
Metagenomic Investigation of Intestinal Microbiota of Insectivorous Synanthropic Bats: Densoviruses, Antibiotic Resistance Genes, and Functional Profiling of Gut Microbial Communities

 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
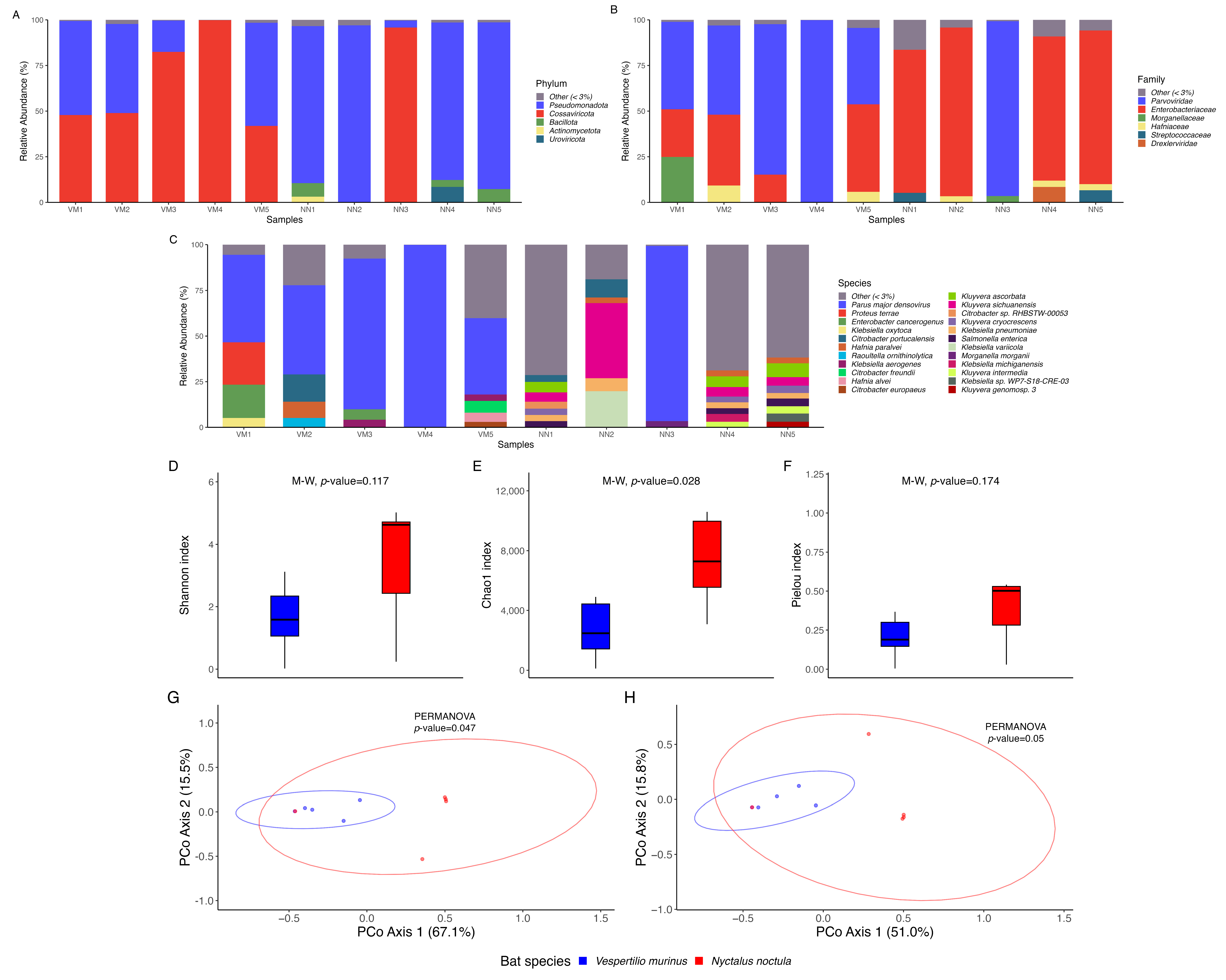
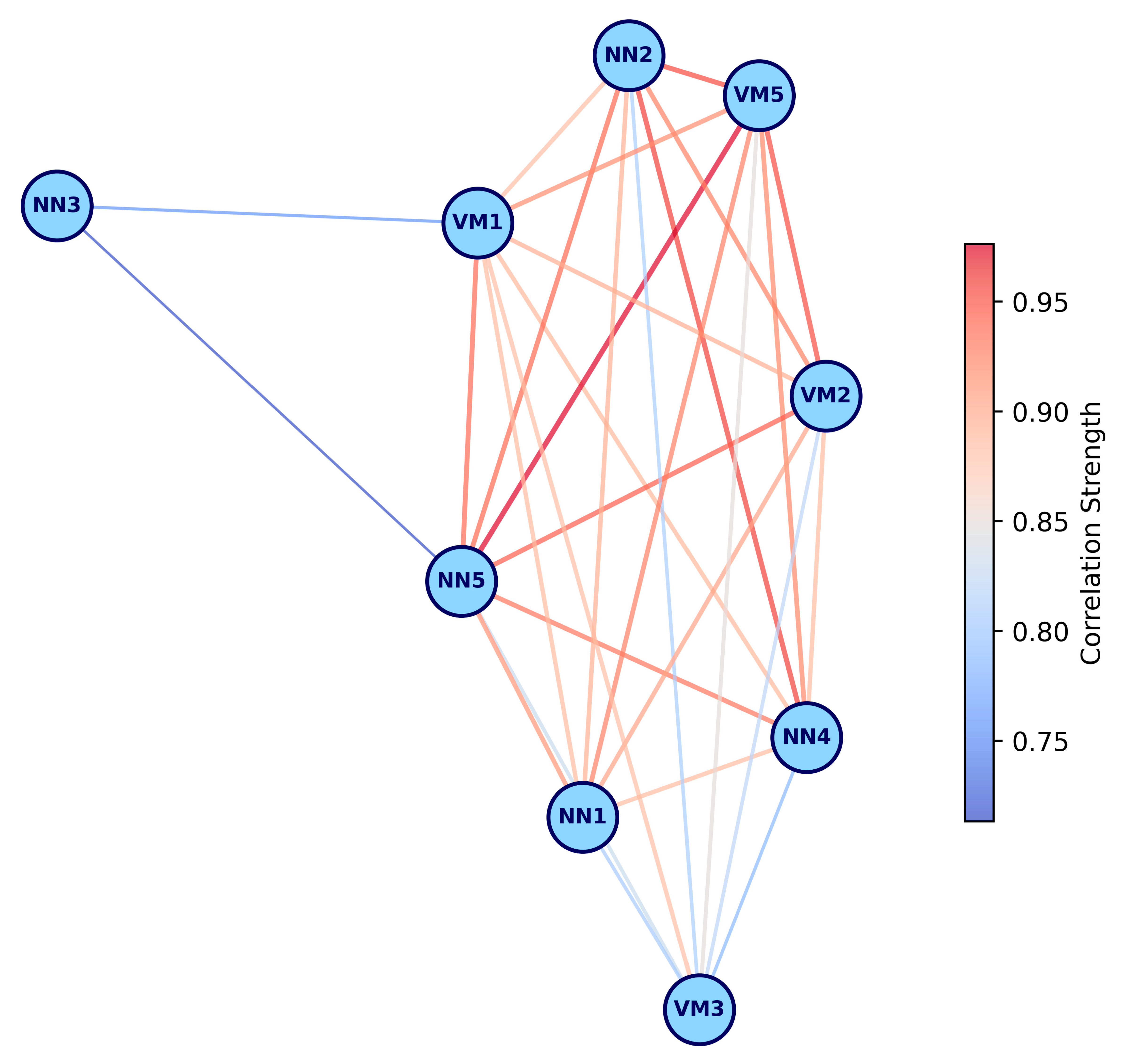
2.1. Microbial Community Composition and Diversity Analysis
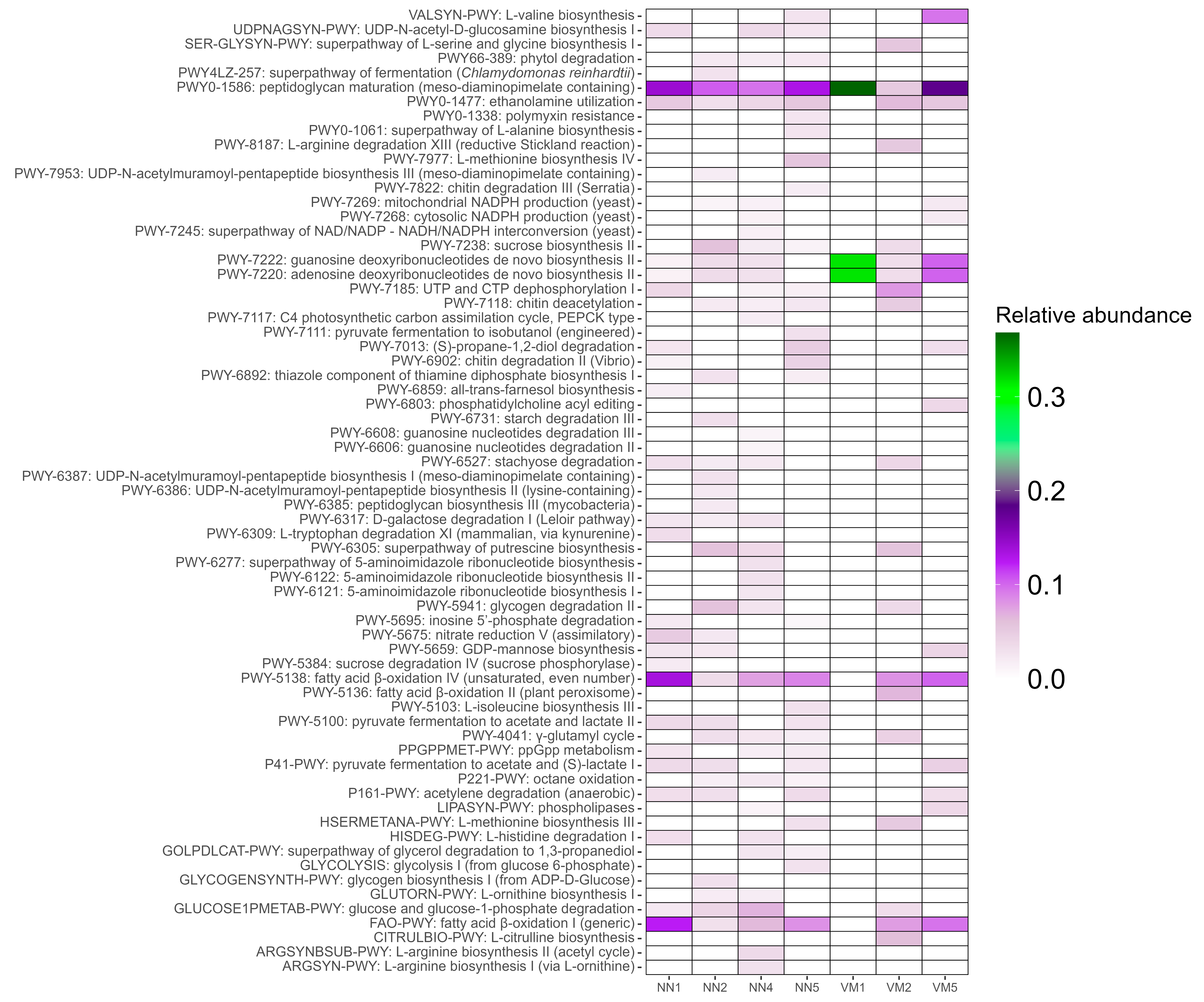
2.2. Functional and Metabolic Profiling
2.3. Antimicrobial Resistance and Virulence Genes
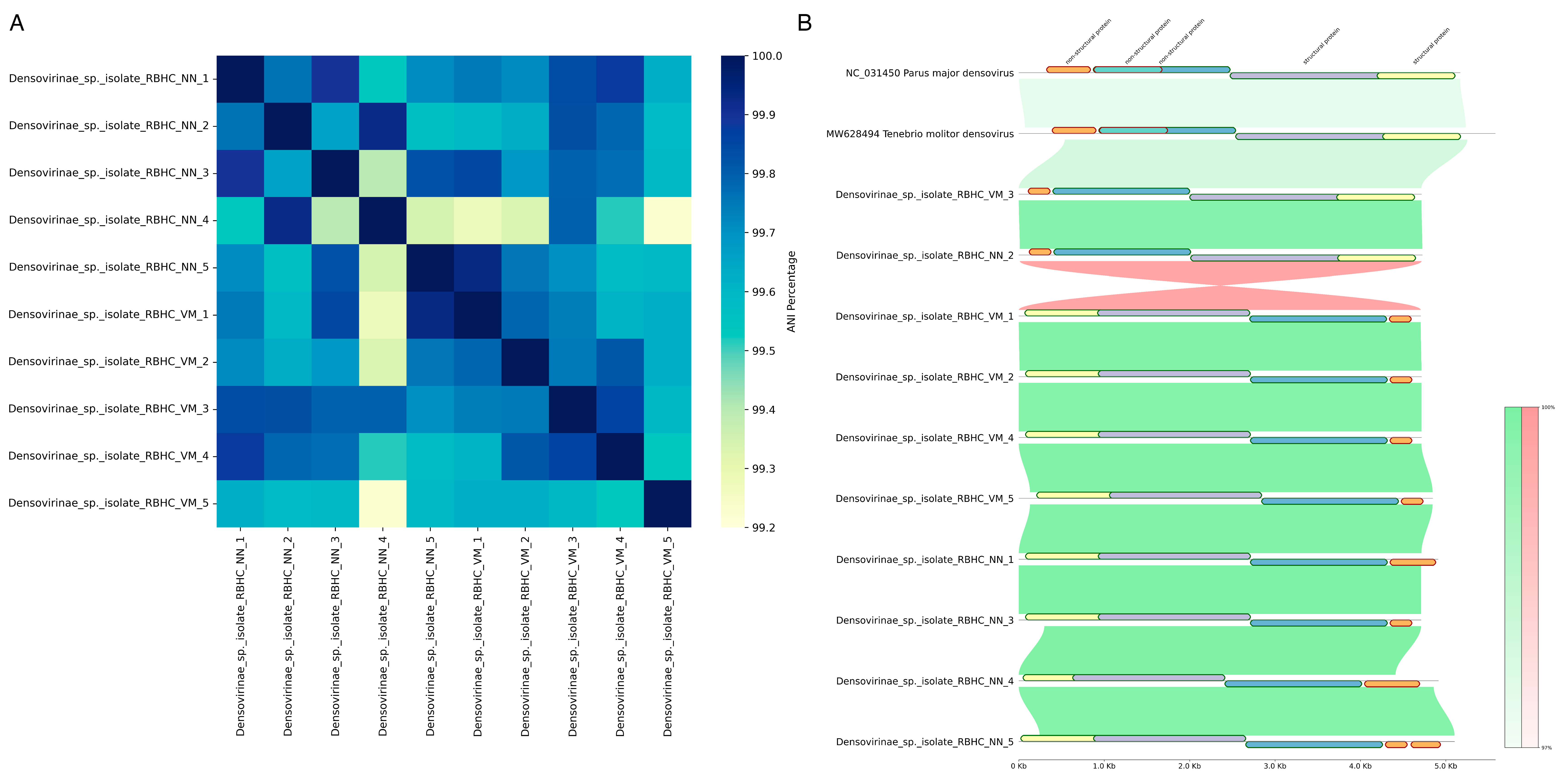
2.4. Genomic Characterization and Phylogenetic Placement of Identified Densoviruses
3. Discussion
4. Materials and Methods
4.1. Sampling
4.2. DNA Extraction and High-Throughput Sequencing
4.3. Quality Control of Raw Reads and Taxonomic Classification
4.4. Diversity and Differential Abundance Analysis
4.5. Metagenome Assembly and Functional Profiling
4.6. Genomic Characterization and Phylogenetic Analysis of Densoviruses
4.7. Data Analysis and Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARGs | Antibiotic Resistance Genes |
| VFDB | Virulence Factors of Bacterial Database |
| CARD | Comprehensive Antibiotic Resistance Database |
| PCoA | Principal Coordinates Analysis |
| ANI | Average Nucleotide Identity |
| ORFs | Open Reading Frames |
| NSP | Non-Structural Protein |
| SP | Structural Protein |
| RND | Resistance-Nodulation-Cell Division |
| MFS | Major Facilitator Superfamily |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | KEGG Orthologous |
| PERMANOVA | Permutational Multivariate Analysis of Variance |
| DNA | Deoxyribonucleic Acid |
| RNA | Ribonucleic Acid |
References
- Pena, S.A.; Alencastre-Santos, A.B.; da Silva, J.B.; Correia, L.L.; Urbieta, G.L.; Graciolli, G.; Palheta, L.R.; Vieira, T.B. Bats (Mammalia, Chiroptera) and Bat Flies (Diptera, Streblidae) from the Cazumbá-Iracema and Chico Mendes Reserve, Western Brazilian Amazon. Parasitol. Res. 2023, 122, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, E.; Cacciotto, C.; Zobba, R.; Pittau, M.; Martella, V.; Alberti, A. Bat Ecology and Microbiome of the Gut: A Narrative Review of Associated Potentials in Emerging and Zoonotic Diseases. Animals 2024, 14, 3043. [Google Scholar] [CrossRef]
- Ohlopkova, O.V.; Stolbunova, K.A.; Popov, I.V.; Popov, I.V.; Kabwe, E.; Davidyuk, Y.N.; Stepanyuk, M.A.; Moshkin, A.D.; Kononova, Y.V.; Lukbanova, E.A.; et al. Detection of Brno Loanvirus (Loanvirus brunaense) in Common Noctule Bats (Nyctalus Noctula) in Southern Russia. Braz. J. Microbiol. 2025, 56, 675–682. [Google Scholar] [CrossRef]
- Popov, I.V.; Ohlopkova, O.V.; Donnik, I.M.; Zolotukhin, P.V.; Umanets, A.; Golovin, S.N.; Malinovkin, A.V.; Belanova, A.A.; Lipilkin, P.V.; Lipilkina, T.A.; et al. Detection of Coronaviruses in Insectivorous Bats of Fore-Caucasus, 2021. Sci. Rep. 2023, 13, 2306. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Baker, M.L.; Kulcsar, K.; Misra, V.; Plowright, R.; Mossman, K. Novel Insights Into Immune Systems of Bats. Front. Immunol. 2020, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Demian, W.L.; Cormier, O.; Mossman, K. Immunological Features of Bats: Resistance and Tolerance to Emerging Viruses. Trends Immunol. 2024, 45, 198–210. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. Why Do Bats Live so Long?-Possible Molecular Mechanisms. Biogerontology 2020, 21, 1–11. [Google Scholar] [CrossRef]
- Carrillo Gaeta, N.; Cavalcante Brito, J.E.; Nunes Batista, J.M.; Gagete Veríssimo de Mello, B.; Dias, R.A.; Heinemann, M.B. Bats Are Carriers of Antimicrobial-Resistant Staphylococcaceae in Their Skin. Antibiotics 2023, 12, 331. [Google Scholar] [CrossRef]
