1. Introduction
Glioblastoma (GBM) is the most common and aggressive malignant brain tumor, originating from neural progenitors or glial precursors within the neuroepithelial tissue of the brain. Classified as a grade IV tumor according to the World Health Organization (WHO) brain tumor classification, GBM exhibits significant cellular heterogeneity, invasiveness, and resistance to conventional therapies. Apart from radiation exposure and certain genetic predispositions, the precise etiology of GBM remains poorly understood, complicating both early diagnosis and effective treatment strategies. Current standard-of-care therapy combines radiotherapy with temozolomide chemotherapy, but despite this multimodal approach, patient prognosis remains dismal, with median survival rarely exceeding 15 months post-diagnosis [
1].
A major challenge in treating GBM arises from the extensive molecular heterogeneity and genetic instability observed within and between individual tumors. Comprehensive genome-wide studies have consistently identified multiple genetic alterations, including amplifications, deletions, or mutations in key oncogenes and tumor suppressors, such as
EGFR,
NF1,
PDGFRA,
IDH1,
BRAF,
TP53, and
PTEN. Furthermore, dysregulated signaling pathways, notably NF-κB and PI3K–AKT–mTOR, are frequently implicated in GBM pathogenesis [
1,
2]. Despite extensive research, targeted therapies aimed at these molecular abnormalities have generally failed in clinical trials, highlighting the complexity and redundancy of GBM signaling networks and the urgent need for novel therapeutic targets and biomarkers [
3]. Given these challenges, a deeper understanding of GBM’s molecular regulators is critical. Identifying key molecular drivers of tumorigenesis and progression can provide valuable insights into novel treatment strategies. Molecular profiling has the potential to refine diagnostic classifications, predict therapeutic responses, and improve patient outcomes through precision medicine approaches. Therefore, elucidating the complex molecular landscape underlying GBM remains a priority in neuro-oncology research.
GBM’s molecular heterogeneity poses a major barrier to the discovery of consistent and generalizable therapeutic targets. Individual transcriptomic studies frequently suffer from methodological variability, small sample sizes, and dataset-specific biases, often resulting in contradictory findings. Meta-analysis offers a solution to these challenges by integrating multiple independent transcriptomic datasets to derive consensus molecular signatures. This approach increases statistical power, enhances reproducibility, and minimizes artifacts, thereby enabling the identification of robust disease-associated genes across diverse patient populations. Focusing on transcription factors (TFs) within this framework is particularly advantageous. As central regulators of gene expression programs, TFs orchestrate numerous cellular processes, including those that drive tumor proliferation, invasion, and resistance to therapy. Perturbing TFs can therefore lead to widespread shifts in downstream signaling networks. Identifying TFs that are consistently dysregulated in GBM has the potential to uncover master regulators of glioma biology and reveal novel therapeutic opportunities. Applying meta-analysis to prioritize transcriptional regulators addresses both the need for statistical rigor and biological relevance, making it a strategic approach for studying complex tumors, such as GBM.
The early growth response (EGR) family consists of four zinc-finger transcription factors—
EGR1,
EGR2,
EGR3, and
EGR4—that regulate gene expression in response to a variety of external stimuli. These TFs play key roles in controlling cell proliferation, differentiation, apoptosis, and neural development. Among them,
EGR3 has been less extensively studied in cancer biology despite its established function in neurodevelopment and psychiatric disorders, such as schizophrenia and bipolar disorder [
4]. Mechanistically,
EGR3 exerts its transcriptional effects through GC-rich response elements, and its activity is tightly regulated by negative feedback from NAB1 and NAB2 corepressors [
5,
6]. Although
EGR3 has been implicated in breast and gastric cancers [
2], its role in GBM remains unresolved. Multiple prior studies have yielded conflicting findings. Tang et al. reported that
EGR3 was upregulated in GBM tissues and cell lines and that its knockdown suppressed proliferation, migration, and invasion while promoting apoptosis—suggesting a potential oncogenic function [
7]. Conversely, Shen et al. observed that
EGR3 was downregulated in glioma tissues and that its overexpression impaired proliferation and colony formation, indicating a possible tumor-suppressive role [
8]. More recently, Knudsen et al. performed large-scale immunohistochemical and spatial profiling analyses and revealed that
EGR3 expression was enriched at the tumor periphery relative to the core, a region often associated with glioma cell infiltration [
2]. High
EGR3 expression was also associated with poor survival in MGMT-methylated patients, implicating
EGR3 in tumor progression and potentially in cell migration [
2]. Separately, Qin et al. identified
EGR3 among six differentially expressed TFs enriched in GBM samples compared to adjacent tissues. Although
EGR3 was reported as downregulated in tumor tissues, high
EGR3 expression correlated with shorter recurrence-free survival, again suggesting a functional role in disease progression [
9]. These discrepancies likely reflect the context-dependent nature of transcription factor activity. Differences in tumor subtype, cellular origin, epigenetic regulation, or the local tumor microenvironment may all influence
EGR3 function. Transcription factors often operate in complex, tissue-specific networks, and their effects can vary depending on co-regulators, expression thresholds, and spatial distribution. In light of these conflicting data and the central regulatory role of TFs in tumor biology, it is essential to reassess
EGR3 in GBM through integrated transcriptomic analysis and experimental validation. A clearer understanding of
EGR3 may reconcile existing inconsistencies and clarify whether it contributes to glioma proliferation, migration, or therapeutic resistance.
