
Bispecific Antibodies in Solid Tumors: Advances and Challenges

 , ,
, , 
Abstract
1. Introduction
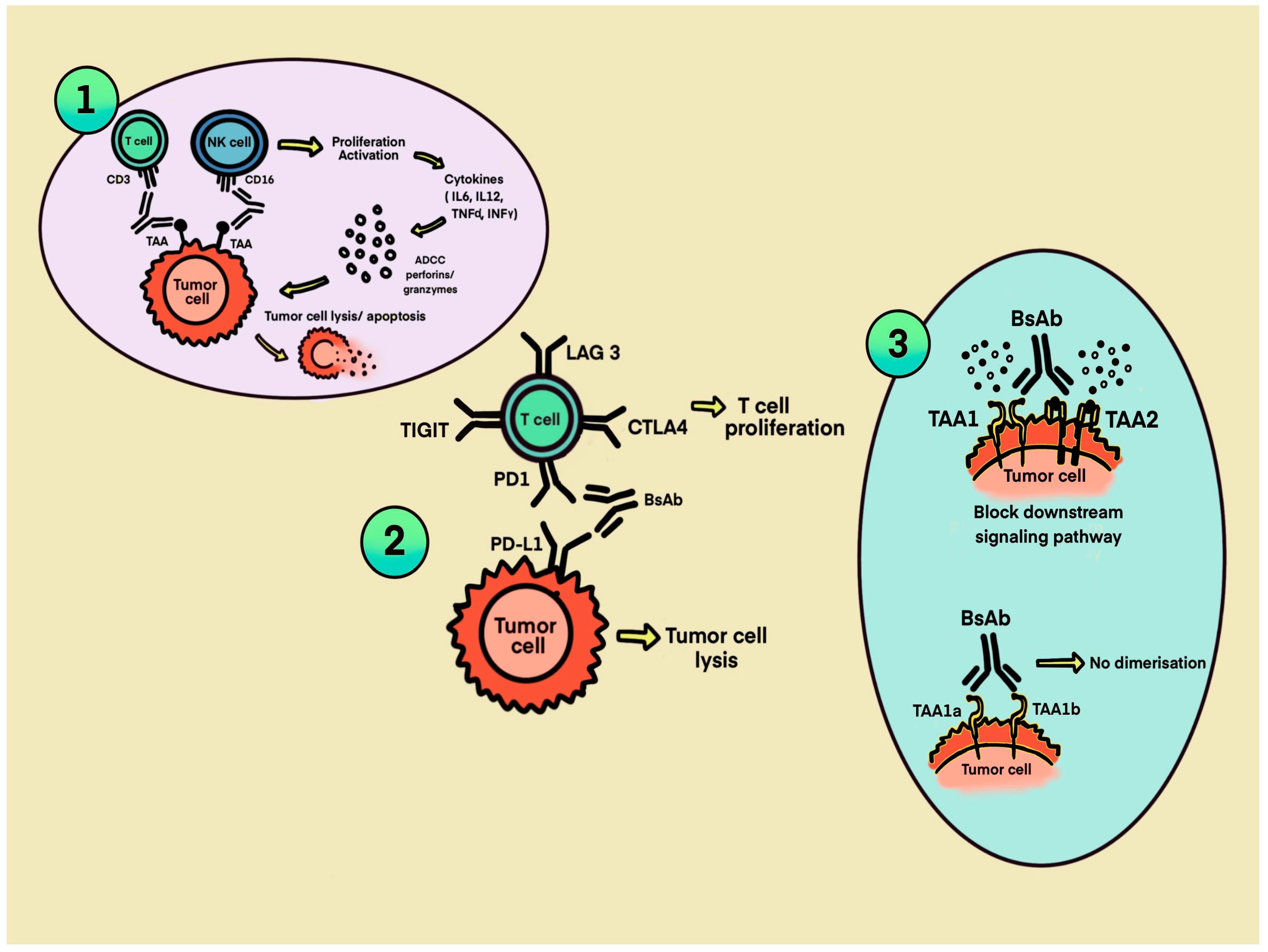
2. Mechanism of BsAbs
3. Current FDA-Approved BsAbs in Solid Tumors
3.1. Tebentafusp (gp100 Targeting)
3.2. Amivantamab (EGFR-Targeting)
3.3. Tarlatamab (DLL3-Targeting)
3.4. Zenocutuzumab (HER2-Targeting)
3.5. Zanidatamab (HER2-Targeting)
4. Current BsAbs in Solid Tumors Under Investigation
4.1. HER2-Targeting BsAbs
4.2. PSMA Targeting BsAbs
4.3. Claudin 18.2-Targeting BsAbs
4.4. CEA-Targeting BsAbs
4.5. EpCAM-Targeting BsAbs
4.6. GPC3-Targeting BsAbs
4.7. HLA-G-Targeting BsAbs
4.8. Immune Checkpoint-Targeting BsAbs
4.8.1. PD-1/CTLA-4-Targeting BsAbs
4.8.2. PD-1/TIGIT-Targeting BsAbs
4.8.3. PD-1/IL2-Targeting BsAbs
4.8.4. PD-1/ICOS-Targeting BsAb
4.8.5. PD-1/LAG3-Targeting BsAbs
4.8.6. PD-L1/PD-1 BsAbs
4.8.7. PD-L1/TIM-3-Targeting BsAbs
4.8.8. PD-L1/CD47-Targeting BsAbs
4.8.9. PD-L1/4-1BB-Targeting BsAbs
4.8.10. PD-L1/OX40-Targeting BsAbs
4.9. EGFR-Targeting BsAbs
4.9.1. EGFR/cMET Targeting BsAbs
4.9.2. EGFR/LGR5-Targeting BsAbs
4.9.3. EGFR/CD28-Targeting BsAbs
4.9.4. EGFR/CD16a-Targeting BsAbs
4.9.5. EGFR/CD3-Targeting BsAbs
4.10. VEGF-Targeting BsAbs
4.11. DLL4/VEGF-Targeting BsAbs
4.12. GD2-Targeting BsAbs
4.13. MUC16-Targeting BsAbs
4.14. 5T4-Targeting BsAbs
4.15. B7-H3-Targeting BsAbs
4.16. TGF-β/PDL1-Targeting BsAbs
4.17. Other BsAbs
5. Challenges in Clinical Utilization of BsAbs in Solid Tumors
5.1. Toxicities
5.1.1. On-Target, Off-Tumor Toxicities
- Antigen density discrimination: BsAbs can be engineered to preferentially bind to cells with high antigen density, sparing normal tissues with low antigen expression [176].
- Affinity modulation: Tuning the binding affinity of BsAbs toward TAAs is critical; lower-affinity antibodies can preferentially bind tumor cells with abundant antigens while avoiding low-expression normal tissues. However, excessive affinity reduction can compromise therapeutic efficacy, highlighting the need for precise molecular engineering [179].
- Switchable BsAb formats: Recent advances have introduced BsAbs that are activated only in the presence of tumor-specific enzymatic activities, such as tumor-associated proteases cleaving an inhibitory domain. These “prodrug” designs further improve tumor selectivity by restricting T-cell activation to the TME [180].
5.1.2. Cytokine Release Syndrome
- Step-up dosing regimens involve administering a lower priming dose followed by gradual escalation to therapeutic levels, allowing for progressive T-cell desensitization and reducing the peak cytokine surge. Although step-up dosing is not mandated by prescribing labels, clinicians should consider monitoring patients for 1 to 2 h following administration when dosing occurs in the outpatient setting. If signs or symptoms of CRS develop, prompt admission to the hospital or transfer to the emergency department should be considered based on clinical severity [184].
- Pre-medications, including corticosteroids and IL-6 receptor antagonists such as tocilizumab, are employed prophylactically or reactively to dampen the inflammatory response without abrogating antitumor activity [185]. Immunosuppressive therapies should be considered to attenuate the immune response in cases of severe CRS. Accurate grading of these toxicities is critical, as it guides clinicians in initiating appropriate interventions. Mild-to-moderate CRS can often be managed with supportive care measures, including intravenous fluids, antihistamines, and antipyretics. In contrast, patients who develop severe CRS require close monitoring and management in an intensive care unit setting [184]. Treatment with dexamethasone at a dose of 8 mg intravenously or orally every 8 h for 3 days is recommended, followed by a gradual steroid taper over the subsequent 4 days. If patients exhibit an inadequate clinical response to corticosteroid therapy or have grade 3 to 4 CRS, administration of tocilizumab, an IL-6 receptor antagonist, at a dose of 8 mg/kg intravenously is indicated. Tocilizumab has demonstrated efficacy in reversing severe manifestations of CRS by targeting the IL-6-mediated inflammatory cascade [184,186].
5.2. Tissue Penetration and Resistance to BsAb Therapy
5.2.1. Tumor Microenvironment
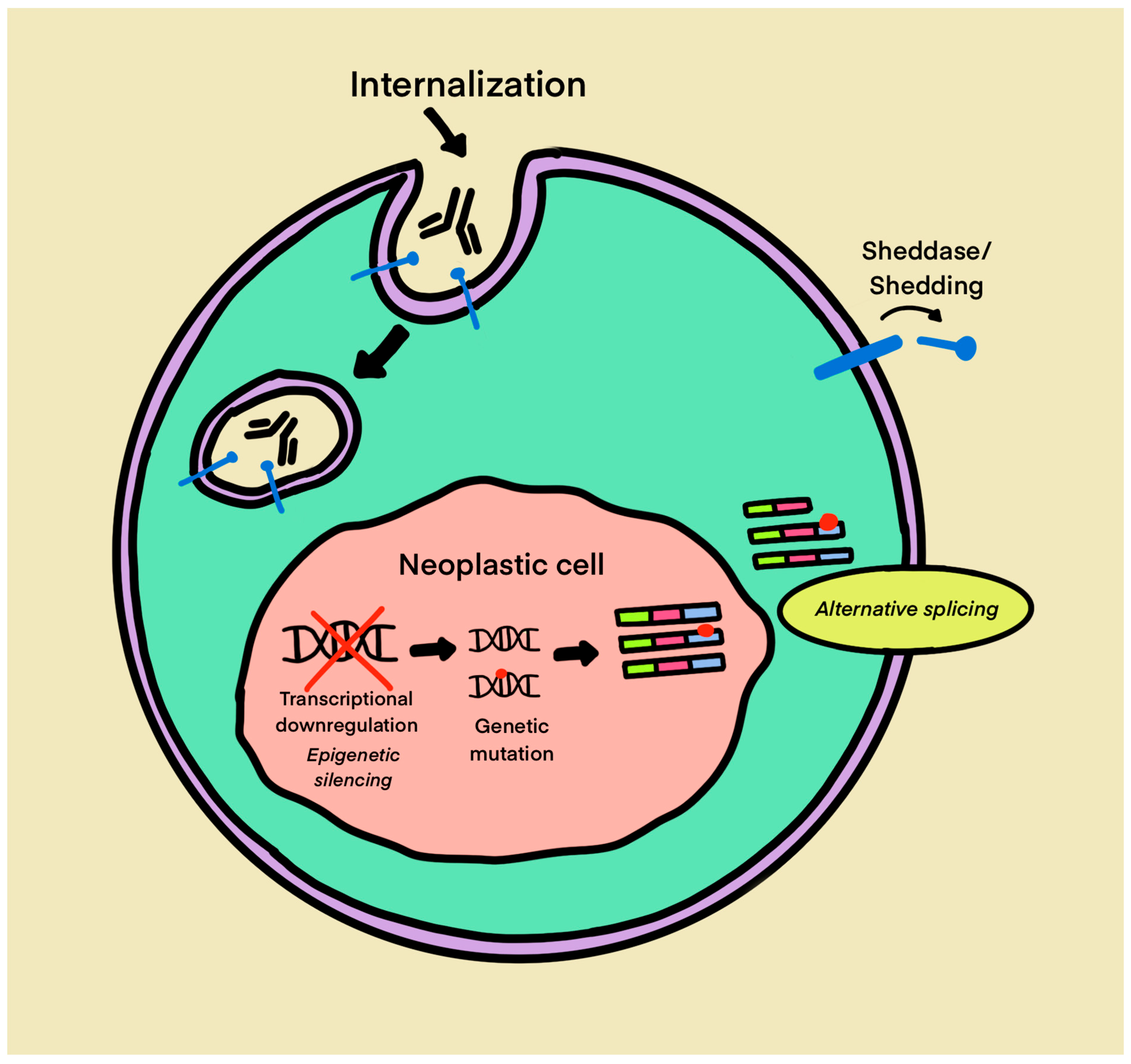
5.2.2. Loss of Target Antigen Expression and Resistance to BsAb Therapy
5.2.3. Anti-Drug Antibodies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Herrera, M.; Pretelli, G.; Desai, J.; Garralda, E.; Siu, L.L.; Steiner, T.M.; Au, L. Bispecific antibodies: Advancing precision oncology. Trends Cancer 2024, 10, 893–919. [Google Scholar] [CrossRef]
- Keam, S.J. Ozoralizumab: First Approval. Drugs 2023, 83, 87–92. [Google Scholar] [CrossRef]
- Shirley, M. Faricimab: First Approval. Drugs 2022, 82, 825–830. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, Q. Clinical Progresses and Challenges of Bispecific Antibodies for the Treatment of Solid Tumors. Mol. Diagn. Amp Ther. 2024, 28, 669–702. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Kim, E.S. Emicizumab-kxwh: First Global Approval. Drugs 2018, 78, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yi, M.; Zhu, S.; Wang, H.; Wu, K. Recent advances and challenges of bispecific antibodies in solid tumors. Exp. Hematol. Oncol. 2021, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Chon, K.; Larkins, E.; Chatterjee, S.; Mishra-Kalyani, P.S.; Aungst, S.; Wearne, E.; Subramaniam, S.; Li, Y.; Liu, J.; Sun, J.; et al. FDA Approval Summary: Amivantamab for the Treatment of Patients with Non-Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations. Clin. Cancer Res. 2023, 29, 3262–3266. [Google Scholar] [CrossRef]
- FDA.gov. FDA Approves Amivantamab-Vmjw with Carboplatin and Pemetrexed for Non-Small Cell Lung Cancer with EGFR Exon 19 Deletions or L858R Mutations. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-amivantamab-vmjw-carboplatin-and-pemetrexed-non-small-cell-lung-cancer-egfr-exon-19#:~:text=On%20September%2019%2C%202024%2C%20the%20FDA%20approved%20amivantamab-vmjw,locally%20advanced%20or%20metastatic%20non-small%20cell%20lung%20cancer (accessed on 3 December 2024).