- Devnath, P.; Karah, N.; Graham, J.P.; Rose, E.S.; Asaduzzaman, M. Evidence of Antimicrobial Resistance in Bats and Its Planetary Health Impact for Surveillance of Zoonotic Spillover Events: A Scoping Review. Int. J. Environ. Res. Public Health 2022, 20, 243. [Google Scholar] [CrossRef]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-Borne Virus Diversity, Spillover and Emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Levy, J.I.; Pekar, J.E.; Goldstein, S.A.; Singh, R.; Hensel, Z.; Gangavarapu, K.; Rogers, M.B.; Moshiri, N.; Garry, R.F.; et al. Genetic Tracing of Market Wildlife and Viruses at the Epicenter of the COVID-19 Pandemic. Cell 2024, 187, 5468–5482.e11. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Sun, J.; Li, D.; Wang, N.; Wang, L.; Zhang, C.; Meng, X.; Ji, X.; Suchard, M.A.; Zhang, X.; et al. Emerging Viruses: Cross-Species Transmission of Coronaviruses, Filoviruses, Henipaviruses, and Rotaviruses from Bats. Cell Rep. 2022, 39, 110969. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, L.M.S.; Bueno-Rocha, I.D.; Oliveira, G.; Pires, E.S.; Vasconcelos, S.; Nunes, G.L.; Frizzas, M.R.; Togni, P.H.B. Going out for Dinner—The Consumption of Agriculture Pests by Bats in Urban Areas. PLoS ONE 2021, 16, e0258066. [Google Scholar] [CrossRef] [PubMed]
- Shilton, L.A.; Altringham, J.D.; Compton, S.G.; Whittaker, R.J. Old World Fruit Bats Can Be Long–Distance Seed Dispersers through Extended Retention of Viable Seeds in the Gut. Proc. R. Soc. Lond. B 1999, 266, 219–223. [Google Scholar] [CrossRef]
- Stewart, A.B.; Srilopan, S.; Wayo, K.; Hassa, P.; Dudash, M.R.; Bumrungsri, S. Bat Pollinators: A Decade of Monitoring Reveals Declining Visitation Rates for Some Species in Thailand. Zoological Lett. 2024, 10, 5. [Google Scholar] [CrossRef]
- Boyles, J.G.; Cryan, P.M.; McCracken, G.F.; Kunz, T.H. Economic Importance of Bats in Agriculture. Science 2011, 332, 41–42. [Google Scholar] [CrossRef]
- Martinez, S.; Sullivan, A.; Hagan, E.; Goley, J.; Epstein, J.H.; Olival, K.J.; Saylors, K.; Euren, J.; Bangura, J.; Zikankuba, S.; et al. Living Safely With Bats: Lessons in Developing and Sharing a Global One Health Educational Resource. Glob. Health Sci. Pract. 2022, 10, e2200106. [Google Scholar] [CrossRef]
- Frank, E.G. The Economic Impacts of Ecosystem Disruptions: Costs from Substituting Biological Pest Control. Science 2024, 385, eadg0344. [Google Scholar] [CrossRef]
- Hoyt, J.R.; Langwig, K.E.; Sun, K.; Parise, K.L.; Li, A.; Wang, Y.; Huang, X.; Worledge, L.; Miller, H.; White, J.P.; et al. Environmental Reservoir Dynamics Predict Global Infection Patterns and Population Impacts for the Fungal Disease White-Nose Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 7255–7262. [Google Scholar] [CrossRef]
- Minnis, A.M.; Lindner, D.L. Phylogenetic Evaluation of Geomyces and Allies Reveals No Close Relatives of Pseudogymnoascus Destructans, Comb. Nov., in Bat Hibernacula of Eastern North America. Fungal Biol. 2013, 117, 638–649. [Google Scholar] [CrossRef]
- Urbina, J.; Chestnut, T.; Allen, J.M.; Levi, T. Pseudogymnoascus Destructans Growth in Wood, Soil and Guano Substrates. Sci. Rep. 2021, 11, 763. [Google Scholar] [CrossRef]
- Popov, I.V.; Popov, I.V.; Krikunova, A.A.; Lipilkina, T.A.; Derezina, T.N.; Chikindas, M.L.; Venema, K.; Ermakov, A.M. Gut Microbiota Composition of Insectivorous Synanthropic and Fructivorous Zoo Bats: A Direct Metagenomic Comparison. Int. J. Mol. Sci. 2023, 24, 17301. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-T.; Shi, S.-H.; Jiang, Y.-L.; Zhao, L.; Chen, H.-L.; Huang, K.-Y.; Yang, G.-L.; Wang, C.-F. Genetic Characterization of a Densovirus Isolated from Great Tit (Parus Major) in China. Infect. Genet. Evol. 2016, 41, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Armién, A.G.; Polon, R.; Rejmanek, D.; Moeller, R.B.; Crossley, B.M. Outbreak of Densovirus with High Mortality in a Commercial Mealworm (Tenebrio Molitor) Farm: A Molecular, Bright-Field, and Electron Microscopic Characterization. Vet. Pathol. 2023, 60, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Tijssen, P.; Pénzes, J.J.; Yu, Q.; Pham, H.T.; Bergoin, M. Diversity of Small, Single-Stranded DNA Viruses of Invertebrates and Their Chaotic Evolutionary Past. J. Invertebr. Pathol. 2016, 140, 83–96. [Google Scholar] [CrossRef]
- Dhar, A.K.; Cruz-Flores, R.; Caro, L.F.A.; Siewiora, H.M.; Jory, D. Diversity of Single-Stranded DNA Containing Viruses in Shrimp. Virusdisease 2019, 30, 43–57. [Google Scholar] [CrossRef]