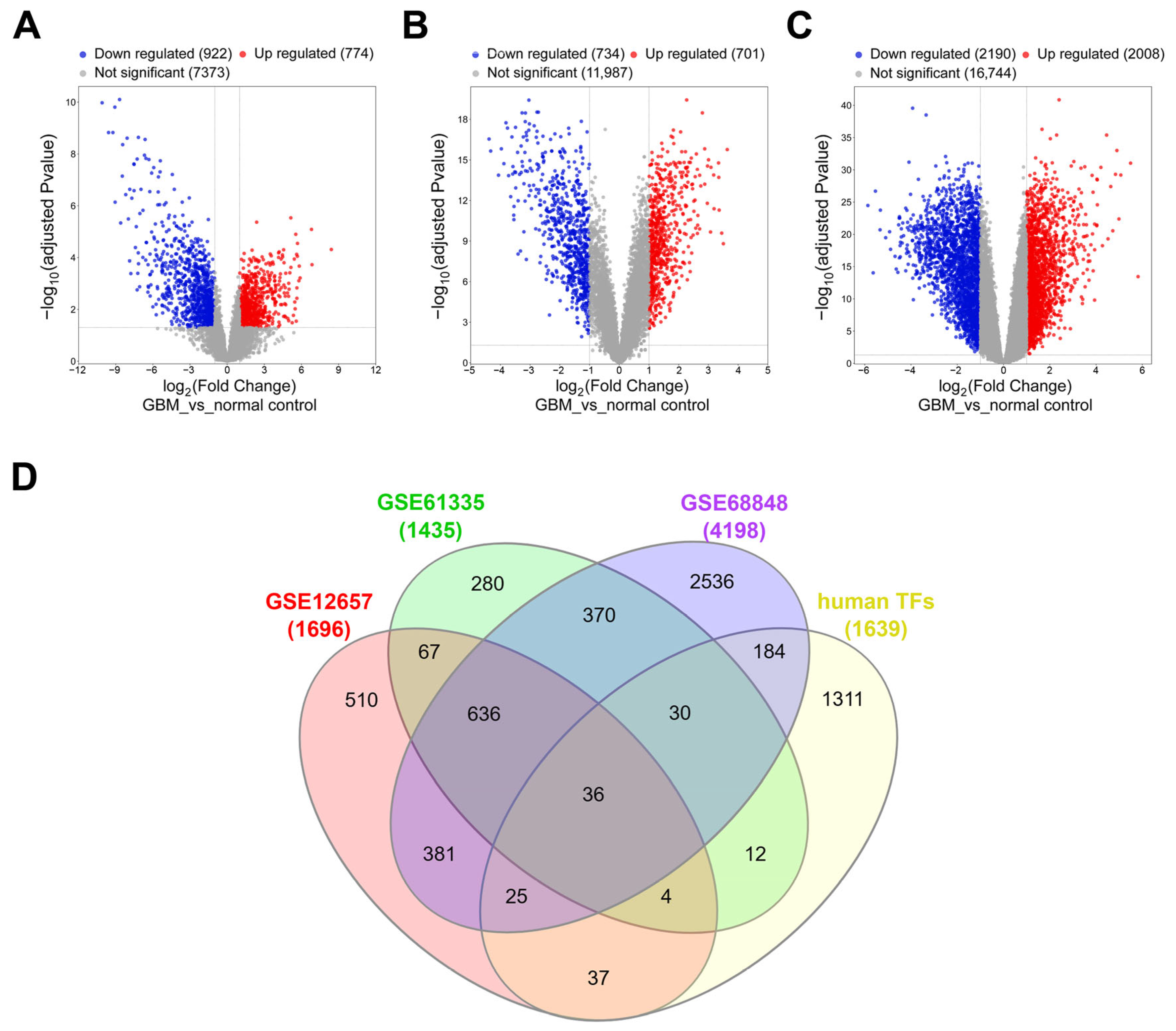
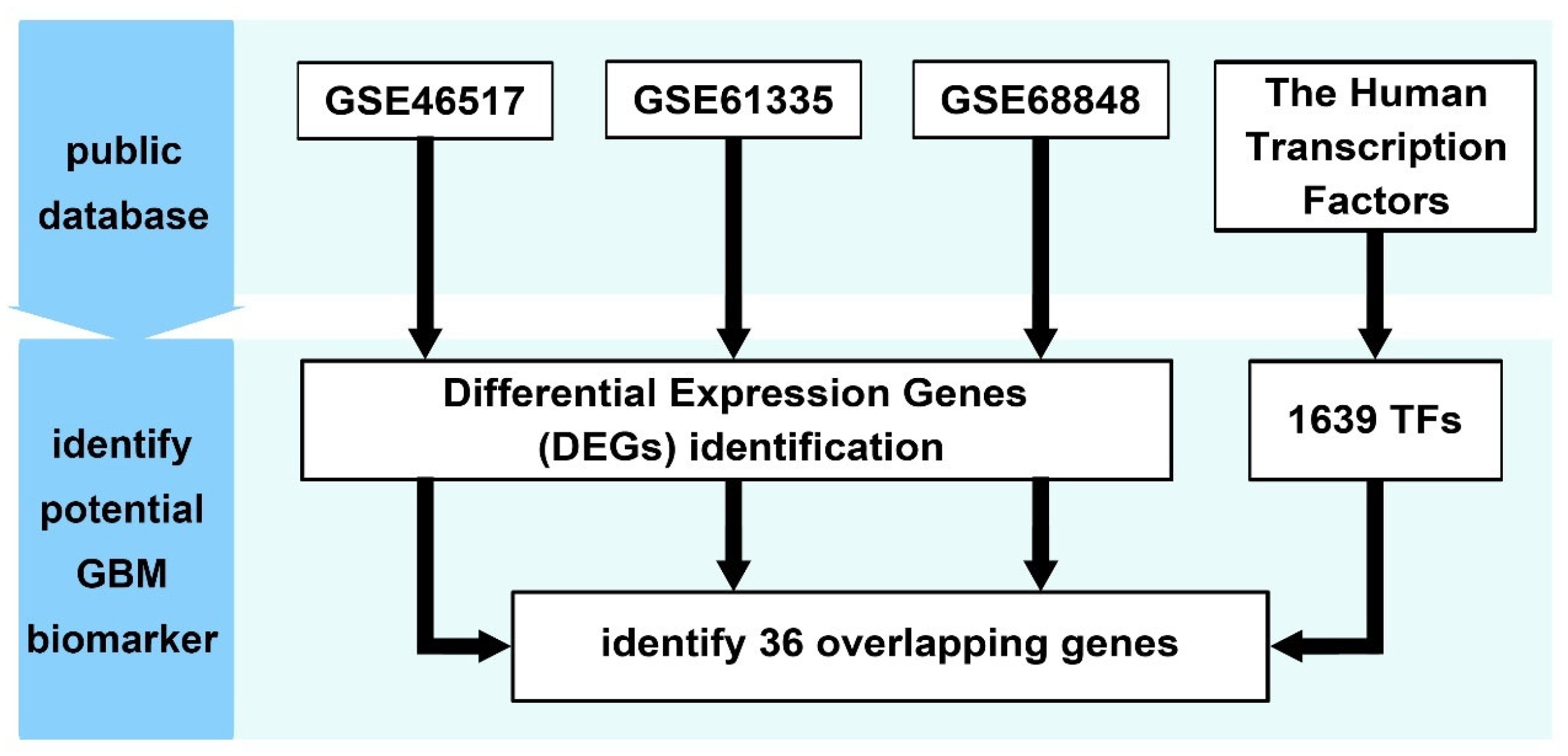
Despite accumulating evidence implicating EGR3 in glioblastoma, its functional role remains unresolved, with previous studies reporting contradictory effects on tumor cell proliferation and progression. To address this uncertainty, we performed a meta-analysis of three independent transcriptomic datasets to identify transcription factors consistently dysregulated in GBM. Among the 36 candidates identified, EGR3 was selected for further investigation based on its differential expression and prior implications in glioma biology. We assessed the functional impact of EGR3 using gain- and loss-of-function approaches in GBM cell lines and performed integrative transcriptomic analysis to explore its downstream regulatory network. These findings provide insights into the transcriptional programs associated with EGR3 and its potential contribution to GBM pathophysiology.