- Dhillon, S. Tarlatamab: First Approval. Drugs 2024, 84, 995–1003. [Google Scholar] [CrossRef]
- Dhillon, S. Tebentafusp: First Approval. Drugs 2022, 82, 703–710. [Google Scholar] [CrossRef]
- FDA.gov. FDA Grants Accelerated Approval to Zenocutuzumab-Zbco for Non-Small Cell Lung Cancer and Pancreatic Adenocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-zenocutuzumab-zbco-non-small-cell-lung-cancer-and-pancreatic (accessed on 25 December 2024).
- FDA.gov. FDA Grants Accelerated Approval to Zanidatamab-Hrii for Previously Treated Unresectable or Metastatic HER2-Positive Biliary Tract Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-zanidatamab-hrii-previously-treated-unresectable-or-metastatic-her2 (accessed on 29 November 2024).
- Dhillon, S. Ivonescimab: First Approval. Drugs 2024, 84, 1135–1142. [Google Scholar] [CrossRef]
- Keam, S.J. Cadonilimab: First Approval. Drugs 2022, 82, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C. First-Line Cadonilimab Plus Chemo Wins Approval in China for Advanced Gastric/GEJ Adenocarcinoma. Available online: https://www.onclive.com/view/first-line-cadonilimab-plus-chemo-wins-approval-in-china-for-advanced-gastric-gej-adenocarcinoma (accessed on 3 December 2024).
- van de Donk, N.W.C.J.; Zweegman, S. T-cell-engaging bispecific antibodies in cancer. Lancet 2023, 402, 142–158. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C.; Piperno-Neumann, S.; Rutkowski, P.; Baurain, J.F.; Schlaak, M.; Butler, M.O.; Sullivan, R.J.; Dummer, R.; Kirkwood, J.M.; Orloff, M.; et al. Three-Year Overall Survival with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2023, 389, 2256–2266. [Google Scholar] [CrossRef]
- Middleton, M.R.; Steven, N.M.; Evans, T.J.; Infante, J.R.; Sznol, M.; Mulatero, C.; Hamid, O.; Shoushtari, A.N.; Shingler, W.; Johnson, A.; et al. Safety, pharmacokinetics and efficacy of IMCgp100, a first-in-class soluble TCR-antiCD3 bispecific t cell redirector with solid tumour activity: Results from the FIH study in melanoma. J. Clin. Oncol. 2016, 34, 3016. [Google Scholar] [CrossRef]
- Hamid, O.; Hassel, J.C.; Shoushtari, A.N.; Meier, F.; Bauer, T.M.; Salama, A.K.S.; Kirkwood, J.M.; Ascierto, P.A.; Lorigan, P.C.; Mauch, C.; et al. Tebentafusp in combination with durvalumab and/or tremelimumab in patients with metastatic cutaneous melanoma: A phase 1 study. J. Immunother. Cancer 2023, 11, e006747. [Google Scholar] [CrossRef]
- Zhou, C.; Tang, K.J.; Cho, B.C.; Liu, B.; Paz-Ares, L.; Cheng, S.; Kitazono, S.; Thiagarajan, M.; Goldman, J.W.; Sabari, J.K.; et al. Amivantamab plus Chemotherapy in NSCLC with EGFR Exon 20 Insertions. N. Engl. J. Med. 2023, 389, 2039–2051. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.Y.; Kim, S.W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Cho, B.C.; Lu, S.; Felip, E.; Spira, A.I.; Girard, N.; Lee, J.S.; Lee, S.H.; Ostapenko, Y.; Danchaivijitr, P.; Liu, B.; et al. Amivantamab plus Lazertinib in Previously Untreated EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2024, 391, 1486–1498. [Google Scholar] [CrossRef]
- Yang, J.C.H.; Kim, Y.J.; Lee, S.H.; Liu, B.; Ostapenko, Y.V.; Lu, S.; Alip, A.; Korbenfeld, E.P.; Dias, J.; Danchaivijitr, P.; et al. 4O: Amivantamab plus lazertinib vs osimertinib in first-line (1L) EGFR-mutant (EGFRm) advanced NSCLC: Final overall survival (OS) from the phase III MARIPOSA study. J. Thorac. Oncol. 2025, 20, S6–S8. [Google Scholar] [CrossRef]
- Ahn, M.J.; Cho, B.C.; Felip, E.; Korantzis, I.; Ohashi, K.; Majem, M.; Juan-Vidal, O.; Handzhiev, S.; Izumi, H.; Lee, J.S.; et al. Tarlatamab for Patients with Previously Treated Small-Cell Lung Cancer. N. Engl. J. Med. 2023, 389, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Fan, J.; Oh, D.-Y.; Choi, H.J.; Kim, J.W.; Chang, H.-M.; Bao, L.; Sun, H.-C.; Macarulla, T.; Xie, F.; et al. Zanidatamab for HER2-amplified, unresectable, locally advanced or metastatic biliary tract cancer (HERIZON-BTC-01): A multicentre, single-arm, phase 2b study. Lancet Oncol. 2023, 24, 772–782. [Google Scholar] [CrossRef]
- Pant, S.; Fan, J.; Oh, D.-Y.; Choi, H.J.; Kim, J.W.; Chang, H.-M.; Bao, L.; Sun, H.-C.; Macarulla, T.; Xie, F.; et al. Zanidatamab in previously-treated HER2-positive (HER2+) biliary tract cancer (BTC): Overall survival (OS) and longer follow-up from the phase 2b HERIZON-BTC-01 study. J. Clin. Oncol. 2024, 42, 4091. [Google Scholar] [CrossRef]
- Schram, A.; Goto, K.; Kim, D.W.; Hollebecque, A.; Rha, S.Y.; Nishino, K.; Duruisseaux, M.; Umemoto, K.; Park, J.O.; Leighl, N.; et al. 1315MO Durable efficacy of zenocutuzumab, a HER2 x HER3 bispecific antibody, in advanced NRG1 fusion-positive (NRG1+) non-small cell lung cancer (NSCLC). Ann. Oncol. 2023, 34, S756–S757. [Google Scholar] [CrossRef]
- Schram, A.; Macarulla, T.; Cleary, J.; Neuzillet, C.; Arnold, D.; Reiss, K.A.; Bekaii-Saab, T.; Rodon, J.; Goto, K.; Rha, S.Y.; et al. 1618P Durable efficacy of zenocutuzumab, a HER2 x HER3 bispecific antibody, in advanced NRG1 fusion positive (NRG1+) pancreatic ductal adenocarcinoma (PDAC). Ann. Oncol. 2023, 34, S895–S896. [Google Scholar] [CrossRef]
- FDA.gov. FDA Approves Amivantamab-Vmjw for EGFR Exon 20 Insertion-Mutated Non-Small Cell Lung Cancer Indications. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-amivantamab-vmjw-egfr-exon-20-insertion-mutated-non-small-cell-lung-cancer-indications (accessed on 25 December 2024).
- Drugs.com. FDA Approves Rybrevant (Amivantamab-Vmjw) Plus Lazcluze (Lazertinib) for PATIENTS with EGFR-Mutated Advanced Lung Cancer. Available online: https://www.drugs.com/newdrugs/fda-approves-rybrevant-amivantamab-vmjw-plus-lazcluze-lazertinib-patients-egfr-mutated-advanced-6357.html (accessed on 25 December 2024).
