.
Table 2.
List of ADME properties, violation, and drug-likeness of selected compounds.
3. Materials and Methods
3.1. Chemistry
Podophyllotoxin (1) was obtained from (Sigma-Aldrich, St. Louis, MO, USA). Other chemicals were also obtained from Sigma-Aldrich. Etoposide-Ebewe (Ebewe Pharma GmbH, Unterach am Attersee, Austria), a ready-to-use infusion solution at a concentration of 20 mg/mL, was used in the study. The 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE apparatus, operating at 500 MHz and 300 MHz (1HNMR), and 125 MHz and 75 MHz (13C NMR), using tetramethylsilane (TMS) as an internal standard, with chemical shifts given in ppm. High-resolution mass spectra were recorded on Quatro LC AMD 604 apparatus using TOF MS ES + method. Melting points were measured on a micro melting point Fisher-John apparatus without correction. Optical rotations were measured with an Autopol IV Rudolph Research Analytical apparatus. X-ray crystallographic data were collected on Bruker ApexII Ultra. Column chromatography was carried out on silica gel SilicaFlash P60 230–400 mesh ASTM (Dichrom GmbH, Haltern am See, Germany), with the indicated eluents. Thin-layer chromatography (TLC) analysis used precoated Si-gel plates (Merck Kieselgel 60 GF254, 0.25 mm) (art. 5554). Anhydrous sodium sulfate was used as a drying agent. The organic solvents (MeOH, n-hexane, CHCl3, CH2Cl2, EtOAc) used in column chromatography were of the standard grade for purification.
3.1.1. General Method of Synthesis
Compounds 4–6 were synthesized in accordance with the procedure previously described [15]. In brief, podophyllotoxin 1 was subjected to oxidation, followed by hydrolysis to form compound 4. The reduction of the ketone group and subsequent Swern oxidation were carried out to afford compound 6.
Compound 4
For synthesis of compound 4, podophyllotoxin 1 (2.05 g, 4.94 mmol, 1 eq) was dissolved in 40 mL of dichloromethane and placed on the ice bath. PDC (2.09 g, 5.55 mmol, 1.1 eq) was added and the mixture was stirred at 0 °C for 3 h. The reaction mixture was quenched with distilled water and extracted with dichloromethane. The organic extracts were collected, dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude product was purified with column chromatography using n-hexane-EtOAc (7:3) as the eluent. The NMR of the product was identical to the literature4. The resulting ketone (1.6 g, 3.9 mmol, 1 eq) was dissolved in MeOH and placed in an ice bath. Concentrated H2SO4 (15.8 mmol, 4 eq) was added dropwise and then the reaction mixture was stirred under reflux for 1 h. The solvent was evaporated and quenched with sodium bicarbonate, followed by extraction with EtOAc. The organic layer was dried over anhydrous Na2SO4 and evaporated. The crude product was purified by column chromatography to afford compound 4 with 75% yield. Prolongation of the reaction time for more than 2 h caused the formation of compound 5 as the main product. Compound 4 was isolated as white amorphous solid. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.53 (s, 1H), 6.58 (s, 1H), 6.11 (s, 2H), 6.04 (dd, J = 1.2, 4.2 Hz, 2H), 4.59 (d, J = 5.4 Hz, 1H), 4.23 (ddd, J = 3.0, 6.6, 10.8 Hz, 1H), 3.80 (s, 3H), 3.73 (s, 3H), 3.73 (s, 3H), 3.66 (s, 3H), 3.63 (dd, J = 6.0, 12.0 Hz, 1H), 3.06 (ddd, J = 3.0, 4.8, 12.9 Hz, 1H), 2.55 (t, J = 6.6 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 197.8, 171.6, 153.4, 153.4, 148.2, 140.9, 137.7, 134.2, 127.2, 108.6, 106.2, 106.0, 102.3, 61.9, 61.0, 56.3, 52.0, 47.3, 46.7, 45.4.
Compound 5 was isolated as white powder. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.54 (s, 1H), 6.59 (s, 1H), 6.09 (s, 1H), 6.02 (dd, J = 1.2, 4.2 Hz, 2H), 4.58 (d, J = 5.1 Hz, 1H), 4.22 (dd, J = 2.4, 9.0 Hz, 1H), 3.80 (m, 1H), 3.79 (s, 3H), 3.72 (s, 6H), 3.68 (s, 3H), 3.54 (dd, J = 3.3, 9.3 Hz, 1H), 3.25 (s, 3H), 2.96 (dt, J = 3.0, 12 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 195.5, 171.9, 153.3, 153.0, 148.1, 140.3, 137.5, 134.4, 127.7, 108.4, 106.4, 106.0, 102.1, 70.4, 61.0, 56.5, 56.2, 51.9, 47.2, 46.1, 44.4. MS (EI) (m/z) for C24H26O9 [M + Na]+ = calc. 481.14735 found 481.14690.
Compound 6
For the synthesis of compound 6, initially, compound 4 was reacted with sodium borohydride, which afforded a 1,3-diol, and the NMR-data were in accordance with those previously described [15]. Next, the 1,3-diol was oxidized via Swern oxidation using oxalyl chloride, DMSO, and triethylamin. Compound 6 was purified by column chromatography with DCM-EtOAc (9:1–7:3) as the eluent and collected as yellow powder in 64% yield. The NMR data were identical to the literature [15]. 1H NMR (300 MHz, CDCl3): δ (ppm) = 9.60 (s, 1H), 7.36 (s, 1H), 7.27 (s, 2H), 6.89 (s, 1H), 6.62 (s, 1H), 6.46 (s, 1H), 6.00 (dd, J = 1.2, 4.6 Hz, 1H), 4.41 (dd, J = 0.9, 8.1 Hz, 1H), 3.98 (d, J = 7.8 Hz, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.81 (s, 3H), 3.40 (s, 3H).