3. Discussion
This study investigates the role of EGR3 in GBM, an aggressive and treatment-resistant brain tumor. Through integrative transcriptomic analysis of public datasets, EGR3 was identified as a transcription factor consistently associated with GBM. Functional assays demonstrated that EGR3 promotes GBM cell viability and growth. By combining transcriptomic data with experimental validation, this study provides a systematic approach to characterizing transcriptional regulators relevant to GBM pathobiology.
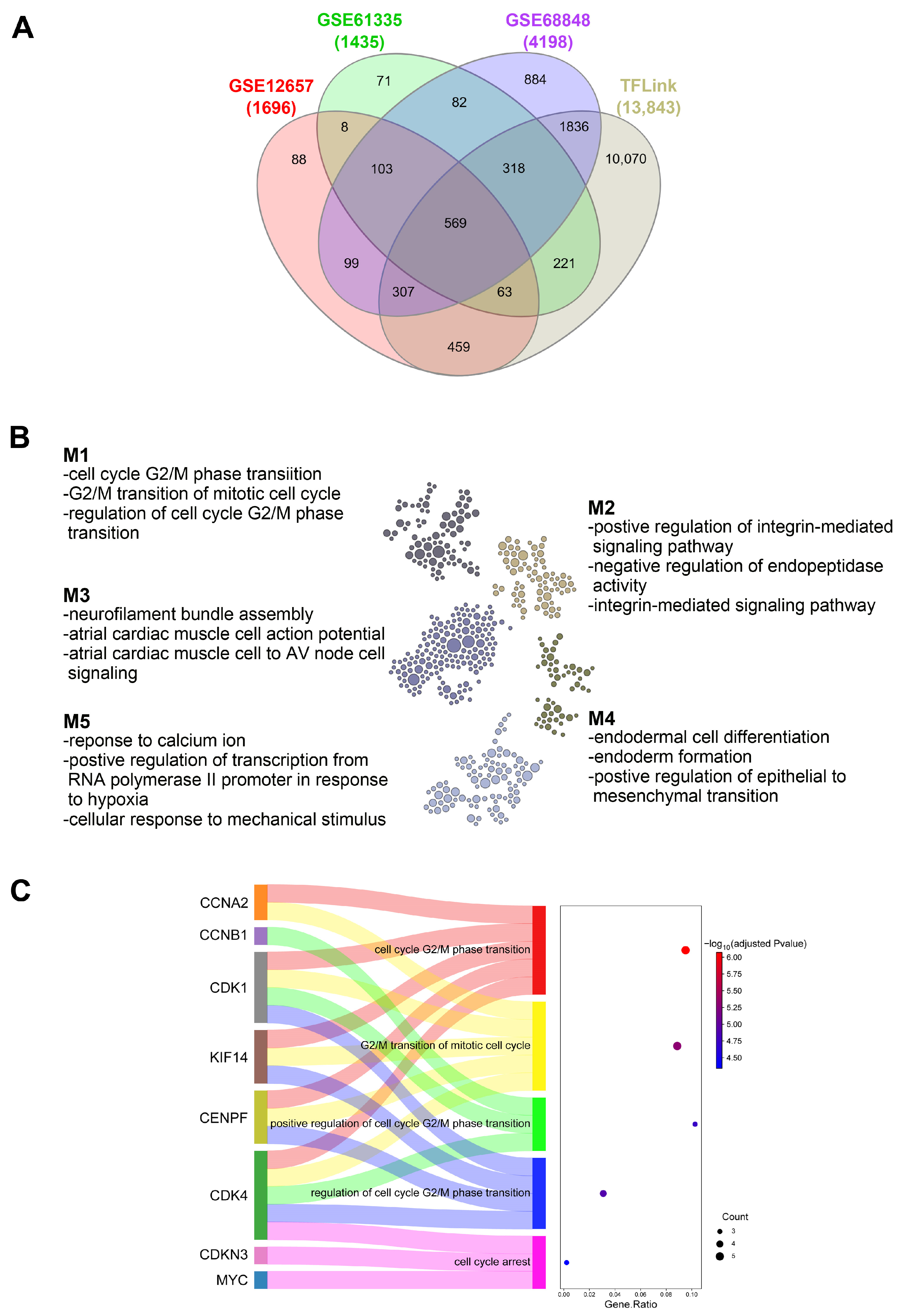
Our findings indicate that EGR3 promotes glioblastoma GBM cell growth, accompanied by increased expression of two oncogenic regulators, MYC and CDK1. Both genes are frequently overexpressed in GBM and are known to contribute to tumor cell proliferation. MYC functions as a key transcriptional regulator of cell growth and metabolic reprogramming and has been consistently associated with high-grade gliomas, including GBM [
12]. CDK1, a central mediator of the G2/M phase transition, plays a direct role in mitotic entry and has been linked to poor prognosis in GBM [
13]. The observed upregulation of
MYC and
CDK1 following EGR3 overexpression suggests that EGR3 may influence cell proliferation by modulating transcriptional programs governing cell cycle progression and metabolism. Together, these results support the existence of an EGR3-associated regulatory program involving MYC and CDK1, while also suggesting that additional downstream targets may contribute to the broader oncogenic landscape in GBM. Although MYC and CDK1 were prioritized based on their consistent upregulation and well-established roles in glioma biology, it is likely that EGR3 governs a wider transcriptional network influencing multiple aspects of tumor progression. Additional candidate targets identified from transcriptomic analysis may play important roles in processes such as invasion, resistance to apoptosis, or therapy response. Future investigations using genome-wide approaches—such as chromatin immunoprecipitation sequencing (ChIP-seq) or transcriptomic profiling following EGR3 perturbation—will be critical to fully define the EGR3 regulatory network and its multifaceted role in GBM progression.