- Paz-Ares, L.G.; Spira, A.I.; Han, J.Y.; Shih, J.Y.; Mascaux, C.; Basu Roy, U.; Zugazagoitia, J.; Kim, Y.J.; Chiu, C.H.; Kim, S.W.; et al. 1269P Preventing infusion-related reactions with intravenous amivantamab: Updated results from SKIPPirr, a phase II study. Ann. Oncol. 2024, 35, S812. [Google Scholar] [CrossRef]
- Leighl, N.B.; Akamatsu, H.; Lim, S.M.; Cheng, Y.; Minchom, A.R.; Marmarelis, M.E.; Sanborn, R.E.; Chih-Hsin Yang, J.; Liu, B.; John, T.; et al. Subcutaneous Versus Intravenous Amivantamab, Both in Combination With Lazertinib, in Refractory Epidermal Growth Factor Receptor-Mutated Non-Small Cell Lung Cancer: Primary Results From the Phase III PALOMA-3 Study. J. Clin. Oncol. 2024, 42, 3593–3605. [Google Scholar] [CrossRef]
- Moik, F.; Absenger, G.; Wurm, R.; Hochmair, M.J.; Ay, C. Prevention and Treatment of Venous Thromboembolism Associated with Amivantamab-Based Therapies in Patients with Lung Cancer—Provisional Clinical Opinion Based on Existing Clinical Practice Guidelines. Cancers 2025, 17, 259. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Champiat, S.; Lai, W.V.; Izumi, H.; Govindan, R.; Boyer, M.; Hummel, H.D.; Borghaei, H.; Johnson, M.L.; Steeghs, N.; et al. Tarlatamab, a First-in-Class DLL3-Targeted Bispecific T-Cell Engager, in Recurrent Small-Cell Lung Cancer: An Open-Label, Phase I Study. J. Clin. Oncol. 2023, 41, 2893–2903. [Google Scholar] [CrossRef]
- Paz-Ares Rodriguez, L.; Felip, E.; Ahn, M.-J.; Blackhall, F.H.; Borghaei, H.; Cho, B.C.; Johnson, M.L.; Ramalingam, S.S.; Reck, M.; Jiang, T.; et al. PP01.72 Randomized Phase-3 Study: Tarlatamab, a DLL3-targeting Bispecific T-cell engager (BiTE), Compared to Standard-of-Care in Relapsed Small Cell Lung Cancer (DeLLphi-304). J. Thorac. Oncol. 2024, 19, e34. [Google Scholar] [CrossRef]
- Kim, D.-W.; Schram, A.M.; Hollebecque, A.; Nishino, K.; Macarulla, T.; Rha, S.Y.; Duruisseaux, M.; Liu, S.V.; Al Hallak, M.N.; Umemoto, K.; et al. The phase I/II eNRGy trial: Zenocutuzumab in patients with cancers harboring NRG1 gene fusions. Future Oncol. 2024, 20, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Petit, T.; Pistilli, B.; Goncalves, A.; Ferreira, A.A.; Dalenc, F.; Cardoso, F.; Mita, M.M.; Manso, L.; Karim, S.M.; et al. Abstract OT2-15-01: Updated analysis of MCLA-128 (zenocutuzumab), trastuzumab, and vinorelbine in patients (pts) with HER2 positive/amplified (HER2+) metastatic breast cancer (MBC) who progressed on previous anti-HER2 ADCs. Cancer Res. 2022, 82, OT2-15-01. [Google Scholar] [CrossRef]
- Tabernero, J.; Shen, L.; Elimova, E.; Ku, G.; Liu, T.; Shitara, K.; Lin, X.; Boyken, L.; Li, H.; Grim, J.; et al. HERIZON-GEA-01: Zanidatamab + chemo ± tislelizumab for 1L treatment of HER2-positive gastroesophageal adenocarcinoma. Future Oncol. 2022, 18, 3255–3266. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Bai, L.-Y.; Jung, M.; Ying, J.; Im, Y.H.; Oh, D.-Y.; Cho, J.Y.; Oh, S.C.; Chao, Y.; Li, H.; et al. Zanidatamab (zani), a HER2-targeted bispecific antibody, in combination with chemotherapy (chemo) and tislelizumab (TIS) as first-line (1L) therapy for patients (pts) with advanced HER2-positive gastric/gastroesophageal junction adenocarcinoma (G/GEJC): Preliminary results from a phase 1b/2 study. J. Clin. Oncol. 2022, 40 (Suppl. S16), 4032. [Google Scholar] [CrossRef]
- Valero, V.; Mouabbi, J.; Alonzo, H.; Ilheme, A.N.; Murthy, R.; Huang, X.; Qiao, W.; Patel, M.; Pohlmann, P.R.; Rauch, G.; et al. 132P Neoadjuvant zanidatamab for stage I node-negative HER2- positive breast cancer (BC). ESMO Open 2023, 8, 10147. [Google Scholar] [CrossRef]
- Lumish, M.; Chui, M.H.; Zhou, Q.; Iasonos, A.; Sarasohn, D.; Cohen, S.; Friedman, C.; Grisham, R.; Konner, J.; Kyi, C.; et al. A phase 2 trial of zanidatamab in HER2-overexpressed advanced endometrial carcinoma and carcinosarcoma (ZW25-IST-2). Gynecol Oncol 2024, 182, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Xu, J.; Ying, J.; Liu, R.; Wu, J.; Ye, F.; Xu, N.; Zhang, Y.; Zhao, R.; Xiang, X.; Wang, J.; et al. KN026 (anti-HER2 bispecific antibody) in patients with previously treated, advanced HER2-expressing gastric or gastroesophageal junction cancer. Eur. J. Cancer 2023, 178, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, D.; Cai, L.; Yao, H.; Yan, M.; Wang, X.; Shen, W.; Du, Y.; Pang, H.; Lai, X.; et al. First-in-human HER2-targeted Bispecific Antibody KN026 for the Treatment of Patients with HER2-positive Metastatic Breast Cancer: Results from a Phase I Study. Clin. Cancer Res. 2022, 28, 618–628. [Google Scholar] [CrossRef]
- Wu, J.; Yang, B.; Ma, L.; Zhang, M.; Wang, K.; Chen, Y.; Fan, Z.; Zhang, J.; Xia, S. Abstract OT2-16-04: KN026 in combination with docetaxel as neoadjuvant treatment for HER2-positive early or locally advanced breast cancer: A single arm, multicenter, phase 2 study. Cancer Res. 2023, 83, OT2-16-04-OT12-16-04. [Google Scholar] [CrossRef]
- Singh, R.; Kim, Y.H.; Lee, S.J.; Eom, H.S.; Choi, B.K. 4-1BB immunotherapy: Advances and hurdles. Exp. Mol. Med. 2024, 56, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Park, Y.B.; Choi, M.; Shin, J.; Kim, K.B.; Park, J.Y.; Kim, J.; Ahn, K.K.; Lee, Y.; Park, K.; et al. Abstract 6354: The HER2/4-1BB bispecific antibody, YH32367 (ABL105) demonstrates optimal efficacy and safety for HER2-expressing tumors and exhibits synergistic efficacy when combined with anti-PD-L1. Cancer Res. 2024, 84, 6354. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, L.; Fang, W.; Yang, Y.; Huang, Y.; Zou, W.; Wang, Z.; Ding, M.; Peng, Y.; Xiao, S.; et al. SI-B001 plus chemotherapy in patients with locally advanced or metastatic EGFR/ALK wild-type non-small cell lung cancer: A phase II, multicenter, open-label study. J. Clin. Oncol. 2023, 41, 9025. [Google Scholar] [CrossRef]
- Xue, L.; Yang, K.; Fang, M.; Ma, X.; Zou, W.; Ding, M.; Wang, Z.; Peng, Y.; Xiao, S.; Wang, H.; et al. Results from two phase II studies of SI-B001, an EGFR×HER3 bispecific antibody, with/without chemotherapy in patients (pts) with recurrent and metastatic head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2023, 41, 6037. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.; Feng, X.; Yang, D.; Lin, M. Advances in PSMA-targeted therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 11–26. [Google Scholar] [CrossRef]
- Hummel, H.-D.; Kufer, P.; Grüllich, C.; Seggewiss-Bernhardt, R.; Deschler-Baier, B.; Chatterjee, M.; Goebeler, M.-E.; Miller, K.; De Santis, M.; Loidl, W.; et al. Pasotuxizumab, a Bite® Immune Therapy for Castration-Resistant Prostate Cancer: Phase I, Dose-Escalation Study Findings. Immunotherapy 2021, 13, 125–141. [Google Scholar] [CrossRef]
- Zarrabi, K.K.; Narayan, V.; Mille, P.J.; Zibelman, M.R.; Miron, B.; Bashir, B.; Kelly, W.K. Bispecific PSMA antibodies and CAR-T in metastatic castration-resistant prostate cancer. Ther. Adv. Urol. 2023, 15, 17562872231182219. [Google Scholar] [CrossRef]
- Deegen, P.; Thomas, O.; Nolan-Stevaux, O.; Li, S.; Wahl, J.; Bogner, P.; Aeffner, F.; Friedrich, M.; Liao, M.Z.; Matthes, K.; et al. The PSMA-targeting Half-life Extended BiTE Therapy AMG 160 has Potent Antitumor Activity in Preclinical Models of Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2021, 27, 2928–2937. [Google Scholar] [CrossRef] [PubMed]
- Dorff, T.; Horvath, L.G.; Autio, K.; Bernard-Tessier, A.; Rettig, M.B.; Machiels, J.P.; Bilen, M.A.; Lolkema, M.P.; Adra, N.; Rottey, S.; et al. A Phase I Study of Acapatamab, a Half-life Extended, PSMA-Targeting Bispecific T-cell Engager for Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2024, 30, 1488–1500. [Google Scholar] [CrossRef]
- Falchook, G.S.; McKean, M.; Kelly, W.K.; Patel, M.R.; Bupathi, M.; Liaw, B.C.-H.; Garmezy, B.; Vuu, I.; Smith, K.; Gokani, P.; et al. Phase 1 clinical trial of AMG 340, a prostate-specific membrane antigen (PSMA)-targeted T-cell engager with a novel low-affinity CD3 binding domain designed to mitigate toxicity for the treatment of metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2024, 42 (Suppl. S16), e14587. [Google Scholar] [CrossRef]
- Lim, E.A.; Schweizer, M.T.; Chi, K.N.; Aggarwal, R.; Agarwal, N.; Gulley, J.; Attiyeh, E.; Greger, J.; Wu, S.; Jaiprasart, P.; et al. Phase 1 Study of Safety and Preliminary Clinical Activity of JNJ-63898081, a PSMA and CD3 Bispecific Antibody, for Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2023, 21, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.N.; Zhang, J.; Kelly, W.K.; Wise, D.R.; Tsao, K.; Carneiro, B.A.; Falchook, G.S.; Sun, F.; Govindraj, S.; Sims, J.S.; et al. Preliminary results from a phase 1/2 study of co-stimulatory bispecific PSMAxCD28 antibody REGN5678 in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2023, 41 (Suppl. S6), 154. [Google Scholar] [CrossRef]
- Mathias-Machado, M.C.; De Jesus, V.H.F.; Jácome, A.; Donadio, M.D.; Aruquipa, M.P.S.; Fogacci, J.; Cunha, R.G.; Da Silva, L.M.; Peixoto, R.D.A. Claudin 18.2 as a New Biomarker in Gastric Cancer—What Should We Know? Cancers 2024, 16, 679. [Google Scholar] [CrossRef] [PubMed]
- Lordick, F.; Chao, J.; Buxò, E.; Van Laarhoven, H.W.M.; Lima, C.M.R.; Lorenzen, S.; Dayyani, F.; Heinemann, V.; Greil, R.; Stienen, S.; et al. 1496TiP Phase I study evaluating safety and tolerability of AMG 910, a half-life extended bispecific T cell engager targeting claudin-18.2 (CLDN18.2) in gastric and gastroesophageal junction (G/GEJ) adenocarcinoma. Ann. Oncol. 2020, 31, S928–S929. [Google Scholar] [CrossRef]
- Gaspar, M.; Natoli, M.; Castan, L.; Rahmy, S.; Kelton, C.; Mulgrew, K.; Korade, M.; Huhn, O.; Gareth Rees, D.; Sigurdardottir, A.; et al. 1169 AZD5863: A specific, potent, affinity-optimized claudin 18.2 and CD3 binding T cell-engager that elicits low cytokine release and is capable of bystander killing. J. Immunother. Cancer 2023, 11, A1288. [Google Scholar] [CrossRef]
- Natoli, M.; Rahmy, S.; Garçon, F.; Olvera, R.A.; Gerada, C.; Opoku-Ansah, G.; Dallaway, L.; Chinello, L.; Giraldo, N.; Clements, M.; et al. 1073 AZD5863, a CLDN18.2 and CD3 binding T-cell engager, establishes a multi-faceted antitumour immune response and combines effectively with chemo-immunotherapy regimens. J. Immunother. Cancer 2024, 12 (Suppl. S2), A1194. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, G.; Liu, Z.; Xu, J.; Zhao, L.; Li, G.; Li, B.; Zhu, Z.; Peng, W.; Zhu, Y.; et al. Abstract 6706: An anti-claudin 18.2/CD3 bispecific antibody for the treatment of claudin 18.2 positive gastric cancer. Cancer Res. 2024, 84, 6706. [Google Scholar] [CrossRef]
- Nakazawa, T.; Tanaka, H.; Kikuchi, A.; Rashid, R.; Avery, K.N.; Qi, J.; Nisthal, A.; Shimazaki, M.; Shirasuna, K. Abstract 2962: ASP2138, a novel 2+1 format, claudin 18.2 x CD3 bispecific antibody, demonstrates selectivity and activity in preclinical cancer models. Cancer Res. 2023, 83, 2962. [Google Scholar] [CrossRef]
- Wang, Y.; Gong, J.; Qu, X.; Shen, L. 1500 A phase II trial of bispecific antibody Q-1802 in patients with relapsed or refractory solid tumors. J. Immunother. Cancer 2024, 12, A1729. [Google Scholar] [CrossRef]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef]