- Li, Y.; Zádori, Z.; Bando, H.; Dubuc, R.; Fédière, G.; Szelei, J.; Tijssen, P. Genome Organization of the Densovirus from Bombyx Mori (BmDNV-1) and Enzyme Activity of Its Capsid. J. Gen. Virol. 2001, 82, 2821–2825. [Google Scholar] [CrossRef]
- Liu, K.; Li, Y.; Jousset, F.-X.; Zadori, Z.; Szelei, J.; Yu, Q.; Pham, H.T.; Lépine, F.; Bergoin, M.; Tijssen, P. The Acheta Domesticus Densovirus, Isolated from the European House Cricket, Has Evolved an Expression Strategy Unique among Parvoviruses. J. Virol. 2011, 85, 10069–10078. [Google Scholar] [CrossRef]
- Li, J.; Dong, Y.; Sun, Y.; Lai, Z.; Zhao, Y.; Liu, P.; Gao, Y.; Chen, X.; Gu, J. A Novel Densovirus Isolated From the Asian Tiger Mosquito Displays Varied Pathogenicity Depending on Its Host Species. Front. Microbiol. 2019, 10, 1549. [Google Scholar] [CrossRef]
- Impact of Biological Invasions on Ecosystem Services; Vilà, M., Hulme, P.E., Eds.; Springer International Publishing: Cham, Switzerland, 2017; ISBN 978-3-319-45119-0. [Google Scholar]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic Analysis of Viruses from Bat Fecal Samples Reveals Many Novel Viruses in Insectivorous Bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, M.; Mortlock, M.; Weyer, J.; Bezuidt, O.; Seamark, E.C.J.; Kearney, T.; Gleasner, C.; Erkkila, T.H.; Cui, H.; Markotter, W. A Metagenomic Viral Discovery Approach Identifies Potential Zoonotic and Novel Mammalian Viruses in Neoromicia Bats within South Africa. PLoS ONE 2018, 13, e0194527. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat Guano Virome: Predominance of Dietary Viruses from Insects and Plants plus Novel Mammalian Viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Šimić, I.; Zorec, T.M.; Lojkić, I.; Krešić, N.; Poljak, M.; Cliquet, F.; Picard-Meyer, E.; Wasniewski, M.; Zrnčić, V.; Ćukušić, A.; et al. Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity. Viruses 2020, 12, 891. [Google Scholar] [CrossRef]
- Tang, S.; Song, X.; Xue, L.; Wang, X.; Wang, X.; Xu, P.; Ren, G. Characterization and Distribution Analysis of a Densovirus Infecting Myzus Persicae Nicotianae (Hemiptera: Aphididae). J. Econ. Entomol. 2016, 109, 580–587. [Google Scholar] [CrossRef]
- Merrikh, C.N.; Merrikh, H. Gene Inversion Potentiates Bacterial Evolvability and Virulence. Nat. Commun. 2018, 9, 4662. [Google Scholar] [CrossRef]
- Pepin, K.M.; Domsic, J.; McKenna, R. Genomic Evolution in a Virus under Specific Selection for Host Recognition. Infect. Genet. Evol. 2008, 8, 825–834. [Google Scholar] [CrossRef]
- Sanjuán, R.; Pereira-Gómez, M.; Risso, J. Genome Instability in DNA Viruses. In Genome Stability; Elsevier: Amsterdam, The Netherlands, 2016; pp. 37–47. ISBN 978-0-12-803309-8. [Google Scholar]
- Bradwell, K.; Combe, M.; Domingo-Calap, P.; Sanjuán, R. Correlation between Mutation Rate and Genome Size in Riboviruses: Mutation Rate of Bacteriophage Qβ. Genetics 2013, 195, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of Viral Mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Bruemmer, A.; Scholari, F.; Lopez-Ferber, M.; Conway, J.F.; Hewat, E.A. Structure of an Insect Parvovirus (Junonia Coenia Densovirus) Determined by Cryo-Electron Microscopy. J. Mol. Biol. 2005, 347, 791–801. [Google Scholar] [CrossRef]
- Jaroenram, W.; Chaivisuthangkura, P.; Owens, L. One Base Pair Deletion and High Rate of Evolution: Keys to Viral Accommodation of Australian Penaeus Stylirostris Densovirus. Aquaculture 2015, 443, 40–48. [Google Scholar] [CrossRef]
- El-Far, M.; Li, Y.; Fédière, G.; Abol-Ela, S.; Bergoin, M.; Tijssen, P. Abortive Infection of the Insect Parvovirus, Mythimna Loreyi Densovirus (MIDNV) in Mammalian Cells. IOBC Wprs Bulletin 2003, 26, 229–232. [Google Scholar]
- Li, T.; Li, H.; Wu, Y.; Li, S.; Yuan, G.; Xu, P. Identification of a Novel Densovirus in Aphid, and Uncovering the Possible Antiviral Process During Its Infection. Front. Immunol. 2022, 13, 905628. [Google Scholar] [CrossRef]