General Procedure for the Synthesis of Compounds 7–9
Compounds 7–9 were prepared in accordance with the procedure previously described [17,18]. To a round-bottomed flask, compound 4 (0.25 mmol, 1 eq), corresponding 2-aminothiophenol (0.28 mmol, 1.2 eq), Na2S2O5 (0.28 mmol, 1.2 eq), and 2.5 mL DMSO were added. The mixture was stirred at 120 °C for 1.5–2 h under argon atmosphere. The mixture was then cooled to room temperature and water was added, causing the formation of yellow precipitate. The precipitate was collected by filtration and dried, followed by purification using column chromatography with n-hexane-EtOAc (8:2) as eluent. The recrystallization of 7 and 9 was carried out with anti-solvent crystallization method, using chloroform with additional n-hexane.
Compound 7 was obtained as light green needles at 67% yield. m.p.: 168–169 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.95 (ddd, J = 0.6, 1.3, 8.0 Hz, 1H), 7.84 (ddd, J = 0.6, 1.4, 7.9 Hz, 1H), 7.44 (ddd, J = 1.5, 7.0, 8.3 Hz, 2H), 7.35 (ddd, J = 1.0, 6.9, 8.3 Hz, 1H), 6.87 (s, 1H), 6.63 (s, 1H), 6.56 (s, 2H), 5.98 (dd, J = 1.5, 6.6 Hz, 1H), 4.61 (dd, J = 1.2, 7.5 Hz, 1H), 4.43 (d, J = 7.5 Hz, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.84 (s, 3H), 3.43 (s, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 171.8, 167.3, 153.9, 153.5, 153.0, 148.7, 146.7, 137.5, 135.0, 134.6, 133.6, 131.8, 128.7, 127.1, 126.3, 125.5, 123.3, 121.6, 121.5, 109.0, 108.9, 107.7, 106.7, 101.5, 61.1, 56.3, 51.9, 49.2, 48.7. MS (EI) (m/z) for C29H25NO7S [M + H]+ = calc. 532.14232, found 532.14245.
Compound 8 was obtained as a light green powder. Yield 63%.1H NMR (300 MHz, CDCl3): δ (ppm) = 8.21 (t, J = 0.9 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.58 (dt, J = 0.6, 8.4 Hz, 1H), 7.45 (s, 1H), 6.88 (s, 1H), 6.65 (s, 1H), 6.55 (s, 2H), 5.99 (dd, J = 1.5, 6.3 Hz, 1H), 4.62 (dd, J = 0.9, 7.5 Hz, 1H), 4.42 (d, J = 7.8 Hz, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.84 (s, 3H), 3.43 (s, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 171.6, 169.2, 153.4, 153.4, 149.0, 146.6, 134.7, 134.7, 132.0, 128.1, 126.7, 122.2, 108.9, 108.9, 106.6, 101.5, 60.9, 56.2, 51.8, 49.0, 48.5. MS (EI) (m/z) for C30H24NO7F3S [M + H+] = calc. 600.12983, found 600.13097.
Compound 9 was obtained as light yellow needles. Yield 72%. m.p.: 182–183 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.92 (dd, J = 0.6, 2.1 Hz, 1H), 7.74 (dd, J = 0.6, 8.7 Hz, 1H), 7.42 (s, 1H), 7.32 (dd, J = 1.8, 8.4 Hz, 1H), 6.86 (s, 1H), 6.63 (s, 1H), 6.55 (s, 2H), 5.98 (dd, J = 0.9, 6.3 Hz, 2H), 4.60 (dd, J = 0.9, 7.5 Hz, 1H), 4.39 (d, J = 7.5 Hz, 1H), 3.89 (s, 3H), 3.84 (s, 6H), 3.43 (s, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 171.8, 154.8, 153.5, 153.5, 148.9, 146.7, 137.6, 134.9, 134.3, 132.8, 132.3, 132.0, 128.4, 126.9, 125.9, 123.0, 122.2, 109.0, 109.0, 106.7, 106.7, 101.6, 61.1, 56.3, 56.3, 51.9, 49.2, 48.6. MS (EI) (m/z) for C29H24NO7SCl [M + H+] = calc. 566.10292, found 566.10179.
Compound 10
To obtain compound 10, compound 7 (30 mg, 0.056 mmol, 1 eq) and hydrazine hydrate (0.428 mmol, 6 eq) were refluxed in 2-propanol (3 mL) for 2 h. The product was purified by preparative TLC (thin-layer chromatography) with CHCl3-MeOH (97:3) as eluent. The product was collected as colorless needles with 78% yield. The crystallization was performed from chloroform with additional n-hexane. M.p.: 198–200 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.92 (dd, J = 0.6, 8,0 Hz, 1H), 7.82 (dd, J = 0.6, 7.4 Hz, 1H), 7.42 (td, J = 1.2, 7.2 Hz, 1H), 7.40 (s, 1H), 7.33 (td, J = 1.5, 7.0 Hz, 1H), 6.86 (s, 1H), 6.73 (s, 1H), 6.38 (s, 2H), 5.99 (dd, J = 1.2, 3.9 Hz, 2H), 4.67 (d, J = 2.1 Hz, 1H), 4.61 (d, J = 3.9 Hz, 1H), 3.75 (s, 3H), 3.71 (s, 3H), 3.71 (s, 3H), 3.63 (s, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 172.6, 168.0, 153.8, 153.2, 153.2, 148.8, 147.3, 137.9, 137.0, 134.5, 132.0, 131.4, 127.5, 126.3, 126.2, 125.4, 123.6, 123.1, 121.5, 110.0, 108.3, 104.7, 104.7, 101.6, 60.8, 56.2, 52.7, 48.5, 46.7. MS (EI) (m/z) for C29H25NO7S [M + H]+ = calc. 532.13517, found 532.14245.