Although this study supports the role of EGR3 as an oncogenic driver in GBM by promoting cell growth and upregulating
MYC and
CDK1, its function in cancer appears to be complex and context-dependent. Previous studies have reported contradictory findings regarding EGR3′s function in gliomas. For instance, Tang et al. [
7] demonstrated that EGR3 promotes GBM cell proliferation, migration, and invasion, consistent with our findings. In contrast, Shen et al. [
8] reported that EGR3 acts as a tumor suppressor, inhibiting glioma cell proliferation. These conflicting results may be attributed to several factors. First, the cellular context, including differences in cell type, genetic background, and tumor microenvironment, may influence EGR3′s functional role. In some contexts, EGR3 may activate growth-promoting genes, such as MYC and CDK1, as demonstrated in this study, while in others, it may activate or repress genes involved in growth suppression or apoptosis. Second, EGR3 may participate in complex regulatory networks with context-dependent co-factors. As a member of the early growth response gene family, a group of transcription factors known for their context-dependent functions, EGR3 can either activate or repress a diverse array of target genes. The availability of co-factors, chromatin states, and interactions with other signaling pathways can dramatically alter its regulatory role. Finally, the apparent paradox may also reflect differences in experimental design, including the use of distinct cell lines, gene editing methods, and culture conditions. Our study specifically focused on human GBM cell lines, where EGR3 consistently promoted cell viability and growth. These findings may not be generalizable to other tumor types or non-cancerous cells, where EGR3 may function differently. This context-dependent regulatory role of EGR3 highlights the importance of carefully considering experimental models and conditions when interpreting its function. Our results contribute to a more nuanced understanding of EGR3, demonstrating that it can function as an oncogenic driver in the specific context of GBM, while also recognizing that its role may vary in other settings.
A deeper understanding of the conflicting roles of EGR3 in gliomas requires careful consideration of the molecular context in which EGR3 operates. Differences in glioma subtypes, such as classical versus mesenchymal, may result in divergent transcriptional programs and responsiveness to EGR3. Moreover, the tumor microenvironment—including the presence of immune cells, stromal components, and hypoxic conditions—can influence transcription factor function through paracrine signaling and chromatin remodeling. EGR3′s activity is also shaped by co-regulatory proteins, such as other transcription factors or chromatin modifiers, which may differ in abundance or interaction patterns across tumor models. These factors may determine whether EGR3 functions as a transcriptional activator or repressor of oncogenic or tumor-suppressive targets. Furthermore, post-translational modifications of EGR3, such as phosphorylation, could modulate its DNA-binding affinity and transcriptional output in a context-specific manner. Dissecting these layers of regulation in future studies will be essential to reconcile the contradictory findings and define the precise role of EGR3 in gliomagenesis.