- Donnik, I.M.; Popov, I.V.; Sereda, S.V.; Popov, I.V.; Chikindas, M.L.; Ermakov, A.M. Coronavirus Infections of Animals: Future Risks to Humans. Biol. Bull. Russ. Acad. Sci. 2021, 48, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.V.; Mazanko, M.S.; Kulaeva, E.D.; Golovin, S.N.; Malinovkin, A.V.; Aleshukina, I.S.; Aleshukina, A.V.; Prazdnova, E.V.; Tverdokhlebova, T.I.; Chikindas, M.L.; et al. Gut Microbiota of Bats: Pro-Mutagenic Properties and Possible Frontiers in Preventing Emerging Disease. Sci. Rep. 2021, 11, 21075. [Google Scholar] [CrossRef]
- Chao, A.; Colwell, R.K. Thirty Years of Progeny from Chao’s Inequality: Estimating and Comparing Richness with Incidence Data and Incomplete Sampling. SORT 2017, 41, 3–54. [Google Scholar]
- Hill, T.C.J.; Walsh, K.A.; Harris, J.A.; Moffett, B.F. Using Ecological Diversity Measures with Bacterial Communities. FEMS Microbiol. Ecol. 2003, 43, 1–11. [Google Scholar] [CrossRef]
- Aizpurua, O.; Nyholm, L.; Morris, E.; Chaverri, G.; Herrera Montalvo, L.G.; Flores-Martinez, J.J.; Lin, A.; Razgour, O.; Gilbert, M.T.P.; Alberdi, A. The Role of the Gut Microbiota in the Dietary Niche Expansion of Fishing Bats. Anim. Microbiome 2021, 3, 76. [Google Scholar] [CrossRef]
- Dai, W.; Leng, H.; Li, J.; Li, A.; Li, Z.; Zhu, Y.; Li, X.; Jin, L.; Sun, K.; Feng, J. The Role of Host Traits and Geography in Shaping the Gut Microbiome of Insectivorous Bats. mSphere 2024, 9, e0008724. [Google Scholar] [CrossRef]
- Li, J.; Chu, Y.; Yao, W.; Wu, H.; Feng, J. Differences in Diet and Gut Microbiota Between Lactating and Non-Lactating Asian Particolored Bats (Vespertilio Sinensis): Implication for a Connection Between Diet and Gut Microbiota. Front. Microbiol. 2021, 12, 735122. [Google Scholar] [CrossRef]
- Mena Canata, D.A.; Benfato, M.S.; Pereira, F.D.; Ramos Pereira, M.J.; Hackenhaar, F.S.; Mann, M.B.; Frazzon, A.P.G.; Rampelotto, P.H. Comparative Analysis of the Gut Microbiota of Bat Species with Different Feeding Habits. Biology 2024, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.V.; Popov, I.V.; Chebotareva, I.P.; Tikhmeneva, I.A.; Peshkova, D.A.; Krikunova, A.A.; Tkacheva, E.V.; Algburi, A.R.; Abdulhameed, A.M.; Jargalsaikhan, A.; et al. Differences in Gut Microbiota Composition, Diversity, and Predicted Functional Activity between Wild and Captive Zoo Carollia Perspicillata in a One Health Perspective. Braz. J. Microbiol. 2025, 56, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xiao, G.; Liu, H.; Zhao, X.; Sun, C.; Tan, X.; Sun, K.; Liu, S.; Feng, J. Captivity Causes Taxonomic and Functional Convergence of Gut Microbial Communities in Bats. PeerJ 2019, 7, e6844. [Google Scholar] [CrossRef]
- Adams, S.; Knapp, J.P.; Neujahr, A.; Burkey, T.; Miller, P.S.; Fernando, S.C. 276 Investigating the Colonization History of Early-Life Microbiome of Piglets. J. Anim. Sci. 2023, 101, 165–166. [Google Scholar] [CrossRef]
- Adeolu, M.; Alnajar, S.; Naushad, S.; S Gupta, R. Genome-Based Phylogeny and Taxonomy of the “Enterobacteriales”: Proposal for Enterobacterales Ord. Nov. Divided into the Families Enterobacteriaceae, Erwiniaceae Fam. Nov., Pectobacteriaceae Fam. Nov., Yersiniaceae Fam. Nov., Hafniaceae Fam. Nov., Morganellaceae Fam. Nov., and Budviciaceae Fam. Nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [CrossRef] [PubMed]
- Khorsand, B.; Asadzadeh Aghdaei, H.; Nazemalhosseini-Mojarad, E.; Nadalian, B.; Nadalian, B.; Houri, H. Overrepresentation of Enterobacteriaceae and Escherichia Coli Is the Major Gut Microbiome Signature in Crohn’s Disease and Ulcerative Colitis; a Comprehensive Metagenomic Analysis of IBDMDB Datasets. Front. Cell Infect. Microbiol. 2022, 12, 1015890. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, J.; Hu, Q.; Rao, X. Morganella Morganii, a Non-Negligent Opportunistic Pathogen. Int. J. Infect. Dis. 2016, 50, 10–17. [Google Scholar] [CrossRef]
- Ma, J.; Song, X.; Li, M.; Yu, Z.; Cheng, W.; Yu, Z.; Zhang, W.; Zhang, Y.; Shen, A.; Sun, H.; et al. Global Spread of Carbapenem-Resistant Enterobacteriaceae: Epidemiological Features, Resistance Mechanisms, Detection and Therapy. Microbiol. Res. 2023, 266, 127249. [Google Scholar] [CrossRef]
