
The Circadian Rhythm Gene Network Could Distinguish Molecular Profile and Prognosis for Glioblastoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
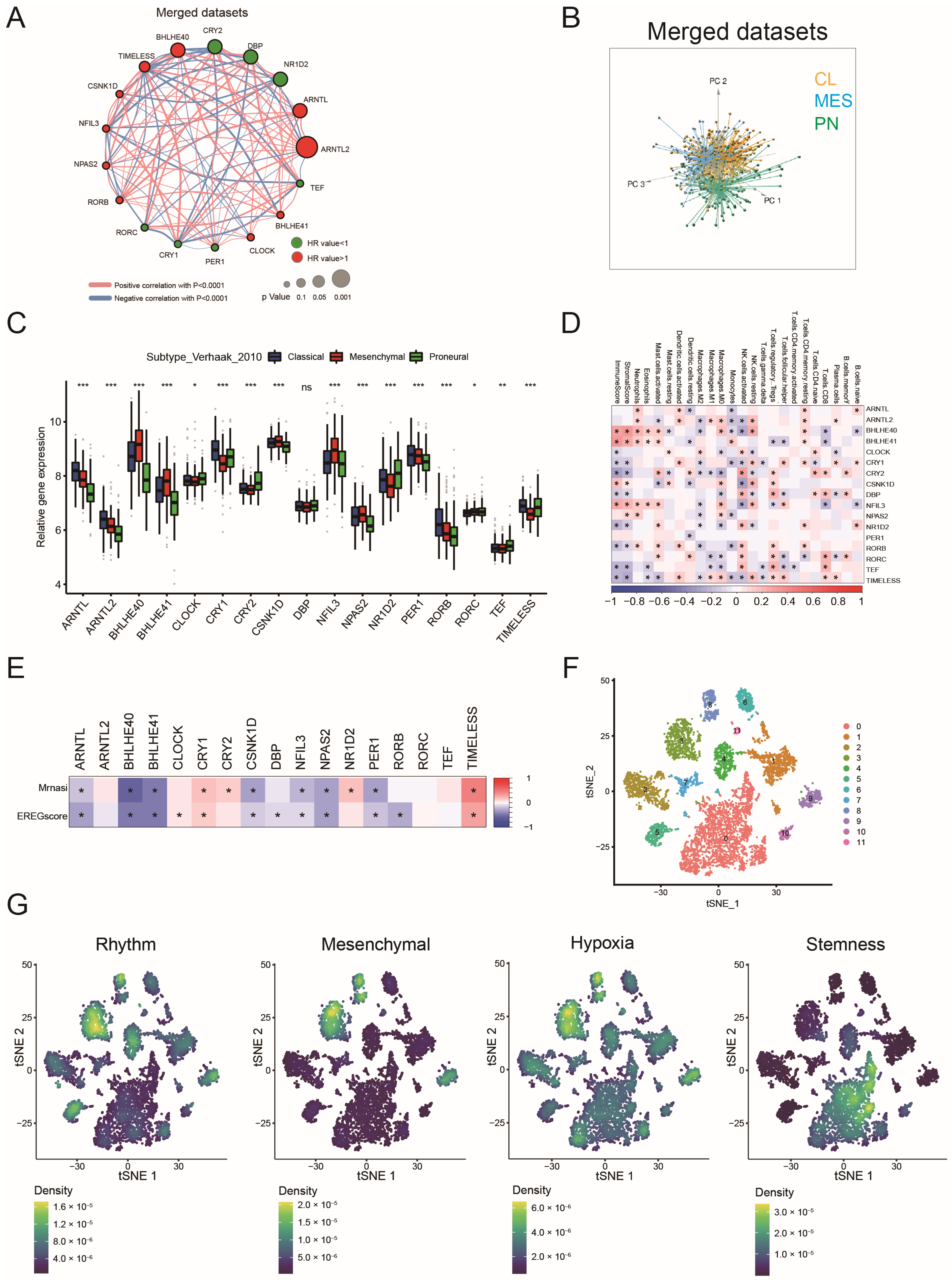