- Pishvaian, M.; Morse, M.A.; McDevitt, J.; Norton, J.D.; Ren, S.; Robbie, G.J.; Ryan, P.C.; Soukharev, S.; Bao, H.; Denlinger, C.S. Phase 1 Dose Escalation Study of MEDI-565, a Bispecific T-Cell Engager that Targets Human Carcinoembryonic Antigen, in Patients With Advanced Gastrointestinal Adenocarcinomas. Clin. Colorectal Cancer 2016, 15, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Melero, I.; Moreno, V.; Steeghs, N.; Marabelle, A.; Rohrberg, K.; Rodriguez-Ruiz, M.E.; Eder, J.P.; Eng, C.; Manji, G.A.; et al. CEA-CD3 bispecific antibody cibisatamab with or without atezolizumab in patients with CEA-positive solid tumours: Results of two multi-institutional Phase 1 trials. Nat. Commun. 2024, 15, 4091. [Google Scholar] [CrossRef]
- Elsayed, A.; Plüss, L.; Nideroest, L.; Rotta, G.; Thoma, M.; Zangger, N.; Peissert, F.; Pfister, S.K.; Pellegrino, C.; Dakhel Plaza, S.; et al. Optimizing the Design and Geometry of T Cell–Engaging Bispecific Antibodies Targeting CEA in Colorectal Cancer. Mol. Cancer Ther. 2024, 23, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Herreros-Pomares, A.; Aguilar-Gallardo, C.; Calabuig-Fariñas, S.; Sirera, R.; Jantus-Lewintre, E.; Camps, C. EpCAM duality becomes this molecule in a new Dr. Jekyll and Mr. Hyde tale. Crit. Rev. Oncol. Hematol. 2018, 126, 52–63. [Google Scholar] [CrossRef]
- Frey, G.; Cugnetti, A.P.; Liu, H.; Xing, C.; Wheeler, C.; Chang, H.; Boyle, W.; Short, J. A novel conditional active biologic anti-EpCAM x anti-CD3 bispecific antibody with synergistic tumor selectivity for cancer immunotherapy. mAbs 2024, 16, 2322562. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab. mAbs 2010, 2, 129–136. [Google Scholar] [CrossRef]
- Knödler, M.; Körfer, J.; Kunzmann, V.; Trojan, J.; Daum, S.; Schenk, M.; Kullmann, F.; Schroll, S.; Behringer, D.; Stahl, M.; et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM × anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Br. J. Cancer 2018, 119, 296–302. [Google Scholar] [CrossRef]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef]
- Kebenko, M.; Goebeler, M.-E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. OncoImmunology 2018, 7, e1450710. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Xu, J.; Liu, R.; Li, N.; Li, G.; Zhang, T.; Zhao, J.; Li, J.; Sun, M.; Wang, K.; et al. An anti-EpCAM x anti-CD3 bispecific antibody, M701, for the treatment of malignant ascites due to epithelial cancer: Interim results of a prospective randomized controlled phase II trial. J. Clin. Oncol. 2024, 42, 12060. [Google Scholar] [CrossRef]
- Melero, I.; Lakhani, N.J.; Powderly, J.; Adjei, A.A.; Lopez, J.S.; Paz-Ares, L.G.; Moreno Candilejo, I.; Thistlethwaite, F.; Rohrberg, K.S.; Jerusalem, G.; et al. 1072TiP Phase I/II dose escalation/expansion trial to evaluate safety and preliminary efficacy of DuoBody-EpCAM×4-1BB (BNT314/GEN1059) alone or in combination with an immune checkpoint inhibitor in patients with malignant solid tumors. Ann. Oncol. 2024, 35, S709–S710. [Google Scholar] [CrossRef]
- Du, K.; Li, Y.; Liu, J.; Chen, W.; Wei, Z.; Luo, Y.; Liu, H.; Qi, Y.; Wang, F.; Sui, J. A bispecific antibody targeting GPC3 and CD47 induced enhanced antitumor efficacy against dual antigen-expressing HCC. Mol. Ther. 2021, 29, 1572–1584. [Google Scholar] [CrossRef]
- Zhou, F.; Shang, W.; Yu, X.; Tian, J. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef]
- Sano, Y.; Azuma, Y.; Tsunenari, T.; Kayukawa, Y.; Shinozuka, J.; Fujii, E.; Amano, J.; Nishito, Y.; Maruyama, T.; Kinoshita, Y.; et al. Combination of T cell-redirecting bispecific antibody ERY974 and chemotherapy reciprocally enhances efficacy against non-inflamed tumours. Nat. Commun. 2022, 13, 5265. [Google Scholar] [CrossRef]
- Safran, H.; Druta, M.; Morse, M.; Lynce, F.; Pintova, S.; Almhanna, K.; Weiss, D.; Gianella-Borradori, A.; Ogita, Y.; Morley, R.; et al. Abstract CT111: Results of a phase 1 dose escalation study of ERY974, an anti-glypican 3 (GPC3)/CD3 bispecific antibody, in patients with advanced solid tumors. Cancer Res. 2021, 81, CT111. [Google Scholar] [CrossRef]
- Komatsu, S.-I.; Kayukawa, Y.; Miyazaki, Y.; Kaneko, A.; Ikegami, H.; Ishiguro, T.; Nakamura, M.; Frings, W.; Ono, N.; Sakata, K.; et al. Determination of starting dose of the T cell-redirecting bispecific antibody ERY974 targeting glypican-3 in first-in-human clinical trial. Sci. Rep. 2022, 12, 12312. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Zwolak, A.; Van De Ven, K.; Versmissen, S.; Brajic, A.; Petley, T.; Weinstock, D.; Aligo, J.; Yi, F.; Jarantow, S.; et al. Abstract ND07: JNJ-78306358: A first-in-class bispecific T cell redirecting HLA-G antibody. Cancer Res. 2022, 82, ND07. [Google Scholar] [CrossRef]
- Geva, R.; Vieito, M.; Ramon, J.; Perets, R.; Pedregal, M.; Corral, E.; Doger, B.; Calvo, E.; Bardina, J.; Garralda, E.; et al. Safety and clinical activity of JNJ-78306358, a human leukocyte antigen-G (HLA-G) x CD3 bispecific antibody, for the treatment of advanced stage solid tumors. Cancer Immunol. Immunother. 2024, 73, 205. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, N.; Li, Z.; Shen, L.; Ji, K.; Zheng, Z.; Liu, D.; Lou, H.; Bai, L.; Liu, T.; et al. Safety and antitumour activity of cadonilimab, an anti-PD-1/CTLA-4 bispecific antibody, for patients with advanced solid tumours (COMPASSION-03): A multicentre, open-label, phase 1b/2 trial. Lancet Oncol. 2023, 24, 1134–1146. [Google Scholar] [CrossRef]
- Wu, X.; Sun, Y.; Yang, H.; Wang, J.; Lou, H.; Li, D.; Wang, K.; Zhang, H.; Wu, T.; Li, Y.; et al. Cadonilimab plus platinum-based chemotherapy with or without bevacizumab as first-line treatment for persistent, recurrent, or metastatic cervical cancer (COMPASSION-16): A randomised, double-blind, placebo-controlled phase 3 trial in China. Lancet 2024, 404, 1668–1676. [Google Scholar] [CrossRef]
- Nakayama, I.; Shitara, K. The current status of immunotherapy and future horizon in the treatment of metastatic and locally advanced gastroesophageal adenocarcinoma. Expert Opin. Biol. Ther. 2024, 24, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Kim, S.W.; Costa, E.C.; Rodríguez, L.M.; Oliveira, J.; Insa Molla, M.A.; Majem, M.; Costa, L.; Su, W.C.; Lee, K.H.; et al. LBA56 MEDI5752 or pembrolizumab (P) plus carboplatin/pemetrexed (CP) in treatment-naïve (1L) non-small cell lung cancer (NSCLC): A phase Ib/II trial. Ann. Oncol. 2022, 33, S1423. [Google Scholar] [CrossRef]
- Scherpereel, A.; Fennell, D.A.; Fujimoto, N.; Marmarelis, M.E.; Tsao, A.; Aerts, J.; Li, X.; Dalvi, T.; Jiang, H.; Krug, L.; et al. P2.14B.05 eVOLVE-Meso: A Global Phase 3 Study of First-Line Volrustomig Plus Chemotherapy in Unresectable Pleural Mesothelioma. J. Thorac. Oncol. 2024, 19, S281. [Google Scholar] [CrossRef]
- Randall, L.; Wu, X.; Mayadev, J.; Takekuma, M.; Estevez-Diz, M.D.P.; Shapira-Frommer, R.; Rimel, B.; Lorusso, D.; Lee, J.-Y.; Zhou, Q.; et al. eVOLVE-Cervical (GOG-3092/ENGOT-cx19/GEICO133-C): A prospective phase III study of volrustomig versus placebo after definitive chemoradiation among women with high-risk, locally advanced cervical cancer. Gynecol. Oncol. 2024, 190, S285. [Google Scholar] [CrossRef]
- Sharma, M.; Sanborn, R.E.; Cote, G.M.; Bendell, J.C.; Kaul, S.; Chen, F.; Berezhnoy, A.; Moore, P.; Bonvini, E.; Sumrow, B.J.; et al. 1020O A phase I, first-in-human, open-label, dose escalation study of MGD019, an investigational bispecific PD-1 x CTLA-4 DART® molecule in patients with advanced solid tumours. Ann. Oncol. 2020, 31, S704–S705. [Google Scholar] [CrossRef]
- Luke, J.J.; Sharma, M.; Chandana, S.R.; Lugowska, I.A.; Szczylik, C.; Zolnierek, J.; Cote, G.M.; Mantia, C.; Dziadziuszko, R.; Sanborn, R.E.; et al. Lorigerlimab, a bispecific PD-1×CTLA-4 DART molecule in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): A phase 1 expansion (exp) cohort. J. Clin. Oncol. 2023, 41 (Suppl. S6), 155. [Google Scholar] [CrossRef]
- Li, Q.; Liu, J.; Zhang, Q.; Ouyang, Q.; Zhang, Y.; Liu, Q.; Sun, T.; Ye, F.; Zhang, B.; Xia, S.; et al. The anti-PD-L1/CTLA-4 bispecific antibody KN046 in combination with nab-paclitaxel in first-line treatment of metastatic triple-negative breast cancer: A multicenter phase II trial. Nat. Commun. 2024, 15, 1015. [Google Scholar] [CrossRef]
- Ma, Y.; Xue, J.; Zhao, Y.; Zhang, Y.; Huang, Y.; Yang, Y.; Fang, W.; Guo, Y.; Li, Q.; Ge, X.; et al. Phase I trial of KN046, a novel bispecific antibody targeting PD-L1 and CTLA-4 in patients with advanced solid tumors. J. Immunother. Cancer 2023, 11, e006654. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.; Li, W.; Li, X.; Fan, Y.; Ma, Z.; Fang, J.; Xie, Q.; Zhuang, W.; Kang, M.; Wang, J.; et al. Efficacy and safety of KN046, a novel bispecific antibody against PD-L1 and CTLA-4, in patients with non-small cell lung cancer who failed platinum-based chemotherapy: A phase II study. Eur. J. Cancer 2023, 190, 112936. [Google Scholar] [CrossRef] [PubMed]
- Chapin, W.J.; Agarwal, P.; DiCicco, L.; Palmer, R.; Cenou, N.; Huang, A.C.C.; Karasic, T.B. Phase II trial of XmAb20717 (vudalimab) in patients with advanced biliary tract cancers. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS4184. [Google Scholar] [CrossRef]
- Hickingbottom, B.; Clynes, R.; Desjarlais, J.; Li, C.; Ding, Y. Preliminary safety and pharmacodynamic (PD) activity of XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in a phase I dose escalation study of patients with selected advanced solid tumors. J. Clin. Oncol. 2020, 38 (Suppl. S15), e15001. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Lee, K.; Malhotra, D.; Pryts, S.; Clancy-Thompson, E.; Omar, B.; Naiman, B.; Overstreet, M.; Taylor, D.; Yang, C.; Mulgrew, K.; et al. 469 Preclinical studies support clinical development of AZD2936, a monovalent bispecific humanized antibody targeting PD-1 and TIGIT. J. Immunother. Cancer 2022, 10, A489. [Google Scholar] [CrossRef]
- Rohrberg, K.S.; Brandão, M.; Alvarez, E.C.; Felip, E.; Gort, E.H.; Hiltermann, T.J.J.N.; Izumi, H.; Kim, D.-W.; Kim, S.-W.; Paz-Ares, L.G.; et al. Safety, pharmacokinetics (PK), pharmacodynamics (PD) and preliminary efficacy of AZD2936, a bispecific antibody targeting PD-1 and TIGIT, in checkpoint inhibitor (CPI)-experienced advanced/metastatic non-small-cell lung cancer (NSCLC): First report of ARTEMIDE-01. J. Clin. Oncol. 2023, 41, 9050. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, C.; Liu, Y.; Wang, R.; Feng, C.; Cai, L.; Chang, S.; Zhao, L. A novel dual mechanism-of-action bispecific PD-1-IL-2v armed by a “βγ-only” interleukin-2 variant. Front. Immunol. 2024, 15, 1369376. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, J.; Wang, H.; Zhang, W.; Liang, X.; Cui, J.; Sun, Y.; Fang, M.; Pan, Y.; Chu, Q.; et al. First-in-class PD-1/IL-2 bispecific antibody fusion protein IBI363 in patients with advanced melanoma: Safety and efficacy results from a phase I study. J. Clin. Oncol. 2024, 42 (Suppl. S16), 9562. [Google Scholar] [CrossRef]
- Ryan, C. FDA Grants Fast Track Designation to IBI363 for Squamous NSCLC. Available online: https://www.onclive.com/view/fda-grants-fast-track-designation-to-ibi363-for-squamous-nsclc (accessed on 20 April 2025).
- Wahner, A. IBI363 Receives FDA Fast Track Designation for Advanced/Metastatic Melanoma. Available online: https://www.onclive.com/view/ibi363-receives-fda-fast-track-designation-for-advanced-metastatic-melanoma (accessed on 20 April 2025).