- Moreira de Gouveia, M.I.; Bernalier-Donadille, A.; Jubelin, G. Enterobacteriaceae in the Human Gut: Dynamics and Ecological Roles in Health and Disease. Biology 2024, 13, 142. [Google Scholar] [CrossRef]
- Shah, T.; Wang, Y.; Wang, Y.; Li, Q.; Zhou, J.; Hou, Y.; Wang, B.; Xia, X. A Comparative Analysis of the Stomach, Gut, and Lung Microbiomes in Rattus Norvegicus. Microorganisms 2023, 11, 2359. [Google Scholar] [CrossRef]
- German, G.J.; DeGiulio, J.V.; Ramsey, J.; Kropinski, A.M.; Misra, R. The TolC and Lipopolysaccharide-Specific Escherichia Coli Bacteriophage TLS-the Tlsvirus Archetype Virus. Phage 2024, 5, 173–183. [Google Scholar] [CrossRef]
- Kumar, P.; Meghvansi, M.K.; Kamboj, D.V. Phenotypic Characterization and Whole-Genome Analysis of a Novel Bacteriophage HCF1 Infecting Citrobacter Amalonaticus and C. Freundii. Front. Microbiol. 2021, 12, 644013. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Li, D.; Lin, W.; Liu, W.; Qin, W.; Xu, L.; Tong, Y. Genomic Analysis of a Novel Active Prophage of Hafnia Paralvei. Arch. Virol. 2022, 167, 2027–2034. [Google Scholar] [CrossRef]
- Muniesa, M.; Jofre, J. Identifying and Analyzing Bacteriophages in Human Fecal Samples: What Could We Discover? Future Microbiol. 2014, 9, 879–886. [Google Scholar] [CrossRef]
- Pilati, G.V.T.; Cadamuro, R.D.; Filho, V.B.; Dahmer, M.; Elois, M.A.; Savi, B.P.; Salles, G.B.C.; Muniz, E.C.; Fongaro, G. Bacteriophage-Associated Antimicrobial Resistance Genes in Avian Pathogenic Escherichia Coli Isolated from Brazilian Poultry. Viruses 2023, 15, 1485. [Google Scholar] [CrossRef]
- Song, S.J.; Sanders, J.G.; Delsuc, F.; Metcalf, J.; Amato, K.; Taylor, M.W.; Mazel, F.; Lutz, H.L.; Winker, K.; Graves, G.R.; et al. Comparative Analyses of Vertebrate Gut Microbiomes Reveal Convergence between Birds and Bats. mBio 2020, 11, e02901-19. [Google Scholar] [CrossRef]
- Popov, I.V.; Berezinskaia, I.S.; Popov, I.V.; Martiusheva, I.B.; Tkacheva, E.V.; Gorobets, V.E.; Tikhmeneva, I.A.; Aleshukina, A.V.; Tverdokhlebova, T.I.; Chikindas, M.L.; et al. Cultivable Gut Microbiota in Synanthropic Bats: Shifts of Its Composition and Diversity Associated with Hibernation. Animals 2023, 13, 3658. [Google Scholar] [CrossRef] [PubMed]
- Mavziutov, A.R.; Bondarenko, V.M.; Zherebtsova, N.I.; Valishin, D.A. [Pathogenicity factors of opportunistic enterobacteria and its role in development of diarrhea]. Zh Mikrobiol. Epidemiol. Immunobiol. 2007, 1, 89–97. [Google Scholar]
- Osei Sekyere, J.; Reta, M.A. Global Evolutionary Epidemiology and Resistome Dynamics of Citrobacter Species, Enterobacter Hormaechei, Klebsiella Variicola, and Proteeae Clones. Environ. Microbiol. 2021, 23, 7412–7431. [Google Scholar] [CrossRef]
- Palusiak, A. Proteus Mirabilis and Klebsiella Pneumoniae as Pathogens Capable of Causing Co-Infections and Exhibiting Similarities in Their Virulence Factors. Front. Cell Infect. Microbiol. 2022, 12, 991657. [Google Scholar] [CrossRef]
- Sanches, M.S.; Rodrigues da Silva, C.; Silva, L.C.; Montini, V.H.; Lopes Barboza, M.G.; Migliorini Guidone, G.H.; Dias de Oliva, B.H.; Nishio, E.K.; Faccin Galhardi, L.C.; Vespero, E.C.; et al. Proteus Mirabilis from Community-Acquired Urinary Tract Infections (UTI-CA) Shares Genetic Similarity and Virulence Factors with Isolates from Chicken, Beef and Pork Meat. Microb. Pathog. 2021, 158, 105098. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hentschel, U.; Kaper, J.B.; Hacker, J. Genome Plasticity in Pathogenic and Nonpathogenic Enterobacteria. Curr. Top. Microbiol. Immunol. 2002, 264, 157–175. [Google Scholar] [PubMed]
- Clarke, T.B.; Davis, K.M.; Lysenko, E.S.; Zhou, A.Y.; Yu, Y.; Weiser, J.N. Recognition of Peptidoglycan from the Microbiota by Nod1 Enhances Systemic Innate Immunity. Nat. Med. 2010, 16, 228–231. [Google Scholar] [CrossRef]
- Gupta, D.; Jing, X.; Dziarski, R. Peptidoglycan Recognition Protein 3-Regulated Microbiome Maintains Healthy Levels of Treg Cells and Protects Mice from Colitis (INC1P.346). J. Immunol. 2015, 194, 54.3. [Google Scholar] [CrossRef]