Emerging evidence from other tumor types supports the highly context-dependent function of EGR3. In nasopharyngeal carcinoma, EGR3 is activated under hypoxia and contributes to immunosuppression and tumor growth through regulation of IL10 and TGFB1 in regulatory B cells [
14]. In contrast, in breast cancer cells treated with a curcumin analog, EGR3 upregulation was linked to inhibition of cell migration, implicating a tumor-suppressive role [
15]. Similarly, in hepatocellular carcinoma [
16], canine mammary cancer [
17], and prostate cancer [
18], EGR3 appears to inhibit growth and metastasis, often through suppression of the epithelial–mesenchymal transition or induction of apoptosis-related pathways. A distinct mechanism was also reported in hepatocellular carcinoma, where EGR3 promoted Fas ligand expression to drive apoptosis [
16]. However, in tamoxifen-resistant breast cancer, EGR3 was upregulated and directly promoted expression of
MCL1, contributing to therapy resistance [
19]. These results suggest that EGR3 may shift between pro- or anti-tumorigenic functions depending on the oncogenic signaling landscape or therapeutic context. Pan-cancer transcriptomic analyses further reinforce the importance of tumor context in determining EGR3 function. A comprehensive bioinformatics study revealed that EGR3 expression is altered across multiple cancers and correlates with immune infiltration, tumor mutational burden, and clinical prognosis [
20]. In nasopharyngeal [
21] and liver cancer [
22], oncogenic microRNAs, such as miR-483-5p and miR-210, directly target EGR3 to promote tumor progression and metastasis. In leukemia, EGR3 expression was shown to be elevated at relapse and associated with immune and lineage differentiation pathways [
23,
24]. In prostate cancer, EGR3 suppresses metastasis by inducing the expression of tumor suppressor genes, such as ZFP36 and SOCS3 [
18], while additional evidence suggests that EGR3 loss correlates with relapse and reduced survival [
25]. A study in gastric cancer also reported that decreased EGR3 expression is associated with poor prognosis, further confirming its suppressive potential in some tumor types [
26]. Collectively, these findings underscore that EGR3 function is shaped by a convergence of tumor-intrinsic and -extrinsic factors—including genetic alterations, transcriptional networks, hormonal cues, and microenvironmental signals. This mechanistic diversity necessitates careful delineation of EGR3′s role in each tumor setting. Future research employing integrative multi-omics approaches across defined cancer subtypes will be critical to identify shared and unique EGR3-driven pathways, ultimately informing context-appropriate therapeutic strategies.
While this study provides valuable mechanistic insights into the role of EGR3 in GBM, a key limitation is the reliance on in vitro models. The functional assays were conducted in established human GBM cell lines, which, although widely used and experimentally tractable, do not fully recapitulate the complex tumor microenvironment, cellular heterogeneity, and immune interactions observed in patient-derived glioblastomas. As such, the translatability of our findings to clinical settings remains to be validated. In future work, in vivo models—such as orthotopic xenografts or genetically engineered mouse models—will be essential to evaluate the impact of EGR3 modulation on tumor growth, invasion, and therapeutic response within the native brain microenvironment. These models will also provide a platform to assess the feasibility of targeting the EGR3–MYC–CDK1 regulatory axis as a potential therapeutic strategy. Furthermore, incorporating patient-derived glioma stem-like cells may help to better reflect interpatient heterogeneity and therapeutic resistance mechanisms.
4. Materials and Methods
4.1. Transcriptomic Dataset Collection and DEG Analysis
Three independent transcriptomic datasets, GSE12657, GSE61335, and GSE68848, were retrieved from the Gene Expression Omnibus (GEO) database. GSE12657 and GSE68848 were analyzed using the GEO2R web tool (National Center for Biotechnology Information, Bethesda, MD, USA), while GSE61335 underwent preprocessing and differential expression analysis using the “affy” package (version 1.82.0, Technical University of Denmark, Kongens Lyngby, Denmark) in R. For all datasets, differentially expressed genes (DEGs) were defined by an adjusted p-value < 0.05 and an absolute log2 fold change (|log2FC|) ≥ 1. DEGs were cross-referenced with a curated list of 1639 human transcription factors to identify consistently dysregulated TFs across all three datasets.
4.2. Survival Analysis
Survival associations for selected EGR3 target genes were evaluated using Kaplan–Meier data from the Human Protein Atlas. Gene expression levels were stratified into high and low expression groups, and survival curves were generated using the SRplot online analysis platform (Central South University, Changsha, Hunan, China). Log-rank tests were used to assess statistical significance.