- Akce, M.; Hu-Lieskovan, S.; Reilley, M.; Strauss, J.F.; Specht, J.M.; Stein, M.N.; Wang, J.S.; Choe, J.H.; Leidner, R.; Davar, D.; et al. A phase 1 multiple-ascending dose study to evaluate the safety and tolerability of XmAb23104 (PD-1 x ICOS) in subjects with selected advanced solid tumors (DUET-3). J. Clin. Oncol. 2022, 40 (Suppl. S16), 2604. [Google Scholar] [CrossRef]
- Jacob, S.; Daud, A. Phase Ib/II study of XmAb23104 (PD1 X ICOS) and XmAb22841 (CTLA-4 X LAG3) combination in metastatic melanoma refractory to prior immune checkpoint inhibitor therapy with and without CNS disease. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS9595. [Google Scholar] [CrossRef]
- Luke, J.J.; Patel, M.R.; Blumenschein, G.R.; Hamilton, E.; Chmielowski, B.; Ulahannan, S.V.; Connolly, R.M.; Santa-Maria, C.A.; Wang, J.; Bahadur, S.W.; et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: A phase 1 trial. Nat. Med. 2023, 29, 2814–2824. [Google Scholar] [CrossRef]
- Ren, Z.; Guo, Y.; Bai, Y.; Ying, J.; Meng, Z.; Chen, Z.; Gu, S.; Zhang, J.; Liang, J.; Hou, X.; et al. Tebotelimab, a PD-1/LAG-3 bispecific antibody, in patients with advanced hepatocellular carcinoma who had failed prior targeted therapy and/or immunotherapy: An open-label, single-arm, phase 1/2 dose-escalation and expansion study. J. Clin. Oncol. 2023, 41 (Suppl. S4), 578. [Google Scholar] [CrossRef]
- Lu, S.; Chen, Y.; Fang, M.; Zou, Z.; Wu, D.; Luo, Z.; Zhang, J.; Chen, J.; Huang, G.; Pan, H.; et al. Abstract CT208: Tebotelimab, a PD-1/LAG-3 bispecific antibody, in patients with untreated, unresectable, recurrent or metastatic, mucosal melanoma: An open-label, single-arm, Phase 1 study. Cancer Res. 2023, 83, CT208. [Google Scholar] [CrossRef]
- Ruan, D.-Y.; Wei, X.-L.; Liu, F.-R.; Hu, X.-C.; Zhang, J.; Ji, D.-M.; Huang, D.-Z.; Zhao, Y.-Q.; Pan, H.-M.; Liao, W.-J.; et al. The first-in-class bispecific antibody IBI318 (LY3434172) targeting PD-1 and PD-L1 in patients with advanced tumors: A phase Ia/Ib study. J. Hematol. Amp Oncol. 2024, 17, 118. [Google Scholar] [CrossRef]
- Albu, D.I.; Wolf, B.J.; Qin, Y.; Wang, X.; Daniel Ulumben, A.; Su, M.; Li, V.; Ding, E.; Angel Gonzalo, J.; Kong, J.; et al. A bispecific anti-PD-1 and PD-L1 antibody induces PD-1 cleavage and provides enhanced anti-tumor activity. OncoImmunology 2024, 13, 2316945. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Geng, Q.; Jiao, P. Anti-PD-L1-Based Bispecific Antibodies Targeting Co-Inhibitory and Co-Stimulatory Molecules for Cancer Immunotherapy. Molecules 2024, 29, 454. [Google Scholar] [CrossRef]
- Sauer, N.; Janicka, N.; Szlasa, W.; Skinderowicz, B.; Kołodzińska, K.; Dwernicka, W.; Oślizło, M.; Kulbacka, J.; Novickij, V.; Karłowicz-Bodalska, K. TIM-3 as a promising target for cancer immunotherapy in a wide range of tumors. Cancer Immunol. Immunother. 2023, 72, 3405–3425. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Bivi, N.; Calderon, B.; Shimizu, T.; Delafontaine, B.; Liu, Z.T.; Szpurka, A.M.; Copeland, V.; Hodi, F.S.; Rottey, S.; et al. Safety and Immunogenicity of LY3415244, a Bispecific Antibody Against TIM-3 and PD-L1, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 2773–2781. [Google Scholar] [CrossRef]
- Oldenborg, P.A. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol 2013, 2013, 614619. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Y.; Chu, Q.; Duan, J.; Wan, R.; Wang, Z.; Zhao, J.; Li, H.; Guo, Y.; Chen, Y.; et al. Abstract CT513: Phase I study of IBI322 (anti-CD47/PD-L1 bispecific antibody) monotherapy therapy in patients with advanced solid tumors in China. Cancer Res. 2022, 82, CT513. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, J.; LI, H.; Qian, W.; Xiao, X.; Cai, Q.; Liu, Y.; Zhang, Y.; Zhang, L.; Qin, L.; et al. S216: CD47/PD-L1 Bispecific Antibody (Ibi322) In Anti-Pd-1 Or Pd-L1 Treatment-Resistant Classical Hodgkin Lymphoma: A Phase I Study. HemaSphere 2023, 7, e8102841. [Google Scholar] [CrossRef]
- Garralda, E.; Oberoi, A.; Velasco, G.d.; Victoria, I.; Pesantez, D.; Santamaría, I.E.; Moreno, V.; Boni, V.; Cervantes, A.; Gambardella, V.; et al. First-in-human study (FIH) of FS222, a next-generation tetravalent PD-L1/CD137 bispecific antibody: Safety, pharmacodynamics (PD), and antitumor activity in patients (pts) with advanced solid tumors including PD-1 refractory melanoma. J. Clin. Oncol. 2024, 42 (Suppl. S16), 2505. [Google Scholar] [CrossRef]
- Falchook, G.S.; LoRusso, P.; Goldman, J.W.; El-Khoueiry, A.B.; Tolcher, A.W.; Xing, Y.; Henry, J.T.; Keam, B.; Kim, D.-W.; Kim, T.-Y.; et al. Phase 1 trial safety and efficacy of ragistomig, a bispecific antibody targeting PD-L1 and 4-1BB, in advanced solid tumors. J. Clin. Oncol. 2024, 42 (Suppl. S16), 2529. [Google Scholar] [CrossRef]
- Kyi, C.; Dongen, M.V.; Rottey, S.; Bermejo, I.M.; Mittag, D.; Gouveia, D.; Bol, K.; Yan, C.; Joe, A.K.; Laus, G.; et al. Phase I study of MCLA-145, a bispecific antibody targeting CD137 and PD-L1, in solid tumors, as monotherapy or in combination with pembrolizumab. J. Clin. Oncol. 2024, 42 (Suppl. S16), 2520. [Google Scholar] [CrossRef]
- Xu, T.; Wang, P.; Fu, Y.-X.; Guo, K.; Peng, J. Abstract LB021: KN-052, a novel PDL1/OX40 bispecific antibody, exhibits potent antitumor efficacy. Cancer Res. 2023, 83, LB021. [Google Scholar] [CrossRef]
- Li, B.; Wu, X.; Gong, S.; Lv, Z.; Zhang, N.; Zhang, Y.; Naren, G.; Wu, D.; Wu, J.; Liu, F.; et al. A Novel Immunostimulatory Pd-L1/Ox40 Tetravalent Bispecific Antibody For Cancer Immunotherapy. Antib. Ther. 2023, 6, tbad014.008. [Google Scholar] [CrossRef]
- Qing, Z.; Gabrail, N.; Uprety, D.; Rotow, J.; Han, B.; Jänne, P.A.; Nagasaka, M.; Zheng, M.; Zhang, Y.; Yang, G.; et al. 22P EMB-01: An EGFR-cMET bispecific antibody, in advanced/metastatic solid tumors phase I results. Ann. Oncol. 2022, 33, S39–S40. [Google Scholar] [CrossRef]
- Brandao, M.d.R.A.; Zugazagoitia, J.; Dingemans, A.-M.C.; Duruisseaux, M.; Parent, P.; Juan-Vidal, O.; Spira, A.I.; Vicier, C.; Zalcman, G.; Barasa, B.; et al. Efficacy and safety of MCLA-129, an anti-EGFR/c-MET bispecific antibody, in non-small-cell lung cancer (NSCLC) with c-METexon 14 skipping mutations (METex14). J. Clin. Oncol. 2024, 42 (Suppl. S16), 8583. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, J.; Wu, L.; Chen, B.; Wang, Z.-H.; Yao, Y.; Dong, X.; Li, W.; Ren, X.; Hou, X.; et al. Efficacy and safety of MCLA-129, an EGFR/c-MET bispecific antibody, in advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2024, 42 (Suppl. S16), 8604. [Google Scholar] [CrossRef]
- Cohen, E.E.; Fayette, J.; Daste, A.; Even, C.; Tourneau, C.L.; Brana, I.; Saada, E.; Fontana, E.; Iglesias, L.; Kato, S.; et al. Abstract CT012: Clinical activity of MCLA-158 (petosemtamab), an IgG1 bispecific antibody targeting EGFR and LGR5, in advanced head and neck squamous cell cancer (HNSCC). Cancer Res. 2023, 83, CT012. [Google Scholar] [CrossRef]
- García, M.D.; Hollebecque, A.; Garcia-Carbonero, R.; Jungels, C.; Smyth, E.; Kato, S.; Argilés, G.; Martin, C.G.; Magin, M.; Shen, Y.-M.; et al. Abstract CT156: MCLA-158 (petosemtamab), an IgG1 bispecific antibody targeting EGFR and LGR5, in advanced gastric/esophageal adenocarcinoma (GEA). Cancer Res. 2023, 83, CT156. [Google Scholar] [CrossRef]
- Segal, N.H.; Girda, E.; Sohal, D.; Lakhani, N.J.; Olszanski, A.J.; Fong, L.; Kinnaman, M.D.; Han, H.; Skokos, D.; Casey, K.A.; et al. A phase 1/2 study of REGN7075 in combination with cemiplimab (cemi) in patients (pts) with advanced solid tumors: Efficacy and safety results. J. Clin. Oncol. 2024, 42 (Suppl. S16), 2503. [Google Scholar] [CrossRef]
- Gadea, O.S.S.; Christenson, E.; El-Khoueiry, A.B.; Cervantes, A.; Raab, C.; Gaertner, U.; Pietzko, K.; Hintzen, G.; Ravenstijn, P.; Morales-Espinosa, D.; et al. AFM24 in combination with atezolizumab in patients with advanced EGFR-expressing solid tumors: Phase 1/2a study design and rationale. J. Clin. Oncol. 2022, 40 (Suppl. S16), TPS2673. [Google Scholar] [CrossRef]
- Kim, H.R.; Saavedra, O.; Cervantes, A.; Lugowska, I.A.; Oberoi, A.; El-Khoueiry, A.B.; Thomas, J.S.; Rogowski, W.; Lopez, J.S.; Shim, B.Y.; et al. Preliminary results from the phase 2 study of AFM24 in combination with atezolizumab in patients with EGFR wild-type (EGFR-WT) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2024, 42 (Suppl. S16), 2522. [Google Scholar] [CrossRef]
- Salek-Ardakani, S.; DiRaimondo, T.; Budimir, N.; Ma, L.; Shenhav, S.; Cicchini, V.; Wu, H.; Jocic, R.; Roup, F.; Cambell, C.; et al. 1123 Preclinical activity and safety profile or JANX008, a novel EGFR-targeting tumor-activated T cell engager for treatment of solid tumors. J. Immunother. Cancer 2022, 10, A1167. [Google Scholar] [CrossRef]
- Fang, W.; Zhao, Y.; Luo, Y.; Yang, R.; Huang, Y.; He, Z.; Zhao, H.; Li, M.; Li, K.; Song, Q.; et al. Ivonescimab Plus Chemotherapy in Non-Small Cell Lung Cancer With EGFR Variant: A Randomized Clinical Trial. JAMA 2024, 332, 561–570. [Google Scholar] [CrossRef]
- Wu, Y.L.; Wang, Z.; Cheng, Y.; Fang, J.; Meng, X.; Pan, Y.; Zhao, H.; Zhao, Y.; Su, H.; Sun, M.; et al. 1255MO A phase II safety and efficacy study of PM8002/BNT327 in combination with chemotherapy in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Ann. Oncol. 2024, 35, S804. [Google Scholar] [CrossRef]
- Lobov, I.B.; Renard, R.A.; Papadopoulos, N.; Gale, N.W.; Thurston, G.; Yancopoulos, G.D.; Wiegand, S.J. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2007, 104, 3219–3224. [Google Scholar] [CrossRef]
- Gordon, M.S.; Nemunaitis, J.; Barve, M.; Wainberg, Z.A.; Hamilton, E.P.; Ramanathan, R.K.; Sledge, G.W.; Yue, H.; Morgan-Lappe, S.E.; Blaney, M.; et al. Phase I Open-Label Study Evaluating the Safety, Pharmacokinetics, and Preliminary Efficacy of Dilpacimab in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2021, 20, 1988–1995. [Google Scholar] [CrossRef]