- Tosoni, G.; Conti, M.; Diaz Heijtz, R. Bacterial Peptidoglycans as Novel Signaling Molecules from Microbiota to Brain. Curr. Opin. Pharmacol. 2019, 48, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J. Peptidoglycan-Induced Modulation of Metabolic and Inflammatory Responses. Immunometabolism 2023, 5, e00024. [Google Scholar] [CrossRef]
- Baud, G.L.C.; Prasad, A.; Ellegaard, K.M.; Engel, P. Turnover of Strain-Level Diversity Modulates Functional Traits in the Honeybee Gut Microbiome between Nurses and Foragers. Genome Biol. 2023, 24, 283. [Google Scholar] [CrossRef]
- Murga-Garrido, S.M.; Ulloa-Pérez, E.J.; Díaz-Benítez, C.E.; Orbe-Orihuela, Y.C.; Cornejo-Granados, F.; Ochoa-Leyva, A.; Sanchez-Flores, A.; Cruz, M.; Castañeda-Márquez, A.C.; Plett-Torres, T.; et al. Virulence Factors of the Gut Microbiome Are Associated with BMI and Metabolic Blood Parameters in Children with Obesity. Microbiol. Spectr. 2023, 11, e0338222. [Google Scholar] [CrossRef]
- Watson, A.R.; Füssel, J.; Veseli, I.; DeLongchamp, J.Z.; Silva, M.; Trigodet, F.; Lolans, K.; Shaiber, A.; Fogarty, E.; Runde, J.M.; et al. Metabolic Independence Drives Gut Microbial Colonization and Resilience in Health and Disease. Genome Biol. 2023, 24, 78. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. KEGG: Biological Systems Database as a Model of the Real World. Nucleic Acids Res. 2025, 53, D672–D677. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, J.; Zhu, Y.; Mao, H.; Li, J.; Kong, X.; Zhu, X.; Zhang, J. Investigating Gut Microbiota-Blood and Urine Metabolite Correlations in Early Sepsis-Induced Acute Kidney Injury: Insights from Targeted KEGG Analyses. Front. Cell Infect. Microbiol. 2024, 14, 1375874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Meng, N.; Li, R.; He, Q.; Liang, X.; Zhou, F.; Zheng, W.; Ma, J.; Yu, X.; Tan, K.; et al. Gut Microbe-Metabolite Profiles Are Associated with Microbial Pathways of Longevity in Women: A Cross-Sectional Study Conducted in China. Gerontology 2024, 70, 76–89. [Google Scholar] [CrossRef]
- Warinner, J.; ElSaadani, M.; Rosenau, K.; Kim, J.H.; Hassan, S.A.; Bhogoju, S.; Wempe, L.; Goretsky, T.; Alam, M.A.; Awuah, S.; et al. AUPHOS, A NOVEL DRUG THAT IMPROVES COLITIS BY MICROBIOME INDUCED METABOLIC CHANGES. Inflamm. Bowel Dis. 2023, 29, S49. [Google Scholar] [CrossRef]
- Xu, L.; Wu, Y.; Yang, X.; Pang, X.; Wu, Y.; Li, X.; Liu, X.; Zhao, Y.; Yu, L.; Wang, P.; et al. The Fe-S Cluster Biosynthesis in Enterococcus Faecium Is Essential for Anaerobic Growth and Gastrointestinal Colonization. Gut Microbes 2024, 16, 2359665. [Google Scholar] [CrossRef]
- El-Demerdash, A.S.; Kamel, S.A.; Elariny, E.Y.T.; Henidi, H.; Mahran, Y.; Alahdal, H.; Saleh, A.M.; Ibrahim, R.A. Natural Inhibitors of Salmonella MDR Efflux Pumps AcrAB and AcrD: An Integrated In Silico, Molecular, and In Vitro Investigation. Int. J. Mol. Sci. 2024, 25, 12949. [Google Scholar] [CrossRef]
- Yap, P.S.X.; Ahmad Kamar, A.; Chong, C.W.; Yap, I.K.S.; Teh, C.S.J. Whole Genome Analysis of Multidrug-Resistant Citrobacter Freundii B9-C2 Isolated from Preterm Neonate’s Stool in the First Week. J. Glob. Antimicrob. Resist. 2020, 21, 246–251. [Google Scholar] [CrossRef]
- Albarri, O.; AlMatar, M.; Var, I.; Köksal, F. Antimicrobial Resistance of Clinical Klebsiella Pneumoniae Isolates: Involvement of AcrAB and OqxAB Efflux Pumps. Curr. Mol. Pharmacol. 2024, 17, e310323215266. [Google Scholar] [CrossRef]
- Kang, H.-W.; Woo, G.-J. Increase of Multidrug Efflux Pump Expression in Fluoroquinolone-Resistant Salmonella Mutants Induced by Ciprofloxacin Selective Pressure. Res. Vet. Sci. 2014, 97, 182–186. [Google Scholar] [CrossRef]
- Schuster, S.; Vavra, M.; Schweigger, T.M.; Rossen, J.W.A.; Matsumura, Y.; Kern, W.V. Contribution of AcrAB-TolC to Multidrug Resistance in an Escherichia Coli Sequence Type 131 Isolate. Int. J. Antimicrob. Agents 2017, 50, 477–481. [Google Scholar] [CrossRef]