4.3. Cell Culture and EGR3 Overexpression
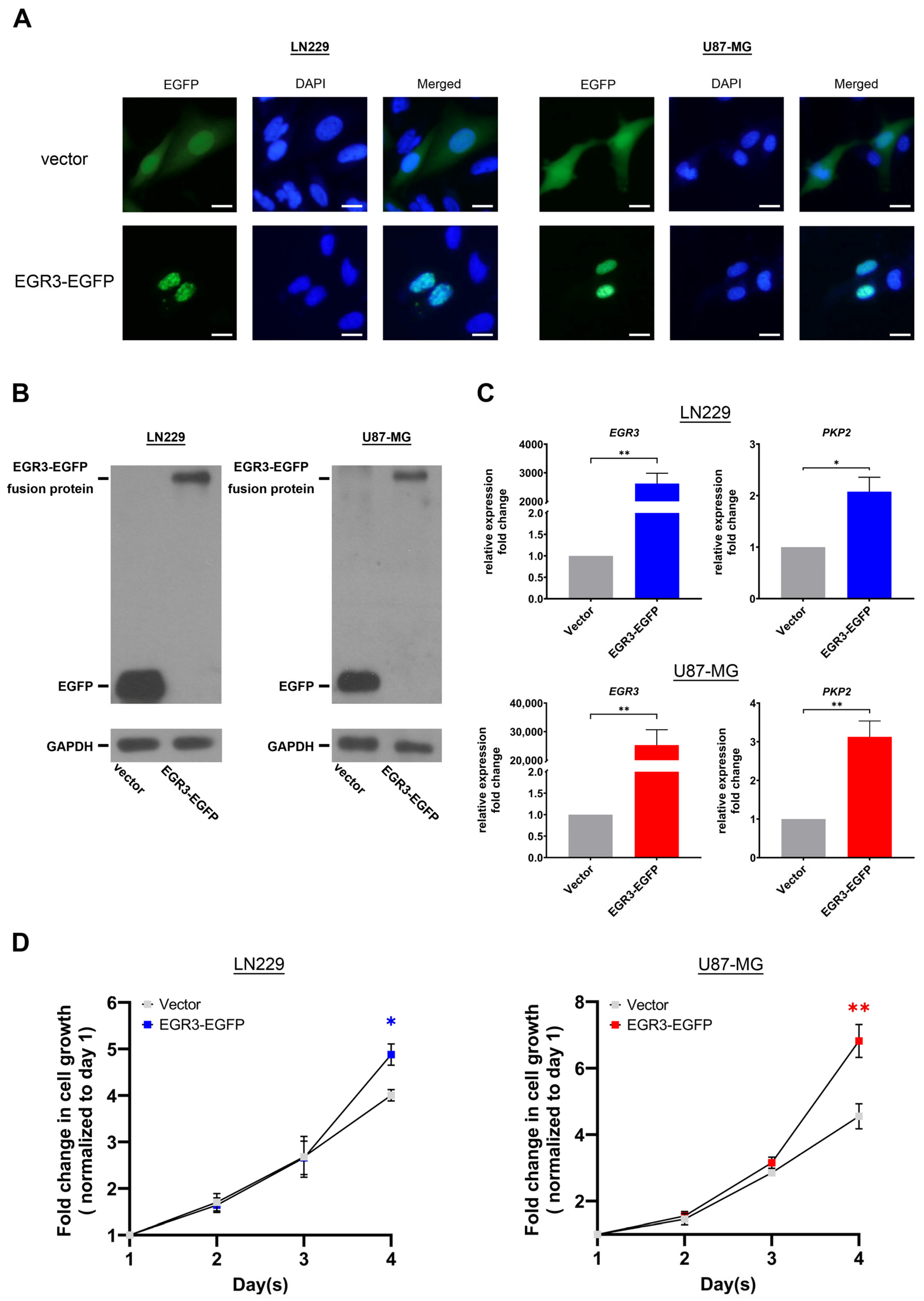
LN229 and U87-MG human glioblastoma cell lines were maintained in high-glucose Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Waltham, MA, USA; contributor: UNI-ONWARD Corp., New Taipei City, Taiwan) supplemented with 10% fetal bovine serum (FBS; Avantor, Radnor, Pennsylvania, USA; contributor: Blossom Biotechnologies, Inc., Taipei City, Taiwan), a 1% antibiotic-antimycotic solution (Gibco, Waltham, MA, USA; contributor: UNI-ONWARD Corp., New Taipei City, Taiwan), and 4.5 g/L of D-glucose. Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. For overexpression experiments, cells were transfected with a plasmid encoding EGR3-GFP or a GFP control using the DreamFect Gold transfection reagent (OZ Biosciences, Marseille, France; contributor: Harmony Biosolution Inc., New Taipei City, Taiwan).