Compound 11
For the synthesis of compound 11, compound 7 (50 mg, 0.09 mmol) was dissolved in 5% KOH solution in methanol and stirred at room temperature for 3 days. The reaction mixture was then neutralized with 2 M HCl aq, methanol (25 mL) was added, and the mixture was evaporated to dryness. The residue was extracted with chloroform. The organic phase was dried with anhydrous MgSO4 and the solvent was evaporated. The product was purified with column chromatography using n-hexane and ethyl acetate at a ratio of 4:1 as eluent. Product 11 was obtained as a yellow solid (31.5 mg, 67% yield). 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.95 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.44 (t, J = 8.3 Hz, 1H), 7.35 (t, J = 7.7 Hz, 1H), 7.32 (s, 1H), 6.82 (s, 1H), 6.51 (s, 2H), 6.48 (s, 1H), 5.96 (s, 1H), 5.95 (s, 1H), 4.15 (q, J = 6.4 Hz, 1H), 3.82 (s, 3H), 3.81 (s, 6H), 3.39 (dd, J = 7.8 Hz, 1H), 3.18 (dd, J = 8.6 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 168.7, 154.0, 153.4, 148.4, 146.7, 139.2, 136.9, 134.5, 134.5, 130.8, 130.5, 127.4, 126.4, 125.3, 12.3, 123.0, 121.6, 109.0, 108.1, 105.6, 101.4, 61.0, 56.2, 45.1, 33.3. 13C NMR (75 MHz, CDCl3, DEPT): δ (ppm) = 101.3, 33.2. MS (EI) (m/z) for C27H23NO5S [M + H]+ = calc. 474.13697, found 474.13788.
Compound 12
To the solution of compound 7 (66.4 mg, 0.125 mmol, 1 eq) in methanol (25 mL), 3 mL of 5% KOH in MeOH solution was added and the mixture was stirred at room temperature for 2 h to form a light green precipitation. The solvent was evaporated into dryness under reduced pressure. DCM (1 mL) was added and the reaction mixture was placed in an ice bath, followed by the addition of oxalyl chloride (0.13 mmol) and DMF (1 µL) as a catalyst. The mixture was stirred for 2 h at 0–10 °C and then concentrated into dryness, followed by the addition of NH2NH2· H2O (0.25 mmol, 2 eq), acetic acid (1 µL), and THF (2 mL) and refluxing for 2.5 h. Water (5 mL) was added and the mixture was neutralized with HCl 2N. The mixture was extracted with EtOAc and the organic phase was dried over anhydrous Na2SO4. The product was purified by column chromatography or preparative TLC with CHCl3-MeOH (98:2) as eluent. Product 12 was collected as white powder with 34% yield. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.17 (s, 1H), 8.00 (ddd, J = 0.6, 1.2, 8.0 Hz, 1H), 7.92 (ddd, J = 0.6, 1.35, 7.9 Hz, 1H), 7.48 (td, J = 1.2, 7.7 Hz, 1H), 7.39 (td, J = 1.2, 7.2 Hz, 1H), 7.20 (s, 1H), 6.95 (s, 1H), 6.62 (s, 2H), 6.08 (s, 2H), 3.95 (s, 3H), 3.90 (s, 6H), 3.64 (s, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) = 169.7, 166.2, 154.2, 153.1, 149.8, 149.1, 138.3, 137.9, 135.4, 132.8, 130.9, 130.7, 129.2, 128.7, 126.8, 126.5, 125.6, 123.7, 121.7, 107.7, 107.8, 104.6, 103.7, 101.9, 61.2, 56.4, 52.4. MS (EI) (m/z) for C29H23NO7S [M + Na]+ = calc. 552.10927, found 552.10874.
Compound 13
To synthesize compound 13, compound 7 (0.06 mmol, 1 eq) was dissolved in dichloromethane (2 mL) and placed in a dry ice bath, followed by dropwise addition of DIBAL-H solution 1.0 M in dichloromethane (0.18 mmol, 3 eq). The reaction was carried out for 30 min under argon atmosphere, maintaining the temperature from −78 °C to −55 °C. Then, MeOH (3 mL) and Rochelle salt solution (10 mL) were added. The mixture was stirred for 1 h and then extracted with ethyl acetate and the organic phase was dried over anhydrous Na2SO4 and evaporated. The crude product was initially purified using column chromatography with CHCl3-MeOH (99:1) and then by preparative TLC in the same system. Product 13 was isolated as a yellowish powder with 8% yield. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.91 (ddd, J = 0.6, 1.2, 8.1 Hz, 1H), 7.86 (ddd, J = 0.6, 1.5, 8.1 Hz, 1H), 7.45 (td, J = 1.5, 7.2 Hz, 1H), 7.39 (dd, J = 1.5, 8.1 Hz, 1H), 7.35 (s, 1H), 6.82 (s, 1H), 6.73 (s, 1H), 6.61(s, 2H), 5.98 (d, J = 6.0 Hz, 1H), 4.46 (d, J = 6.6 Hz, 1H), 3.97 (d, J = 1.8 Hz, 2H), 3.87 (s, 3H), 3.85 (s, 6H), 3.61 (q, J = 3.9 Hz, 1H). MS (EI) (m/z) for C28H25NO6S [M + H]+ = calc. 504.14745, found 504.14754.