- Baron, S.; Le Devendec, L.; Lucas, P.; Larvor, E.; Jové, T.; Kempf, I. Characterisation of Plasmids Harbouring Extended-Spectrum Cephalosporin Resistance Genes in Escherichia Coli from French Rivers. Vet. Microbiol. 2020, 243, 108619. [Google Scholar] [CrossRef] [PubMed]
- Bonanno Ferraro, G.; Bonomo, C.; Brandtner, D.; Mancini, P.; Veneri, C.; Briancesco, R.; Coccia, A.M.; Lucentini, L.; Suffredini, E.; Bongiorno, D.; et al. Characterisation of Microbial Communities and Quantification of Antibiotic Resistance Genes in Italian Wastewater Treatment Plants Using 16S rRNA Sequencing and Digital PCR. Sci. Total Environ. 2024, 933, 173217. [Google Scholar] [CrossRef] [PubMed]
- Glenn, L.M.; Englen, M.D.; Lindsey, R.L.; Frank, J.F.; Turpin, J.E.; Berrang, M.E.; Meinersmann, R.J.; Fedorka-Cray, P.J.; Frye, J.G. Analysis of Antimicrobial Resistance Genes Detected in Multiple-Drug-Resistant Escherichia Coli Isolates from Broiler Chicken Carcasses. Microb. Drug Resist. 2012, 18, 453–463. [Google Scholar] [CrossRef]
- Rajaei, M.; Moosavy, M.-H.; Gharajalar, S.N.; Khatibi, S.A. Antibiotic Resistance in the Pathogenic Foodborne Bacteria Isolated from Raw Kebab and Hamburger: Phenotypic and Genotypic Study. BMC Microbiol. 2021, 21, 272. [Google Scholar] [CrossRef]
- Singh, N.; Goel, G.; Raghav, M. Insights into Virulence Factors Determining the Pathogenicity of Cronobacter Sakazakii. Virulence 2015, 6, 433–440. [Google Scholar] [CrossRef]
- Wibberg, D.; Szczepanowski, R.; Eikmeyer, F.; Pühler, A.; Schlüter, A. The IncF Plasmid pRSB225 Isolated from a Municipal Wastewater Treatment Plant’s on-Site Preflooder Combining Antibiotic Resistance and Putative Virulence Functions Is Highly Related to Virulence Plasmids Identified in Pathogenic E. Coli Isolates. Plasmid 2013, 69, 127–137. [Google Scholar] [CrossRef]
- Yuan, L.; Li, X.; Du, L.; Su, K.; Zhang, J.; Liu, P.; He, Q.; Zhang, Z.; Peng, D.; Shen, L.; et al. RcsAB and Fur Coregulate the Iron-Acquisition System via entC in Klebsiella Pneumoniae NTUH-K2044 in Response to Iron Availability. Front. Cell Infect. Microbiol. 2020, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Obodoechi, L.O.; Carvalho, I.; Chenouf, N.S.; Martínez-Álvarez, S.; Sadi, M.; Nwanta, J.A.; Chah, K.F.; Torres, C. Antimicrobial Resistance in Escherichia Coli Isolates from Frugivorous (Eidolon Helvum) and Insectivorous (Nycteris Hispida) Bats in Southeast Nigeria, with Detection of CTX-M-15 Producing Isolates. Comp. Immunol. Microbiol. Infect. Dis. 2021, 75, 101613. [Google Scholar] [CrossRef]
- Costa, L.D.F.X.; Grassotti, T.T.; Canani, C.R.; de Lira, A.D.; de Moura, T.M.; Campos, A.A.S.; Frazzon, J.; Frazzon, A.P.G. Diversidade, Perfis de Resistência e Virulência de Enterococcus Spp. Em Fezes de Morcegos Urbanos Tadarida Brasiliensis (Brazilian Free-Tailed Bats). Rev. Bras. De Biociências 2019, 17, 43–52. [Google Scholar]
- Garcês, A.; Correia, S.; Amorim, F.; Pereira, J.E.; Igrejas, G.; Poeta, P. First report on extended-spectrum beta-lactamase (ESBL) producing Escherichia coli from European free-tailed bats (Tadarida teniotis) in Portugal: A one-health approach of a hidden contamination problem. J. Hazard. Mater. 2019, 370, 219–224. [Google Scholar] [CrossRef]
- Selvin, J.; Lanong, S.; Syiem, D.; De Mandal, S.; Kayang, H.; Kumar, N.S.; Kiran, G.S. Culture-Dependent and Metagenomic Analysis of Lesser Horseshoe Bats’ Gut Microbiome Revealing Unique Bacterial Diversity and Signatures of Potential Human Pathogens. Microb. Pathog. 2019, 137, 103675. [Google Scholar] [CrossRef]
- Huang, L.; Dai, W.; Sun, X.; Pu, Y.; Feng, J.; Jin, L.; Sun, K. Diet-Driven Diversity of Antibiotic Resistance Genes in Wild Bats: Implications for Public Health. Microbiol. Res. 2025, 293, 128086. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome Analysis Using the Kraken Software Suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Pielou, E.C. The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Chao, A.; Bunge, J. Estimating the Number of Species in a Stochastic Abundance Model. Biometrics 2002, 58, 531–539. [Google Scholar] [CrossRef]