- Strickler, J.H.; Cubillo, A.; Liang, J.-T.; Matrana, M.; Kozloff, M.; Lowe, T.; Blaney, M.; Sahtout, M.; Naumovski, L.; Wainberg, Z.A. Efficacy and Safety of Dilpacimab (ABT-165) versus Bevacizumab Plus Folfiri in Metastatic Colorectal Cancer: A Phase II Study. Future Oncol. 2022, 18, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Invest New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Corr, B.R.; Culm-Merdek, K.; Mockbee, C.; Youssoufian, H.; Stagg, R.; Naumann, R.W.; Wenham, R.M.; Rosengarten, R.D.; Benjamin, L.; et al. Phase Ib Study of Navicixizumab Plus Paclitaxel in Patients With Platinum-Resistant Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. J. Clin. Oncol. 2022, 40, 2568–2577. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Park, J.O.; Kim, J.W.; Kim, K.-P.; Yoon, J.; Kim, T.-Y.; Kim, S.T.; Park, Y.S.; Lee, J.; Kim, J.-W.; et al. CTX-009 (ABL001), a bispecific antibody targeting DLL4 and VEGF A, in combination with paclitaxel in patients with advanced biliary tract cancer (BTC): A phase 2 study. J. Clin. Oncol. 2023, 41 (Suppl. S4), 540. [Google Scholar] [CrossRef]
- Fontaine, M.; Pilgrim, S.; Schuetz, T. Trial in progress: A phase 2/3 randomized, controlled study of CTX-009 in combination with paclitaxel versus paclitaxel alone in adult patients with unresectable advanced, metastatic or recurrent biliary tract cancers who have received one prior systemic chemotherapy regimen. J. Clin. Oncol. 2023, 41 (Suppl. S4), TPS640. [Google Scholar] [CrossRef]
- Sait, S.; Modak, S. Anti-GD2 immunotherapy for neuroblastoma. Expert. Rev. Anticancer. Ther. 2017, 17, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Yankelevich, M.; Thakur, A.; Modak, S.; Chu, R.; Taub, J.; Martin, A.; Schalk, D.; Schienshang, A.; Whitaker, S.; Rea, K.; et al. Targeting refractory/recurrent neuroblastoma and osteosarcoma with anti-CD3×anti-GD2 bispecific antibody armed T cells. J. Immunother. Cancer 2024, 12, e008744. [Google Scholar] [CrossRef]
- Moore, K.N.; Bouberhan, S.; Hamilton, E.P.; Liu, J.F.; O’Cearbhaill, R.E.; O’Malley, D.M.; Papadamitriou, K.; Schröder, D.; Nieuwenhuysen, E.V.; Yoo, S.-Y.; et al. First-in-human phase 1/2 study of ubamatamab, a MUC16xCD3 bispecific antibody, administered alone or in combination with cemiplimab in patients with recurrent ovarian cancer. J. Clin. Oncol. 2023, 41 (Suppl. S16), TPS5624. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Hong, L.-L.; Ling, Z.-Q. MUC16: Clinical targets with great potential. Clin. Exp. Med. 2024, 24, 101. [Google Scholar] [CrossRef]
- Crawford, A.; Haber, L.; Kelly, M.P.; Vazzana, K.; Canova, L.; Ram, P.; Pawashe, A.; Finney, J.; Jalal, S.; Chiu, D.; et al. A Mucin 16 bispecific T cell-engaging antibody for the treatment of ovarian cancer. Sci. Transl. Med. 2019, 11, 497. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuysen, E.V.; Bouberhan, S.; Papadimitriou, K.; Arend, R.C.; Lee, J.-Y.; O’Cearbhaill, R.E.; O’Malley, D.M.; You, B.; Martin, A.G.; Michels, J.; et al. A phase 1/2 study of ubamatamab (REGN4018), a MUC16×CD3 bispecific antibody, administered alone or in combination with cemiplimab (anti–PD-1) in patients with recurrent ovarian cancer or MUC16+ endometrial cancer. J. Clin. Oncol. 2024, 42 (Suppl. S16), TPS5632. [Google Scholar] [CrossRef]
- Stern, P.L.; Harrop, R. 5T4 oncofoetal antigen: An attractive target for immune intervention in cancer. Cancer Immunol Immunother 2017, 66, 415–426. [Google Scholar] [CrossRef]
- Nelson, M.H.; Fritzell, S.; Miller, R.; Werchau, D.; Van Citters, D.; Nilsson, A.; Misher, L.; Ljung, L.; Bader, R.; Deronic, A.; et al. The Bispecific Tumor Antigen-Conditional 4-1BB x 5T4 Agonist, ALG.APV-527, Mediates Strong T-Cell Activation and Potent Antitumor Activity in Preclinical Studies. Mol. Cancer Ther. 2023, 22, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Getu, A.A.; Tigabu, A.; Zhou, M.; Lu, J.; Fodstad, Ø.; Tan, M. New frontiers in immune checkpoint B7-H3 (CD276) research and drug development. Mol. Cancer 2023, 22, 43. [Google Scholar] [CrossRef]
- Bupathi, M.; Wang, J.S.; Hu-Lieskovan, S.; Piha-Paul, S.A.; Chmielowski, B.; Garmezy, B.; Najjar, Y.G.; Stein, M.N.; Yao, L.; Kanodia, J.; et al. 764 A phase 1, first-in-human (FIH), open-label, dose-finding and expansion study of XmAb808, a B7H3 x CD28 bispecific antibody, in combination with pembrolizumab in patients with advanced solid tumors. J. Immunother. Cancer 2023, 11, A859. [Google Scholar] [CrossRef]
- Holzmayer, S.J.; Liebel, K.; Hagelstein, I.; Salih, H.R.; Märklin, M. The bispecific B7H3xCD3 antibody CC-3 induces T cell immunity against bone and soft tissue sarcomas. Front. Immunol. 2024, 15, 1391954. [Google Scholar] [CrossRef] [PubMed]
- Zekri, L.; Lutz, M.; Prakash, N.; Manz, T.; Klimovich, B.; Mueller, S.; Hoerner, S.; Hagelstein, I.; Engel, M.; Chashchina, A.; et al. An optimized IgG-based B7-H3xCD3 bispecific antibody for treatment of gastrointestinal cancers. Mol. Ther. 2023, 31, 1033–1045. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—an excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef]
- Barlesi, F.; Isambert, N.; Felip, E.; Cho, B.C.; Lee, D.H.; Peguero, J.; Jerusalem, G.; Penel, N.; Saada-Bouzid, E.; Garrido, P.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Non-Small Cell Lung Cancer Resistant or Refractory to Immune Checkpoint Inhibitors. Oncologist 2023, 28, 258–267. [Google Scholar] [CrossRef]
- Cho, B.C.; Daste, A.; Ravaud, A.; Salas, S.; Isambert, N.; McClay, E.; Awada, A.; Borel, C.; Ojalvo, L.S.; Helwig, C.; et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in advanced squamous cell carcinoma of the head and neck: Results from a phase I cohort. J. Immunother. Cancer 2020, 8, e000664. [Google Scholar] [CrossRef]
- Birrer, M.; Li, G.; Yunokawa, M.; Lee, J.-Y.; Kim, B.G.; Oppermann, C.P.; Zhou, Q.; Nishio, S.; Okamoto, A.; Wu, X.; et al. Bintrafusp Alfa for Recurrent or Metastatic Cervical Cancer After Platinum Failure. JAMA Oncol. 2024, 10, 1204. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Lee, J.S.; Wu, Y.-L.; Cicin, I.; Dols, M.C.; Ahn, M.-J.; Cuppens, K.; Veillon, R.; Nadal, E.; Dias, J.M.; et al. Bintrafusp Alfa Versus Pembrolizumab in Patients with Treatment-Naive, Programmed Death-Ligand 1–High Advanced NSCLC: A Randomized, Open-Label, Phase 3 Trial. J. Thorac. Oncol. 2023, 18, 1731–1742. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Zhou, T.; Chen, G.; Huang, Y.; Liu, F.; Liu, Z.; Qu, S.; Lei, Y.; Chen, X.; et al. A phase Ib study of SHR-1701, a bifunctional fusion protein targeting PD-L1 and TGF-β, in patients with recurrent or metastatic nasopharyngeal carcinoma (RM-NPC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 6024. [Google Scholar] [CrossRef]
- Yi, L.; Pan, H.; Ning, Z.; Xu, L.; Zhang, H.; Peng, L.; Liu, Y.; Yang, Y.; Si, W.; Wang, Y.; et al. Clinical and biomarker analyses of SHR-1701 combined with famitinib in patients with previously treated advanced biliary tract cancer or pancreatic ductal adenocarcinoma: A phase II trial. Signal Transduct. Target. Ther. 2024, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tang, D.; Wang, J.; Zhou, Q.; Peng, J.; Lou, H.; Sun, Y.; Cai, Y.; Chen, H.; Yang, J.; et al. SHR-1701, a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, for Recurrent or Metastatic Cervical Cancer: A Clinical Expansion Cohort of a Phase I Study. Clin. Cancer Res. 2022, 28, 5297–5305. [Google Scholar] [CrossRef]
- Zhou, Q.; Pan, Y.; Yang, X.; Zhao, Y.; Han, G.; Pang, Q.; Zhang, Z.; Wang, Q.; Yao, J.; Wang, H.; et al. Neoadjuvant SHR-1701 with or without chemotherapy in unresectable stage III non-small-cell lung cancer: A proof-of-concept, phase 2 trial. Cancer Cell 2024, 42, 1258–1267.e2. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Seth, P.; Winkis, A.; Chevalier, K.; Marthaler, A.; Sproesser, K.; Torti, V.; Shah, N.; Lacy, E.; Frisk, A.-L.; et al. Abstract LB122: JNJ-87890387, a novel ENPP3 bispecific T-cell redirector (ENPP3xCD3) with tumor selectivity through targeting apical surface antigens. Cancer Res. 2024, 84, LB122. [Google Scholar] [CrossRef]
- Pal, S.; Yalamanchili, S.; Li, H.; Kanodia, J.; Clynes, R.; Tang, Z.; Garmezy, B. 667 A phase 1, multiple-dose study to evaluate the safety and tolerability of XmAb®819 (ENPP3 x CD3) in subjects with relapsed or refractory clear cell renal cell carcinoma (RCC). J. Immunother. Cancer 2022, 10, A697. [Google Scholar] [CrossRef]
- Malla, M.; Deshmukh, S.K.; Wu, S.; Samec, T.; Olevian, D.C.; El Naili, R.; El-Rayes, B.; Xiu, J.; Farrell, A.; Lenz, H.-J.; et al. Mesothelin expression correlates with elevated inhibitory immune activity in patients with colorectal cancer. Cancer Gene Ther. 2024, 31, 1547–1558. [Google Scholar] [CrossRef]
- Kurtenbach, S.; Sanchez, M.I.; Kuznetsoff, J.; Rodriguez, D.A.; Weich, N.; Dollar, J.J.; Cruz, A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; et al. PRAME induces genomic instability in uveal melanoma. Oncogene 2024, 43, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Bunk, S.; Hofmann, M.; Pszolla, G.; Hutt, M.; Schwöbel, F.; Unverdorben, F.; Aschmoneit, N.; Wagner, C.; Jaworski, M.; Schuster, H.; et al. IMA402, an Off-the-Shelf, Next-Generation TCR Bispecific (TCER®) for Efficiently Targeting an HLA-Presented Peptide from the Pan-Cancer Antigen PRAME. Blood 2022, 140, 9089–9090. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Qin, X.; Ning, W.; Liu, H.; Liu, X.; Luo, W.; Xia, N. Stepping forward: T-cell redirecting bispecific antibodies in cancer therapy. Acta Pharm. Sin. B 2024, 14, 2361–2377. [Google Scholar] [CrossRef]
- Crawford, A.; Chiu, D. Targeting Solid Tumors Using CD3 Bispecific Antibodies. Mol. Cancer Ther. 2021, 20, 1350–1358. [Google Scholar] [CrossRef]
- Guidi, L.; Etessami, J.; Valenza, C.; Valdivia, A.; Meric-Bernstam, F.; Felip, E.; Curigliano, G. Bispecific Antibodies in Hematologic and Solid Tumors: Current Landscape and Therapeutic Advances. Am. Soc. Clin. Oncol. Educ. Book 2025, 45, e473148. [Google Scholar] [CrossRef]