Compound 14
For the synthesis of compound 14, compound 7 (48 mg, 0.09 mmol), methionine (56 mg, 0.4 mmol), and methanesulfonic acid (2 mL, 30 mmol) were stirred at 40◦C for 45 min. Then, the reaction mixture was neutralized with K2CO3 solution to pH 6 and extracted twice with ethyl acetate. The organic extracts were dried using anhydrous MgSO4, filtered, and evaporated. The product was purified with column chromatography with n-hexane–ethyl acetate (3:2) as eluent. Compound 14 was obtained at 11% yield. 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.97 (d, J = 10.0 Hz, 1H), 7.85 (d, J = 10.0 Hz, 1H), 7.45 (t, J = 7.5 Hz, 2H), 7.36 (t, J = 10.0 Hz, 1H), 6.83 (s, 1H), 6.62 (s, 1H), 6.55 (s, 2H), 4.58 (d, J = 10.0 Hz, 1H), 4.39 (d, J = 5.0 Hz, 2H), 3.86 (s, 6H), 3.44 (s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) = 172.6, 167.7, 153.6, 147.1, 145.3, 142.4, 134.3, 134.1, 133.6, 130.3, 130.2, 127.7, 126.3, 125.8, 125.3, 122.9, 121.4, 116.0, 115.1, 106.2,, 56.3, 56.3, 51.9, 48.9, 48.4. MS (EI) (m/z) for C27H23NO7S [M + H]+ = calc. 506.12680, found 506.12611.
Compound 15
To obtained compound 15, compound 7 (35 mg, 0.06 mmol) was dissolved in DCM (1 mL); then, TMSI (24 μL, 0.21 mmol) was added. The reaction was stirred at −4 °C for 24 h. For work up, water was added and the reaction mixture was extracted three times with DCM. The organic extracts were dried over anhydrous MgSO4 and evaporated. Then, column chromatography was performed with the ethyl acetate–hexane (1:1) system and preparative chromatography in the same system. The pure product in yellow solid form was obtained in 44% yield. 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.96 (d, J = 9.0 Hz, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.45 (d, J = 1.0 Hz, 2H), 7.43 (s, 1H), 7.35 (t, J = 8.3 Hz, 1H), 6.87 (s, 1H), 6.61 (s, 1H), 6.56 (s, 2H), 5.98 (d, J = 4.5 Hz, 1H), 5.96 (d, J = 1.5 Hz, 1H), 5.53 (s, 1H), 4.59 (d, J = 8.5 Hz, 1H), 4.42 (d, J = 7.5 Hz, 1H), 3.87 (s, 6H), 3.43 (s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) = 172.6, 167.3, 154.1, 148.7, 147.4, 146.7, 134.7, 133.5, 132.3, 130.4, 128.9, 127.7, 126.3, 125.4, 123.3, 121.6, 108.9, 108.8, 106.9, 105.0, 101.4, 56.6, 51.7, 49.1, 48.9 ppm. MS (EI) (m/z) for C28H23NO7S [M + H]+ = calc. 518.12680, found 518.12670.
3.1.2. X-Ray Crystallography Analysis of 7, 9 and 10
Crystals of 7, 9, and 10 were examined and selected under a microscope in polarized light. Selected crystals were mounted on a κ-goniometer of an Oxford Xcalibur R diffractometer equipped with a CCD detector, and their diffraction was examined for quality control. The diffracted reflection intensities for the X-ray wavelength of 1.54184 Å were measured separately for each individually aligned single crystal. Full-matrix least-squares refinement on F2 was performed, and absorption corrections were applied using the analytical method [37]. The dual methods from SHELXT [38] were used to solve the structures from the diffraction data. The structure model was then refined using SHELXL-97 [39].
All hydrogen atoms, except the hydrogen atoms in some of the methyl groups, were located from a differential density map. The coordinates of the hydrogen atoms attached to the carbon atoms were treated as fixed contributors using standard geometric criteria of the idealized geometry. X-Seed [40] and Shelxle [41] were used as GUIs. They supported the refinement and visualization of the structure. X-Seed was also used to create artwork [42] in POV-RAY [43]. The final crystal and structural data for compounds 7, 9, and 10 are summarized in CIF format and deposited at the Cambridge Crystallographic Data Centre. All files are available free of charge from the Cambridge Structural Database at https://www.ccdc.cam.ac.uk/structures/ (CCDC 2449015–2449017)—URL accessed on 9 April 2025.
Compound 7 was crystallized by dissolving it in warm chloroform, followed by the dropwise addition of warm n-hexane, allowing for slow crystallization. C29H25NO7S, Mr = 531.56, crystal size 0.459 × 0.102 × 0.050 mm3, orthorhombic, space group P212121, unit cell dimensions a = 11.4293(2) Å, b = 14.7160(3) Å, c = 32.0658(5)Å, V = 5393.26(17) Å3, Z = 8, Z’ = 2, α = β = γ = 90°, calculated density = 1.308 mg/cm3, F(000) = 2224, 121,168 reflections were collected, of which 11,541 are unique (Rint = 0.0528), final R indices (I > 2σI) R1 = 4.68%, wR2 = 12.33%, goodness of fit = 1.045, and Flack parameter = 0.004(5) confirm that the absolute configuration is correct.