- Mann, H.B.; Whitney, D.R. On a Test of Whether One of Two Random Variables Is Stochastically Larger than the Other. Ann. Math. Statist. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Jaccard, P. Étude Comparative de La Distribution Florale Dans Une Portion Des Alpes et Du Jura; Imprimerie Corbaz & Comp.: Lexington, MA, USA, 1901. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; Kenett, R.S., Longford, N.T., Piegorsch, W.W., Ruggeri, F., Eds.; Wiley: Hoboken, NJ, USA, 2017; pp. 1–15. ISBN 978-1-118-44511-2. [Google Scholar]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of Metagenome Assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with bioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and Non-Redundant UniProt Reference Clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Graham, E.D.; Heidelberg, J.F.; Tully, B.J. Potential for Primary Productivity in a Globally-Distributed Bacterial Phototroph. ISME J. 2018, 12, 1861–1866. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis--10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chen, C.; Akyol, T.; Dusa, A.; Yu, G.; Cao, B.; Cai, P. ggVennDiagram: Intuitive Venn Diagram Software Extended. iMeta 2024, 3, e177. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Kans, J. Entrez Direct: E-Utilities on the UNIX Command Line. In Entrez Programming Utilities Help [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2024. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)Orthologs in Large-Scale Analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Xu, S.; Li, L.; Luo, X.; Chen, M.; Tang, W.; Zhan, L.; Dai, Z.; Lam, T.T.; Guan, Y.; Yu, G. Ggtree: A Serialized Data Object for Visualization of a Phylogenetic Tree and Annotation Data. Imeta 2022, 1, e56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Feng, T.; Xu, S.; Gao, F.; Lam, T.T.; Wang, Q.; Wu, T.; Huang, H.; Zhan, L.; Li, L.; et al. Ggmsa: A Visual Exploration Tool for Multiple Sequence Alignment and Associated Data. Brief. Bioinform. 2022, 23, bbac222. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A Fast and Versatile Genome Alignment System. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable Data Analysis with Snakemake. F1000Res 2021, 10, 33. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popov, I.V.; Manakhov, A.D.; Gorobets, V.E.; Diakova, K.B.; Lukbanova, E.A.; Malinovkin, A.V.; Venema, K.; Ermakov, A.M.; Popov, I.V. Metagenomic Investigation of Intestinal Microbiota of Insectivorous Synanthropic Bats: Densoviruses, Antibiotic Resistance Genes, and Functional Profiling of Gut Microbial Communities. Int. J. Mol. Sci. 2025, 26, 5941. https://doi.org/10.3390/ijms26135941
Popov IV, Manakhov AD, Gorobets VE, Diakova KB, Lukbanova EA, Malinovkin AV, Venema K, Ermakov AM, Popov IV. Metagenomic Investigation of Intestinal Microbiota of Insectivorous Synanthropic Bats: Densoviruses, Antibiotic Resistance Genes, and Functional Profiling of Gut Microbial Communities. International Journal of Molecular Sciences. 2025; 26(13):5941. https://doi.org/10.3390/ijms26135941
Chicago/Turabian StylePopov, Ilia V., Andrey D. Manakhov, Vladislav E. Gorobets, Kristina B. Diakova, Ekaterina A. Lukbanova, Aleksey V. Malinovkin, Koen Venema, Alexey M. Ermakov, and Igor V. Popov. 2025. "Metagenomic Investigation of Intestinal Microbiota of Insectivorous Synanthropic Bats: Densoviruses, Antibiotic Resistance Genes, and Functional Profiling of Gut Microbial Communities" International Journal of Molecular Sciences 26, no. 13: 5941. https://doi.org/10.3390/ijms26135941
APA StylePopov, I. V., Manakhov, A. D., Gorobets, V. E., Diakova, K. B., Lukbanova, E. A., Malinovkin, A. V., Venema, K., Ermakov, A. M., & Popov, I. V. (2025). Metagenomic Investigation of Intestinal Microbiota of Insectivorous Synanthropic Bats: Densoviruses, Antibiotic Resistance Genes, and Functional Profiling of Gut Microbial Communities. International Journal of Molecular Sciences, 26(13), 5941. https://doi.org/10.3390/ijms26135941