- Moore, P.A.; Shah, K.; Yang, Y.; Alderson, R.; Roberts, P.; Long, V.; Liu, D.; Li, J.C.; Burke, S.; Ciccarone, V.; et al. Development of MGD007, a gpA33 x CD3-Bispecific DART Protein for T-Cell Immunotherapy of Metastatic Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Jarantow, S.W.; Bushey, B.S.; Pardinas, J.R.; Boakye, K.; Lacy, E.R.; Sanders, R.; Sepulveda, M.A.; Moores, S.L.; Chiu, M.L. Impact of Cell-surface Antigen Expression on Target Engagement and Function of an Epidermal Growth Factor Receptor × c-MET Bispecific Antibody. J. Biol. Chem. 2015, 290, 24689–24704. [Google Scholar] [CrossRef]
- Hinner, M.J.; Aiba, R.S.B.; Jaquin, T.J.; Berger, S.; Dürr, M.C.; Schlosser, C.; Allersdorfer, A.; Wiedenmann, A.; Matschiner, G.; Schüler, J.; et al. Tumor-Localized Costimulatory T-Cell Engagement by the 4-1BB/HER2 Bispecific Antibody-Anticalin Fusion PRS-343. Clin Cancer Res 2019, 25, 5878–5889. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, X.; Wang, X.; Yuan, K.; Wang, G.; Hu, L.; Zhang, G.; Pei, W.; Wang, L.; Sun, C.; et al. Bispecific antibodies in cancer therapy: Target selection and regulatory requirements. Acta Pharm. Sin. B 2023, 13, 3583–3597. [Google Scholar] [CrossRef]
- Huang, S.; van Duijnhoven, S.M.J.; Sijts, A.; van Elsas, A. Bispecific antibodies targeting dual tumor-associated antigens in cancer therapy. J. Cancer Res. Clin. Oncol. 2020, 146, 3111–3122. [Google Scholar] [CrossRef] [PubMed]
- Geiger, M.; Stubenrauch, K.G.; Sam, J.; Richter, W.F.; Jordan, G.; Eckmann, J.; Hage, C.; Nicolini, V.; Freimoser-Grundschober, A.; Ritter, M.; et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat. Commun. 2020, 11, 3196. [Google Scholar] [CrossRef] [PubMed]
- Weddell, J. Mechanistically modeling peripheral cytokine dynamics following bispecific dosing in solid tumors. CPT Pharmacomet. Syst. Pharmacol. 2023, 12, 1726–1737. [Google Scholar] [CrossRef]
- Leclercq-Cohen, G.; Steinhoff, N.; Alberti Servera, L.; Nassiri, S.; Danilin, S.; Piccione, E.; Yanguez, E.; Husser, T.; Herter, S.; Schmeing, S.; et al. Dissecting the Mechanisms Underlying the Cytokine Release Syndrome (CRS) Mediated by T-Cell Bispecific Antibodies. Clin. Cancer Res. 2023, 29, 4449–4463. [Google Scholar] [CrossRef]
- Kamakura, D.; Asano, R.; Yasunaga, M. T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity. Pharmaceuticals 2021, 14, 1172. [Google Scholar] [CrossRef] [PubMed]
- Crombie, J.L.; Graff, T.; Falchi, L.; Karimi, Y.H.; Bannerji, R.; Nastoupil, L.; Thieblemont, C.; Ursu, R.; Bartlett, N.; Nachar, V.; et al. Consensus recommendations on the management of toxicity associated with CD3xCD20 bispecific antibody therapy. Blood 2024, 143, 1565–1575. [Google Scholar] [CrossRef]
- Ludwig, H.; Terpos, E.; van de Donk, N.; Mateos, M.V.; Moreau, P.; Dimopoulos, M.A.; Delforge, M.; Rodriguez-Otero, P.; San-Miguel, J.; Yong, K.; et al. Prevention and management of adverse events during treatment with bispecific antibodies and CAR T cells in multiple myeloma: A consensus report of the European Myeloma Network. Lancet Oncol 2023, 24, e255–e269. [Google Scholar] [CrossRef]
- Cobb, D.A.; Lee, D.W. Cytokine Release Syndrome Biology and Management. Cancer J. 2021, 27, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Middelburg, J.; Kemper, K.; Engelberts, P.; Labrijn, A.F.; Schuurman, J.; van Hall, T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers 2021, 13, 287. [Google Scholar] [CrossRef]
- Li, H.; Er Saw, P.; Song, E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol. Immunol. 2020, 17, 451–461. [Google Scholar] [CrossRef]
- Miao, L.; Newby, J.M.; Lin, C.M.; Zhang, L.; Xu, F.; Kim, W.Y.; Forest, M.G.; Lai, S.K.; Milowsky, M.I.; Wobker, S.E.; et al. The Binding Site Barrier Elicited by Tumor-Associated Fibroblasts Interferes Disposition of Nanoparticles in Stroma-Vessel Type Tumors. ACS Nano 2016, 10, 9243–9258. [Google Scholar] [CrossRef] [PubMed]
- Barakzai, S.; Xu, M.; Veillard, I.; Matoba, Y.; Kononenko, A.; Kim, E.; Bregar, A.; Eisenhauer, E.L.; Penson, R.T.; Bouberhan, S.; et al. Mechanisms of resistance to bispecific T-cell engager therapy for ovarian cancer. J. Clin. Oncol. 2023, 41 (Suppl. S16), 5571. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Hu, S.; Fu, W.; Xu, W.; Yang, Y.; Cruz, M.; Berezov, S.D.; Jorissen, D.; Takeda, H.; Zhu, W. Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk. Cancer Res. 2015, 75, 159–170. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Trial | Patient Population | Number of Patients | Treatment Arm | mOS (Months) | mPFS (Months) | ORR (%) | mDOR (Months) | FDA Approval |
|---|---|---|---|---|---|---|---|---|
| IMCgp100-202 Phase III [18] | Previously untreated metastatic uveal melanoma with HLA-A*02:01 | 252 | tebentafusp | 21.6 | 3.4 | 11 | 11.1 | 25 January 2022 |
| 126 | investigator choice (control group): pembrolizumab, ipilimumab or dacarbazine | 16.9 | 2.9 | 5 | 9.7 | |||
| PAPILLON Phase III [21] | Advanced NSCLC with EGFR exon 20 insertions without prior systemic treatment | 153 | amivantamab-chemotherapy | NE | 11.4 | 73 | 9.7 | 1 March 2024 |
| 155 | chemotherapy | 24.4 | 6.7 | 47 | 4.4 | |||
| CHRYSALIS Phase I [22] | Advanced or metastatic NSCLC with EGFR exon 20 insertions who progressed on or after platinum-based chemotherapy. | 81 | amivantamab | 22.8 | 8.3 | 40 | 11.1 | 21 May 2021 |
| MARIPOSA Phase III [23,24] | Locally advanced or metastatic NSCLC with EGFR exon 19 deletions or exon 20 L858R mutations | 429 | amivantamab-lazertinib | NE | 23.7 | 86 | 25.8 | 20 August 2024 |
| 429 | osimertinib | 36.7 | 16.6 | 85 | 16.8 | |||
| 216 | lazertinib | - | - | - | - | |||
| DeLLphi-301 Phase II [25] | Extensive-stage SCLC with disease progression after platinum-based chemotherapy | 100 (10 mg) | tarlatamab | 14.3 | 4.9 | 40 | - | 16 May 2024 |
| HERIZON-BTC-01 Phase IIb [26,27] | HER2-amplified, unresectable, locally advanced or mBTC patients who progressed on prior gemcitabine therapy | 80 | zanidatamab | 15.5 | 5.5 | 41.3 | 12.9 | 20 November 2024 |
| eNRGy Phase II [28] | Advanced or metastatic NRG1 fusion-positive NSCLC patients who had disease progression following standard-of-care treatment | 65 | zenocutuzumab | - | - | 34 | 12.9 | 4 December 2024 |
| eNRGy Phase II [29] | Advanced or metastatic NRG1 fusion-positive pancreatic adenocarcinoma patients who had disease progression following standard-of-care treatment | 27 | zenocutuzumab | - | - | 44 | 9.1 | 4 December 2024 |
| Target | Agent | Phase | Clinical Trial | Status | Conditions |
|---|---|---|---|---|---|
| Gp100 x CD3 | Tebentafusp | II | NCT06070012 | R | HLA-A*02:01-positive previously untreated metastatic uveal melanoma |
| II | NCT06246149 | R | HLA-A*02:01-positive high-risk uveal melanoma following definitive treatment (as adjuvant treatment) | ||
| II | NCT06414590 | R | Large surgically unresectable (other than complete enucleation of eye) primary uveal melanoma (as neoadjuvant treatment) | ||
| III | NCT05549297 | R | HLA-A*02:01-positive patients with previously treated advanced melanoma (TEBE-AM) | ||
| EGFR x cMET | Amivantamab | I/II | NCT06385080 | R | Recurrent/metastatic head and neck squamous cell carcinoma (HNSCC) |
| I/II | NCT05845671 | R | Advanced NSCLC harboring ALK, ROS1 and RET gene mutations (in combination with TKI) | ||
| II | NCT05299125 | A, NR | Recurrent/metastatic NSCLC with EGFR mutations (in combination with lazertinib and pemetrexed) | ||
| III | NCT06662786 | R | KRAS/NRAS/BRAF wild-type unresectable or metastatic left-sided CRC (in combination with mFOLFOX6 or FOLFIRI) as first line | ||
| III | NCT06750094 | R | Previously treated KRAS/NRAS/BRAF wild-type colon cancer (in combination with FOLFIRI) | ||
| I/II | NCT06083857 | R | MET-altered NSCLC (in combination with tepotinib | ||
| I | NCT06632236 | R | High-grade malignant brain tumors with EGFR amplification | ||
| I/II | NCT05379595 | R | Advanced or metastatic CRC | ||
| II | NCT06667076 | R | EGFR-mutated locally advanced or metastatic NSCLC (in combination with lazertinib compared to amivantamab plus platinum-based chemotherapy) | ||
| DLL3 x CD3 | Tarlatamab | III | NCT06211036 | R | ES-SCLC following treatment with platinum, etoposide and durvalumab |
| III | NCT06117774 | R | LS-SCLC who have not progressed following concurrent chemoradiation | ||
| II | NCT06788938 | R | Advanced DLL3-expressing tumors including neuroendocrine neoplasms | ||
| HER2 x HER3 | Zenocutuzumab | II | NCT02912949 | A, NR | Solid tumors harboring an NRG1 fusion |
| HER2 (2 distinct domains) | Zanidatamab | III | NCT06282575 | R | Advanced HER2-positive BTC (in combination with chemotherapy with or without PD-1/PDL1 inhibitor) |
| II | NCT06043427 | R | HER2-positive advanced GEJ adenocarcinoma who failed at least one prior trastuzumab-containing regimen (in combination with paclitaxel and ramucirumab) | ||
| II | NCT05035836 | R | Early-stage HER2-positive breast cancer | ||
| III | NCT06435429 | R | Metastatic HER2-positive breast cancer | ||
| HER2 x CD3 | Runimotamab (RG6194) | I | NCT03448042 | A, NR | Metastatic HER2-positive breast cancer |
| HER2 (2 distinct domains) | KN026 | II | NCT05985707 | NYR | HER2-positive colorectal and biliary carcinoma |
| III | NCT06747338 | NYR | Early or locally advanced HER2-positive breast cancer (in combination with HB1801 as neoadjuvant treatment) | ||
| II/III | NCT05427383 | R | HER2-positive gastric cancer patients who failed first-line therapy | ||
| HER2 x 4-1BB | YH32367 (ABL105) | I/II | NCT05523947 | R | HER2-positive locally advanced or metastatic solid tumors |
| HER3 x EGFR | Izalontamab (SI-B001) | III | NCT05943795 | R | Previously treated NSCLC (adenocarcinoma and squamous cell carcinoma) in combination with docetaxel |
| II | NCT06668961 | R | Recurrent or metastatic HNSCC (in combination with SI-B003 and platinum-based chemotherapy) | ||
| II | NCT05054439 | R | Recurrent and metastatic HNSCC (in combination with paclitaxel) | ||
| PSMA x CD3 | Acapatamab (AMG 160) | Discontinued | |||
| PSMA x CD3 | AMG 340 | I | NCT04740034 | Completed | mCRPC |
| PSMA x CD3 | JNJ-081/JNJ63898081 | I | NCT03926013 | Completed | Advanced solid tumors |
| PSMA x CD28 | REGN5678 | I/II | NCT03972657 | R | mCRPC and other tumors (in combination with cemiplimab) |