Compound 9 was crystallized by dissolving it in warm chloroform, followed by the dropwise addition of warm n-hexane, allowing for slow crystallization. C29H24ClNO7S, Mr = 566.00, crystal size 0.664 × 0.531 × 0.025 mm3, orthorhombic, space group P212121, unit cell dimensions a = 11.4586(4) Å, b = 14.9377(4) Å, c = 32.3951(13) Å, V = 5544.9(3) Å3, Z = 8, Z’ = 2, α = β = γ = 90°, calculated density = 1.356 mg/cm3, F(000) = 2352, 129,676 reflections were collected, of which 9924 are unique (Rint = 0.1228), final R indices (I > 2σI) R1 = 9.1%, wR2 = 26.88%, goodness of fit = 1.048, and Flack parameter = −0.002(10) confirm the proper choice of absolute structure.
Compound 10 was crystallized by dissolving it in warm chloroform, followed by the dropwise addition of warm n-hexane, allowing for slow crystallization. C29H25NO7S, Mr = 531.56, crystal size 0.272 × 0.120 × 0.034 mm3, monoclinic, space group P21, unit cell dimensions a = 13.7756(4) Å, b = 13.1370(4) Å, c = 14.2759(4) Å, V = 2512.22(13) Å3, Z = 8, Z’ = 2, α = γ = 90° β = 103.492(3)°, calculated density = 1.405 mg/cm3, F(000) = 1112, 44,748 reflections were collected, of which 10,616 are unique (Rint = 0.0506), final R indices (I > 2σI) R1 = 4.58%, wR2 = 11.92%, goodness of fit = 1.029, and Flack parameter = 0.005(10) confirm that the absolute configuration is correct.
3.2. Biological Evaluation
3.2.1. Cell Culture
The synthesized compounds were tested and evaluated for their cytotoxicity using an in vitro cytotoxicity assay. We studied five cancer cell lines and one non-cancerous cell line. The cells were purchased from the American Type Culture Collection (ATCC), United States, including HeLa (cervical), LoVo (colorectal), MCF-7 (breast), SKOV3 (ovarian), B16F10 (murine melanoma), and HaCaT (human keratinocyte). Dulbecco’s modified Eagle’s medium (containing 10% FBS) was used to culture HeLa, MCF-7, B16F10, and HaCaT cells, while RPMI 1640 medium (containing 10% FBS) was used for SKOV3 and LoVo cells. The cells were cultured in 25 cm2 culture flasks (Corning, Corning, NY, USA) in a humidified atmosphere of 5% CO2 at 37 °C and passaged every three days with standard Trypsin-EDTA solution (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). All compounds were initially prepared as stock solutions at a concentration of 10 mM (in DMSO). Serial dilutions were performed in the indicated medium to achieve the required concentration gradient of compounds (20 µM, 10 µM, 5 µM, 2.5 µM, 1.25 µM, 0.625 µM, 0.312 µM). The maximum percentage of DMSO used in the compound solutions was 0.1%, based on the procedure previously described by our group [44].
3.2.2. The Crystal Violet Assay
This assay was performed in reference to the method already described [18]. In brief, the cell lines (HeLa, MCF-7, and SKOV-3) were seeded in the indicated medium in a 96-well culture plate (Nest Biotechnology, Wuxi, China) at a density of 1 × 104 cells per well (100 µL) in quadruplicates and then incubated at 37 °C in a 5% CO2 atmosphere. After 24 h of incubation, 100 µL aliquots of the compounds in medium at increasing concentrations were added to each well, and the cells were further incubated for 48 h. After the incubation period, the cells were washed with PBS twice, and then with 70% ethanol, followed by staining with 0.05% crystal violet solution in water (ChemPur, Piekary Slaskie, Poland) for 20 min. The excess crystal violet was washed out with distilled water several times. The stained cells then solubilized in 1% Triton™ X-100 Surfact-Amps (POCH SA, Gliwice, Poland) or 10% PBS. The absorbance of each well was measured using Fluostar plate reader at 550 nM.
3.2.3. The PrestoBlue Assay
The PrestoBlue assay was performed according to the method previously reported by Strus et al. [44]. The cells (LoVo and B16F10) were seeded in the indicated medium in a 96-well culture plate (Nest Biotechnology) at a density of 1 × 104 cells per well (100 µL) and incubated at 37 °C in a 5% CO2 atmosphere. After 24 h of incubation, 100 µL aliquots of the compounds in medium were added to each well, and the cells were further incubated for 48 h. After 48 h, the old medium was removed, and 100 µL of fresh medium was added. Then, 10 µL of PrestoBlue solution (Thermo Fisher Scientific, Waltham, MA, USA) was added to each well and incubated for 60 min. The fluorescence intensity was measured with excitation at 544 nm and emission at 615 nm using a Fluorstar microplate reader (BMG Labtech, Ortenberg, Germany). The results were expressed as a percentage of the intensity relative to that of control cells (non-treated cells).
3.2.4. Cell Cycle Analysis by Flow Cytometry
For cell cycle analysis, HeLa cells were treated for 24 or 48 h with compounds 7 and 11 at concentrations of 5 µM and 10 µM. After the incubation period, the cells were fixed in a freezing medium (ice-cold 70% ethanol solution in PBS) and stored at −20 °C. The cell suspension was then centrifuged, washed with PBS, and resuspended in PBS containing 200 µg of DNase-free RNase A (Sigma) for 20 min at room temperature. Propidium iodide (Sigma) was added to a final concentration of 5 µg/mL, and the cells were stained for at least 2 h at 4 °C. The analysis was performed using a FACSCalibur flow cytometer (Becton Dickinson, San Diego, CA, USA) and analyzed with CellQuest software, v.3.3.