4.4. CRISPR/Cas9-Mediated Knockout of EGR3
Two single-guide RNAs (sgRNAs) targeting the EGR3 coding sequence were designed using the CHOPCHOP online tool [
27]. sgRNA-4 (5′-GCCGTTCGGACGAGCTGACC-3′, targeting nucleotides 860–879) and sgRNA-5 (5′-CGTGTCTTTCCACGACCCCC-3′, targeting nucleotides 471–490) were individually cloned into a plasmid vector encoding both Cas9 and EGFP. LN229 cells were transfected with the sgRNA constructs using the DreamFect Gold transfection reagent, following the manufacturer’s protocol. After transfection, cells were subjected to puromycin (1 μg/mL) selection until control cells (DreamFect-only group) were completely eliminated, typically within one week. A scramble sgRNA (5′-GCTTAGTTACGCGTGGACGA-3′) was used as a negative control [
28]. Knockout efficiency of EGR3 was validated by evaluating the expression of PKP2, a known downstream target of EGR3, using quantitative real-time PCR (qRT-PCR). The sequences of primers utilized in this study are provided in
Table 1.
4.5. Western Blot Analysis
Protein lysates were prepared using a RIPA buffer supplemented with protease (Sigma-Aldrich, St. Louis, MO, USA; contributor: UNI-ONWARD Corp., New Taipei City, Taiwan) and phosphatase inhibitors (EMD Millipore Corp., Burlington, MA, USA; contributor: UNI-ONWARD Corp., New Taipei City, Taiwan). Samples were separated on SDS-PAGE gels and transferred onto PVDF membranes (EMD Millipore Corp., Burlington, MA, USA; contributor: UNI-ONWARD Corp., New Taipei City, Taiwan). Membranes were probed with primary antibodies against EGR3 (1:6000; Cloud-Clone Corp., Houston, TX, USA; contributor: ASIA-BIOSCIENCE CO., LTD., Taipei City, Taiwan), GFP (1:5000; Proteintech Group, Rosemont, IL, USA; contributor: ABreal Biotech Co., Taipei City, Taiwan), and GAPDH (1:15,000; Proteintech Group, Rosemont, IL, USA; contributor: ABreal Biotech Co., Taipei City, Taiwan), followed by HRP-conjugated secondary antibodies. Signal detection was performed using enhanced chemiluminescence (ECL; Cytiva, Marlborough, Massachusetts, USA; contributor: UNI-ONWARD Corp., New Taipei City, Taiwan).
4.6. Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA; contributor: Life Technologies Co., Ltd., Taipei City, Taiwan) and reverse-transcribed into cDNA using the First Strand Synthesis Kit (BIONOVAS Biotechnology Co., Ltd., Seattle, WA, USA; contributor: WonWon Biotechnology Co., Ltd., Taoyuan City, Taiwan). Quantitative real-time PCR was performed using the SYBR Green Master Mix (ABclonal, Woburn, Massachusetts, USA; contributor: BIOTOOLS CO., LTD., New Taipei City, Taiwan) on an ABI system (Applied Biosystems, Thermo Fisher Scientific, Foster City, CA, USA; contributor: LIFE TECHNOLOGIES CO., LTD., Taipei City, Taiwan). GAPDH served as the internal control. Gene expression levels were calculated using the 2−ΔΔCt method.
4.7. Cell Viability Assay
Cell viability was evaluated using the MTT assay following EGR3 overexpression or knockout. Briefly, 5 × 103 cells per well were seeded into 96-well plates in 200 μL of a complete medium and incubated overnight. Subsequently, 10 μL of the MTT reagent (5 mg/mL) was added directly to each well and incubated for 3 h at 37 °C. After incubation, the supernatant was carefully removed, and 100 μL of DMSO was added to dissolve the formazan crystals. Absorbance was measured at 570 nm, and viability was compared between control and experimental groups.
4.8. Transcriptomic Integration and Functional Enrichment
Putative
EGR3 targets were retrieved from the TFLink database [
11] and cross-referenced with DEGs identified in the meta-analysis. The overlapping gene set was analyzed using HumanBase for functional module discovery with brain-specific settings [
29]. Gene ontology biological process enrichment analysis was performed to identify regulatory modules. A subset of cell cycle-related genes from the top-enriched module was selected for further analysis.
4.9. Statistical Analysis
Statistical analyses were performed using GraphPad Prism version 9.0 (GraphPad Software, Boston, MA, USA) and the SRplot online analysis platform [
30]. qRT-PCR results were analyzed using either one-way analysis of variance (ANOVA) with Dunnett’s post hoc test or a two-tailed unpaired Student’s
t-test, depending on the number of comparison groups. Western blot quantification data were evaluated using one-way ANOVA followed by Dunnett’s multiple comparisons test. Cell viability results from MTT assays were analyzed using two-way ANOVA with Bonferroni correction. Data are presented as means ± standard errors of the mean (SEM). Statistical significance was defined as follows: *,
p < 0.05, **,
p < 0.01, ***,
p < 0.001, and ****,
p < 0.0001.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}