| CLDN18.2 x CD3 | Gresonitamab (AMG 910) | I | NCT04260191 | Terminated | Claudin 18.2-positive gastric and GEJ adenocarcinoma |
| CLDN18.2 x CD3 | AZD5863 | I/II | NCT06005493 | R | Advanced or metastatic solid tumors |
| CLDN18.2 x CD3 | LNF2007 | I | NCT06752447 | NYR | Advanced solid tumors |
| CLDN18.2 x CD3 | ASP2138 | I | NCT05365581 | R | CLDN 18.2-positive advanced gastric/GEJ or pancreatic cancer |
| CLDN18.2 x PD-L1 | Q-1802 | I | NCT04856150 | R | CLDN18.2-expressing advanced or metastatic solid tumors |
| I/II | NCT05964543 | R | Advanced or recurrent metastatic CLDN18.2-positive primary gastric/GEJ adenocarcinoma (in combination with XELOX) | ||
| CEA x CD3 | Cibisatamab (RO6958688) | I/II | NCT03337698 | A, NR | Metastatic NSCLC |
| CEA x CD3 | MEDI565 (AMG211) | I | NCT02291614 | Terminated | Advanced gastrointestinal cancer |
| EpCAM x CD3 | Catumaxomab | Discontinued | |||
| EpCAM x CD3 | Solitomab (AMG 110) | No current clinical trials | |||
| EpCAM x CD3 | BA3182 | I | NCT05808634 | R | Advanced adenocarcinoma |
| EpCAM x 4-1BB | BNT314/GEN1059 | I/II | NCT06150183 | R | Metastatic or advanced malignant solid tumors |
| GPC3 x CD3 | ERY974 | I | NCT05022927 | A, NR | Locally advanced or metastatic HCC |
| GPC3 x CD3 | SAR-4442000 | I/II | NCT05450562 | A, NR | Advanced solid tumors (alone or in combination with atezolizumab) |
| HLA-G x CD3 | JNJ-78306358 | No current clinical trials | |||
| PD-1 x CTLA4 | Cadonilimab (AK104) | II | NCT05932212 | R | Recurrent or metastatic vulvar cancer (alone or in combination with chemotherapy) |
| III | NCT06566755 | R | RAS-mutated or right-sided metastatic microsatellite stable CRC (in combination with chemotherapy and bevacizumab) | ||
| II | NCT06448910 | R | Locally advanced unresectable stage III NSCLC (concurrent chemoradiotherapy) | ||
| III | NCT05489289 | R | Adjuvant therapy in HCC with high-risk recurrence after curative resection | ||
| II/III | NCT06241599 | R | Recurrent or metastatic nasopharyngeal carcinoma (in combination with chemotherapy) | ||
| PD-1 x CTLA4 | Volrustomig (MEDI5752) | III | NCT06097728 | R | Unresectable pleural mesothelioma (in combination with carboplatin and pemetrexed) |
| III | NCT06079671 | R | High-risk locally advanced cervical cancer (FIGO stage IIIC to IVA) | ||
| I | NCT05821231 | R | Metastatic soft tissue sarcoma (in combination with radiation) | ||
| III | NCT06129864 | R | Unresected locally advanced HNSCC patients who have not progressed after receiving definitive concurrent chemoradiotherapy | ||
| III | NCT05984277 | R | mNSCLC and PD-L1 < 50% (in combination with chemotherapy) (eVOLVE-Lung02 trial) | ||
| PD-1 x CTLA4 | Lorigerlimab (MGD019) | II | NCT05848011 | A, NR | mCRPC (in combination with docetaxel) |
| II | NCT05475171 | R | Advanced or metastatic cervical cancer | ||
| II | NCT06730347 | R | Previously treated patients with platinum-resistant ovarian cancer or clear-cell gynecologic cancer | ||
| PD-L1 x CTLA4 | Erfonrilimab (KN046) | II/III | NCT06020352 | R | Neoadjuvant therapy in stage IB-IIIB NSCLC (in combination with axitinib) |
| II | NCT06099821 | R | MSI-H gastrointestinal cancers resistant to PD-1/PDL1 (in combination with regorafenib or apatinib) | ||
| PD-L1 x CTLA4 | Vudalimab (XmAb20717) | II | NCT05005728 | R | mCRPC who progressed on prior therapy (alone or in combination with chemotherapy) |
| I/II | NCT06173505 | R | Advanced NSCLC | ||
| PD-1 x TIGIT | Rilvegostomig (AZD2936) | III | NCT06627647 | R | Metastatic NSCLC patients whose tumors express PD-L1 (≥1%) |
| PD-1 x IL-2 | IBI363 | I | NCT05460767 | R | Advanced solid tumors or lymphoma |
| II | NCT06281678 | R | Advanced solid malignancies (melanoma, NSCLC, CRC, RCC) | ||
| II | NCT06081920 | R | Advanced melanoma | ||
| I | NCT06610799 | R | Advanced or metastatic gastric/GEJ cancer (in combination with capecitabine and oxaliplatin) | ||
| II | NCT06797297 | R | Unresectable or metastatic mucosal or acral melanoma without prior systemic therapy | ||
| PD-1 x ICOS | XmAb23104 | Terminated | No current clinical trials | ||
| PD-1× LAG-3 | Tebotelimab (MGD013) | Terminated | No current clinical trials | ||
| PD-L1 x LAG-3 | ABL501 | No current clinical trials | |||
| PD-L1 x PD-1 | LY3434172 (IBI318) | II | NCT04777084 | R | Advanced NSCLC patients who failed first-line PD-1/PD-L1 inhibitor therapy, advanced NSCLC with EGFR-sensitive mutation/ALK fusion after EGFR-TKI/ALK-TKI treatment resistance, and advanced NSCLC with negative PD-L1 expression EGFR, ALK and ROS1 wild type |
| PD-L1 x PD-1 | CTX8371 | I | NCT06150664 | R | Advanced malignancies |
| PD-L1/TIM-3 | LY3415244 | I | NCT03752177 | Terminated | Advanced solid tumors |
| CD47 x PD-L1 | IBI322 | II | NCT05296603 | R | ES-SCLC patients who failed first-line PD-L1 inhibitors (in combination with lenvatinib) |
| PD-L1 x 4-1BB | FS222 | I | NCT04740424 | R | Previously treated patients with advanced tumors |
| PD-L1 x 4-1BB | MCLA145 | No current clinical trials | |||
| PD-L1 x 4-1BB | ATG101 | I | NCT04986865 | R | Metastatic/advanced solid tumors and mature B cell non-Hodgkin lymphoma |
| PD-L1 x 4-1BB | ABL503 | I | NCT04762641 | R | Locally advanced or metastatic solid tumors |
| PD-L1 x OX40 | KN051 | I | NCT05309512 | Terminated | Advanced solid tumors |
| PD-L1 x OX40 | EMB-09 | I | NCT05263180 | R | Advanced or metastatic solid tumors |
| EGFR x cMET | Bafisontamab (EMB01) | I/II | NCT05498389 | NYR | EGFR mutant NSCLC who progressed on standard treatment |
| I/II | NCT05176665 | R | Advanced or metastatic gastrointestinal cancer (gastric cancer, HCC, cholangiocarcinoma and CRC) | ||
| I/II | NCT03797391 | R | Previously treated EGFR and/or cMET mutated advanced or metastatic solid tumors | ||
| EGFR x cMET | MCLA-129 | I/II | NCT04868877 | R | Advanced NSCLC and other solid tumors |
| EGFR x LGR5 | Petosemtamab (MCLA158) | III | NCT06525220 | R | PD-L1-positive HNSCC |
| III | NCT06496178 | R | Previously treated HNSCC | ||
| EGFR x CD28 | REGN7075 | II | NCT06465329 | R | Operable stage II-IIIB NSCLC |
| EGFR x CD16A | AFM24 | I/II | NCT05109442 | R | EGFR-expressing advanced solid tumors (in combination with atezolizumab) |
| EGFR x CD3 | JANX008 | I | NCT05783622 | R | Advanced or metastatic solid tumor malignancies |
| EGFR x CD3 | CX-904 | I | NCT05387265 | R | Advanced solid tumors |
| VEGFA x PD-1 | Ivonescimab (AK112) | III | NCT06767514 | R | Metastatic NSCLC with high PD-L1 |
| II | NCT06567314 | R | Cutaneous squamous cell carcinoma | ||
| II | NCT06925724 | R | Endometrial and cervical cancers | ||
| II | NCT06848842 | R | Unresectable CRC | ||
| II | NCT06375486 | R | Unresectable HCC | ||
| VEGFA x PD-1 | PM8002 | II | NCT05918107 | R | Unresectable malignant mesothelioma |
| II | NCT05879055 | R | Previously treated neuroendocrine neoplasm | ||
| III | NCT06616532 | R | Previously treated SCLC (in combination with paclitaxel) | ||
| DLL4 x VEGF | Dilpacimab (ABT165) | No current clinical trials | |||
| DLL4 x VEGF | Navicixizumab (OMP-305B83) | II | NCT05453825 | Unknown | Advanced solid tumors |
| III | NCT05043402 | Unknown | Platinum-resistant advanced epithelial ovarian cancer and specific biomarkers who progressed on 2 prior lines | ||
| DLL4 x VEGF | CTX009 (ABL001) | II/III | NCT05506943 | A, NR | Unresectable advanced, metastatic or recurrent BTCs (in combination with paclitaxel) |
| I/II | NCT06548412 | R | Unresectable or metastatic BTC (in combination with gemcitabine, cisplatin and durvalumab) | ||
| GD2 x CD3 | Nivatrotamab | I/II | NCT03860207 | Terminated | Relapsed/refractory neuroblastoma, osteosarcoma and other solid tumor cancers |
| I/II | NCT04750239 | Terminated | Relapsed and recurrent metastatic SCLC | ||
| MUC16 x CD3 | Ubamatamab (REGN4018) | I/II | NCT03564340 | R | Recurrent ovarian cancer or MUC16-positive cancers |
| 5T4 x 4-1BB | ALG.APV-527 | I/II | NCT05934539 | R | Advanced solid tumors who failed standard treatments |
| B7-H3 x CD28 | XmAb808 | I | NCT05585034 | A, NR | Advanced solid tumors (in combination with pembrolizumab) |
| B7-H3 x CD3 | CC-3 | I | NCT05999396 | R | Metastatic CRC |
| TGF-β x PDL1 | Bintrafusp alfa | II | NCT04874311 | R | Advanced sarcoma |
| II | NCT04417660 | R | Thymoma and thymic carcinoma | ||
| NA | NCT04481256 | R | Esophageal or GEJ squamous cell carcinoma | ||
| TGF-β x PDL1 | SHR-1701 | II | NCT05106023 | R | Advanced melanoma (in combination with temozolomide) |
| II | NCT05300269 | R | Locally advanced rectal cancer | ||
| ENPP3 x CD3 | XmAb-819 | I | NCT05433142 | R | Advanced solid tumors |
| ENPP3 x CD3 | JNJ-87890387 | I | NCT06178614 | R | Advanced solid tumors |
| MSLN x CD3 | JNJ-79032421 | I | NCT06255665 | R | Advanced solid tumors |
| PRAME x CD3 | IMA-402 | I/II | NCT05958121 | R | Recurrent or refractory solid tumors |
| PRAME x CD3 | IMC-F106C | III | NCT06112314 | R | Treatment naive HLA-A*02:01-positive advanced melanoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, K.S.; Musleh Ud Din, S.; Dalal, S.; Gonzalez, T.; Dalal, M.; Ferraro, P.; Hussein, A.; Vulfovich, M. Bispecific Antibodies in Solid Tumors: Advances and Challenges. Int. J. Mol. Sci. 2025, 26, 5838. https://doi.org/10.3390/ijms26125838
Shan KS, Musleh Ud Din S, Dalal S, Gonzalez T, Dalal M, Ferraro P, Hussein A, Vulfovich M. Bispecific Antibodies in Solid Tumors: Advances and Challenges. International Journal of Molecular Sciences. 2025; 26(12):5838. https://doi.org/10.3390/ijms26125838
Chicago/Turabian StyleShan, Khine Swe, Saba Musleh Ud Din, Shivani Dalal, Teresita Gonzalez, Misha Dalal, Pablo Ferraro, Atif Hussein, and Michel Vulfovich. 2025. "Bispecific Antibodies in Solid Tumors: Advances and Challenges" International Journal of Molecular Sciences 26, no. 12: 5838. https://doi.org/10.3390/ijms26125838
APA StyleShan, K. S., Musleh Ud Din, S., Dalal, S., Gonzalez, T., Dalal, M., Ferraro, P., Hussein, A., & Vulfovich, M. (2025). Bispecific Antibodies in Solid Tumors: Advances and Challenges. International Journal of Molecular Sciences, 26(12), 5838. https://doi.org/10.3390/ijms26125838