3.2.5. Statistical Analysis
Data analysis was carried out using GraphPad Prism 5.0.3. Calculations were performed using MS-Excel version 2021. The cytotoxic effect was expressed as the relative viability of treated cells (percentage growth control) and was calculated as follows: % relative viability = , where Ae is experimental absorbance/fluorescence intensity, Ac is the absorbance/ fluorescence intensity of growth control (only cells with medium), and Ab is the absorbance/fluorescence intensity of a blank (only medium). The IC50 values were obtained by using the sigmoidal dose–response function in GraphPad Prism. The results were expressed as mean ± standard deviation (SD) [45].
3.3. In Silico Studies
The molecular docking analysis was conducted to predict and evaluate the binding interaction between compounds 7, 11, and 15 with tubulin. The crystal structure of tubulin (PDB ID: 1SA1) [46] was obtained from Protein Data Bank (https://www.rcsb.org/, accessed on 24 December 2024). The molecular docking calculations were performed with SwissDock via AutoDock Vina, a free web-based docking service which minimized the technical barrier to using docking software [31,32]. The ligands or compounds were inputted as SMILE notation, as previously converted by Chemical Sketch Tool (https://www.rcsb.org/chemical-sketch/, accessed on 24 December 2024), while the protein target was provided as the PDB ID. In this analysis, we specified the docking position to the podophyllotoxin binding site on β-tubulin. The docking results were then analyzed and visualized with PyMOL version 2.5.5 and Discovery Studio Visualizer 2021.
3.4. ADME Prediction
The SwissADME web tool (http://www.swissadme.ch/, accessed on 9 April 2025), a free website to predict the physicochemical properties of small molecules, was used to study the pharmacokinetics and drug-likeness of the most potent compounds [35]. The 2D chemical structures were input based on the molecular sketcher on ChemAxon’s Marvin JS or typing/pasting SMILE notation.
4. Conclusions
Based on previous results concerning the synthesis and determination of the favorable pharmacological properties of the benzothiazole derivative of podophyllotoxin (7), further derivatives were obtained and subjected to meticulous evaluation of their biological activity. It was confirmed that the presence of a dioxolane ring and free-rotating 3′,4′,5′-trimethoxyphenil group are important for the activity and also the stereochemistry of the methyl ester fragment with the R configuration, which alongside the unsubstituted benzothiazole moiety are substantial for cytotoxicity. Interestingly, compound 11, which did not have an ester substituent at C-8′, showed a strong antimitotic effect against cancer cells via G2/M cell cycle arrest. In addition, the ADMET prediction indicated its strong GI absorption and, taking into account the fact that Lipinski’s Rule of Five is fulfilled, it seems that good prospects are opening up for the construction of new effective anticancer derivatives.
Author Contributions
Conceptualization, I.M.-B. and Z.C.; investigation, P.N.R., Z.M. and A.O.; methodology, P.N.R., Z.M., I.M.-B. and Z.C.; writing—original draft preparation, P.N.R. and Z.M.; writing—review and editing, I.M.-B., Z.C., P.N.R. and Z.M.; X-ray analysis, A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the University of Warsaw in the form of grant BOB-661-1622/2024.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available on request due to restrictions, e.g., privacy or ethical considerations. The data presented in this study are available on request from the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Yu, X.; Che, Z.; Xu, H. Recent Advances in the Chemistry and Biology of Podophyllotoxins. Chem. Eur. J. 2017, 23, 4467–4526. [Google Scholar] [CrossRef] [PubMed]
- Prow, T.W.; Grice, J.E.; Lin, L.L.; Faye, R.; Butler, M.; Becker, W.; Wurm, E.M.T.; Yoong, C.; Robertson, T.A.; Soyer, H.P.; et al. Nanoparticles and Microparticles for Skin Drug Delivery. Adv. Drug Deliv. Rev. 2011, 63, 470–491. [Google Scholar] [CrossRef] [PubMed]
- Canel, C.; Moraes, R.M.; Dayan, F.E.; Ferreira, D. Podophyllotoxin. Phytochemistry 2000, 54, 115–120. [Google Scholar] [CrossRef]
- Motyka, S.; Jafernik, K.; Ekiert, H.; Sharifi-Rad, J.; Calina, D.; Al-Omari, B.; Szopa, A.; Cho, W.C. Podophyllotoxin and Its Derivatives: Potential Anticancer Agents of Natural Origin in Cancer Chemotherapy. Biomed. Pharmacother. 2023, 158, 114145. [Google Scholar] [CrossRef]
- Fan, H.; Zhu, Z.; Xian, H.; Wang, H.; Chen, B.; Tang, Y.-J.; Tang, Y.; Liang, X. Insight Into the Molecular Mechanism of Podophyllotoxin Derivatives as Anticancer Drugs. Front. Cell Dev. Biol. 2021, 9, 709075. [Google Scholar] [CrossRef]
- Strus, P.; Sadowski, K.; Ploch, W.; Jazdzewska, A.; Oknianska, P.; Raniszewska, O.; Mlynarczuk-Bialy, I. The Effects of Podophyllotoxin Derivatives on Noncancerous Diseases: A Systematic Review. Int. J. Mol. Sci. 2025, 26, 958. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, W.; Gao, P.; Guan, X.; Wang, B.; Zhang, L.; Yao, Y.; Guo, Y.; Wang, Y.; Jiang, S.; et al. Global, Regional, and National Burden of Breast, Cervical, Uterine, and Ovarian Cancer and Their Risk Factors among Women from 1990 to 2021, and Projections to 2050: Findings from the Global Burden of Disease Study 2021. BMC Cancer 2025, 25, 330. [Google Scholar] [CrossRef]
- Wunderlich, K.; Suppa, M.; Gandini, S.; Lipski, J.; White, J.M.; Del Marmol, V. Risk Factors and Innovations in Risk Assessment for Melanoma, Basal Cell Carcinoma, and Squamous Cell Carcinoma. Cancers 2024, 16, 1016. [Google Scholar] [CrossRef]
- Xiao, J.; Gao, M.; Sun, Z.; Diao, Q.; Wang, P.; Gao, F. Recent Advances of Podophyllotoxin/Epipodophyllotoxin Hybrids in Anticancer Activity, Mode of Action, and Structure-Activity Relationship: An Update (2010–2020). Eur. J. Med. Chem. 2020, 208, 112830. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, T.; Jin, X.; Lin, K.; Dai, X.; Gao, T.; Huang, G.; Fan, M.; Ma, L.; Liu, Z.; et al. Synthesis and Biological Evaluation of Cytotoxic Activity of Novel Podophyllotoxin Derivatives Incorporating Piperazinyl-Cinnamic Amide Moieties. Bioorganic Chem. 2022, 123, 105761. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.Á.; Miguel Del Corral, J.M.; García, P.A.; Rojo, M.V.; De La Iglesia-Vicente, J.; Mollinedo, F.; Cuevas, C.; San Feliciano, A. Synthesis and Biological Evaluation of New Podophyllic Aldehyde Derivatives with Cytotoxic and Apoptosis-Inducing Activities. J. Med. Chem. 2010, 53, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rakesh, K.P.; Shantharam, C.S.; Manukumar, H.M.; Asiri, A.M.; Marwani, H.M.; Qin, H.-L. Podophyllotoxin Derivatives as an Excellent Anticancer Aspirant for Future Chemotherapy: A Key Current Imminent Needs. Bioorg. Med. Chem. 2018, 26, 340–355. [Google Scholar] [CrossRef]
- Negi, A.S.; Gautam, Y.; Alam, S.; Chanda, D.; Luqman, S.; Sarkar, J.; Khan, F.; Konwar, R. Natural Antitubulin Agents: Importance of 3,4,5-Trimethoxyphenyl Fragment. Bioorg. Med. Chem. 2015, 23, 373–389. [Google Scholar] [CrossRef]
- Kamal, A.; Srinivasa Reddy, T.; Polepalli, S.; Shalini, N.; Reddy, V.G.; Subba Rao, A.V.; Jain, N.; Shankaraiah, N. Synthesis and Biological Evaluation of Podophyllotoxin Congeners as Tubulin Polymerization Inhibitors. Bioorg. Med. Chem. 2014, 22, 5466–5475. [Google Scholar] [CrossRef]
- Nerella, S.; Kankala, S.; Paidakula, S.; Gavaji, B. Synthesis of D-Ring Modified Acid Hydrazide Derivatives of Podophyllotoxin and Their Anticancer Studies as Tubulin Inhibiting Agents. Bioorganic Chem. 2020, 94, 103384. [Google Scholar] [CrossRef] [PubMed]
- Lisiecki, K.; Czarnocki, Z. Flow Photochemistry as a Tool for the Total Synthesis of (+)-Epigalcatin. Org. Lett. 2018, 20, 605–607. [Google Scholar] [CrossRef]
- Strus, P.; Borensztejn, K.; Szczepankiewicz, A.A.; Lisiecki, K.; Czarnocki, Z.; Nieznanska, H.; Wojcik, C.; Bialy, L.P.; Mlynarczuk-Bialy, I. Novel Podophyllotoxin and Benzothiazole Derivative Induces Transitional Morphological and Functional Changes in HaCaT Cells. Toxicol. Vitr. 2021, 73, 105144. [Google Scholar] [CrossRef]
- Lisiecki, K.; Krawczyk, K.K.; Roszkowski, P.; Maurin, J.K.; Czarnocki, Z. Formal Synthesis of (−)-Podophyllotoxin through the Photocyclization of an Axially Chiral 3,4-Bisbenzylidene Succinate Amide Ester–a Flow Photochemistry Approach. Org. Biomol. Chem. 2016, 14, 460–469. [Google Scholar] [CrossRef]
- Hernández, Á.P.; Díez, P.; García, P.A.; Miguel Del Corral, J.M.; Pérez-Andrés, M.; Díez, D.; San Feliciano, A.; Fuentes, M.; Castro, M.Á. New Hybrids Derived from Podophyllic Aldehyde and Diterpenylhydroquinones with Selectivity toward Osteosarcoma Cells. ACS Med. Chem. Lett. 2018, 9, 328–333. [Google Scholar] [CrossRef]
- Huynh, T.-K.-C.; Nguyen, T.-H.-A.; Tran, N.-H.-S.; Nguyen, T.-D.; Hoang, T.-K.-D. A Facile and Efficient Synthesis of Benzimidazole as Potential Anticancer Agents. J. Chem. Sci. 2020, 132, 84. [Google Scholar] [CrossRef]
- Xi, W.; Sun, H.; Bastow, K.F.; Xiao, Z.; Lee, K.-H. Identification of Novel 4′-O-Demethyl-Epipodophyllotoxin Derivatives as Antitumor Agents Targeting Topoisomerase II. Molecules 2022, 27, 5029. [Google Scholar] [CrossRef]
- Guminski, Y.; Grousseaud, M.; Cugnasse, S.; Imbert, T. Practical Demethylation of Podophyllotoxin and Efficient Preparation of 4-Amino-4-Deoxy-4′-Demethylepipodophyllotoxin. Synth. Commun. 2012, 42, 2780–2789. [Google Scholar] [CrossRef]
- Hao, S.-Y.; Feng, S.-L.; Wang, X.-R.; Wang, Z.; Chen, S.-W.; Hui, L. Novel Conjugates of Podophyllotoxin and Coumarin: Synthesis, Cytotoxicities, Cell Cycle Arrest, Binding CT DNA and Inhibition of Topo IIβ. Bioorg. Med. Chem. Lett. 2019, 29, 2129–2135. [Google Scholar] [CrossRef]
- Daley, L.; Meresse, P.; Bertounesque, E.; Monneret, C. A One-Pot, Efficient Synthesis of the Potent Cytotoxic Podophyllotoxin Derivative NPF. Tetrahedron Lett. 1997, 38, 2673–2676. [Google Scholar] [CrossRef]
- Escudero-Martínez, J.M.; Pérez-Pertejo, Y.; Reguera, R.M.; Castro, M.Á.; Rojo, M.V.; Santiago, C.; Abad, A.; García, P.A.; López-Pérez, J.L.; San Feliciano, A.; et al. Antileishmanial Activity and Tubulin Polymerization Inhibition of Podophyllotoxin Derivatives on Leishmania Infantum. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 272–285. [Google Scholar] [CrossRef] [PubMed]
- García, P.A.; Hernández, Á.-P.; Gómez-Zurita, M.A.; Miguel Del Corral, J.M.; Gordaliza, M.; Francesch, A.; San Feliciano, A.; Castro, M.Á. Cytotoxic Cyclolignans Obtained by the Enlargement of the Cyclolignan Skeleton of Podophyllic Aldehyde, a Selective Podophyllotoxin-Derived Cyclolignan. Molecules 2024, 29, 1442. [Google Scholar] [CrossRef]
- Tronina, T.; Bartmańska, A.; Popłoński, J.; Rychlicka, M.; Sordon, S.; Filip-Psurska, B.; Milczarek, M.; Wietrzyk, J.; Huszcza, E. Prenylated Flavonoids with Selective Toxicity against Human Cancers. Int. J. Mol. Sci. 2023, 24, 7408. [Google Scholar] [CrossRef]
- Desbene, S.; Giorgi-Renault, S. Drugs That Inhibit Tubulin Polymerization: The Particular Case of Podophyllotoxin and Analogues. Curr. Med. Chem.-Anti-Cancer Agents 2012, 2, 71–90. [Google Scholar] [CrossRef]
- Sackett, D.L. Podophyllotoxin, Steganacin and Combretastatin: Natural Products That Bind at the Colchicine Site of Tubulin. Pharmacol. Ther. 1993, 59, 163–228. [Google Scholar] [CrossRef]
- Bugnon, M.; Röhrig, U.F.; Goullieux, M.; Perez, M.A.S.; Daina, A.; Michielin, O.; Zoete, V. SwissDock 2024: Major Enhancements for Small-Molecule Docking with Attracting Cavities and AutoDock Vina. Nucleic Acids Res. 2024, 52, W324–W332. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.-W.; Huang, W.-J.; Liu, Y.-H.; Liu, Q.-G.; Song, J.; Hu, T.; Chen, P.; Zhang, S.-Y. Design, Synthesis and Biological Evaluation of 1,2,3-Triazole Benzothiazole Derivatives as Tubulin Polymerization Inhibitors with Potent Anti-Esophageal Cancer Activities. Eur. J. Med. Chem. 2024, 265, 116118. [Google Scholar] [CrossRef]
- Song, J.; Gao, Q.-L.; Wu, B.-W.; Zhu, T.; Cui, X.-X.; Jin, C.-J.; Wang, S.-Y.; Wang, S.-H.; Fu, D.-J.; Liu, H.-M.; et al. Discovery of Tertiary Amide Derivatives Incorporating Benzothiazole Moiety as Anti-Gastric Cancer Agents in Vitro via Inhibiting Tubulin Polymerization and Activating the Hippo Signaling Pathway. Eur. J. Med. Chem. 2020, 203, 112618. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro Software System, Version 1.171.38.35a; Rigaku Corporation: Oxford, UK, 2015.
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.J. X-Seed 4: Updates to a Program for Small-Molecule Supramolecular Crystallography. J. Appl. Crystallogr. 2020, 53, 1141–1146. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Atwood, J.L.; Barbour, L.J. Molecular Graphics: From Science to Art. Cryst. Growth Des. 2003, 3, 3–8. [Google Scholar] [CrossRef]
- Persistence of Vision Pty. Ltd. Available online: http://www.povray.org/ (accessed on 9 April 2025).
- Strus, P.; Sadowski, K.; Kostro, J.; Szczepankiewicz, A.A.; Nieznańska, H.; Niedzielska, M.; Zlobin, A.; Nawar Ra’idah, P.; Molęda, Z.; Szawkało, J.; et al. Cellular Distribution and Ultrastructural Changes in HaCaT Cells, Induced by Podophyllotoxin and Its Novel Fluorescent Derivative, Supported by the Molecular Docking Studies. Int. J. Mol. Sci. 2024, 25, 5948. [Google Scholar] [CrossRef] [PubMed]
- Młynarczuk-Biały, I.; Roeckmann, H.; Kuckelkorn, U.; Schmidt, B.; Umbreen, S.; Gołąb, J.; Ludwig, A.; Montag, C.; Wiebusch, L.; Hagemeier, C.; et al. Combined Effect of Proteasome and Calpain Inhibition on Cisplatin-Resistant Human Melanoma Cells. Cancer Res. 2006, 66, 7598–7605. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-like Domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).