2.1. Landscape of Circadian Rhythm Gene Expression in GBM
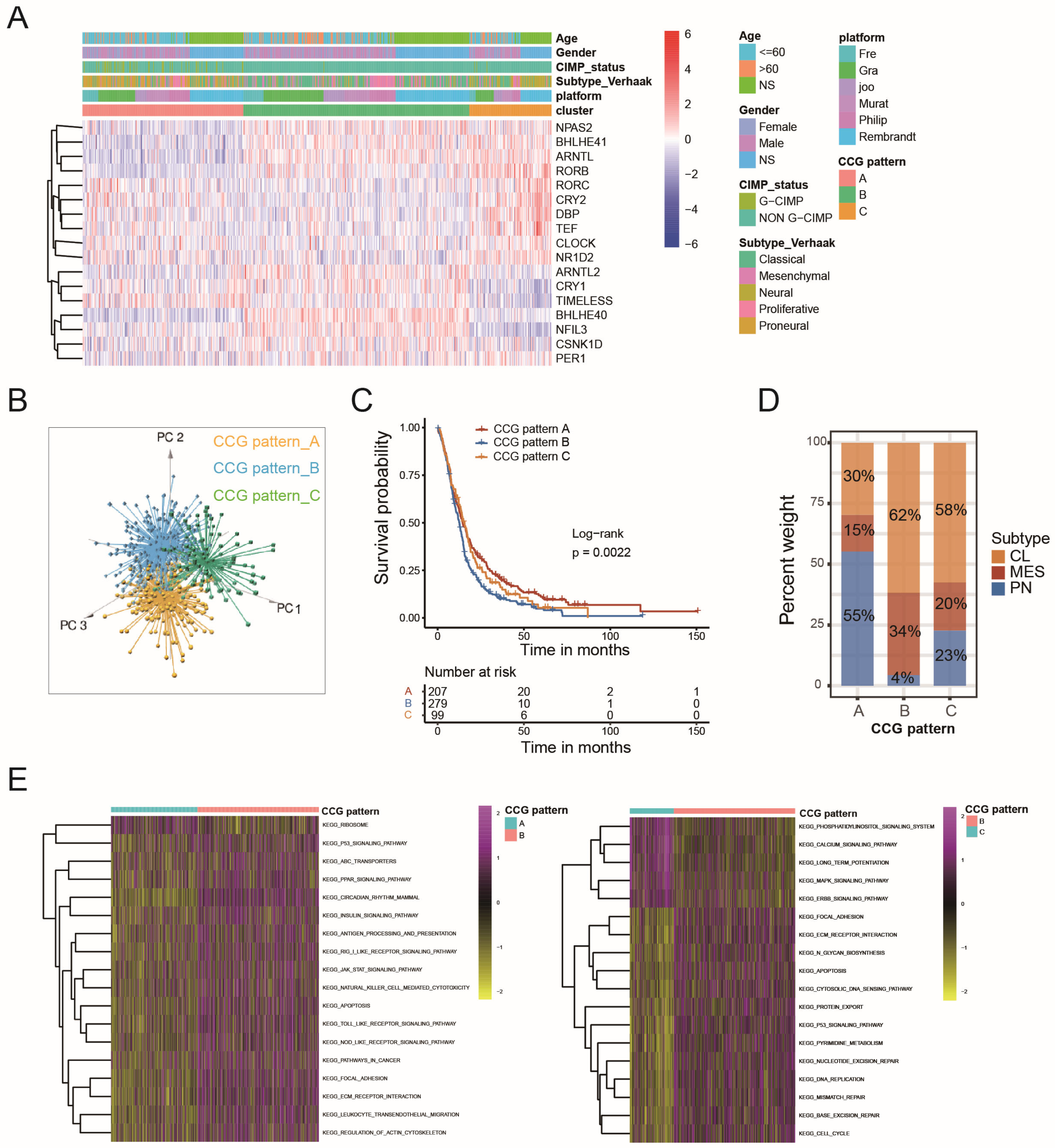
2.2. Circadian Core-Gene Patterns and Circadian-Related Gene Patterns Could Stratify GBM Patients According to Molecular Profile and Prognosis
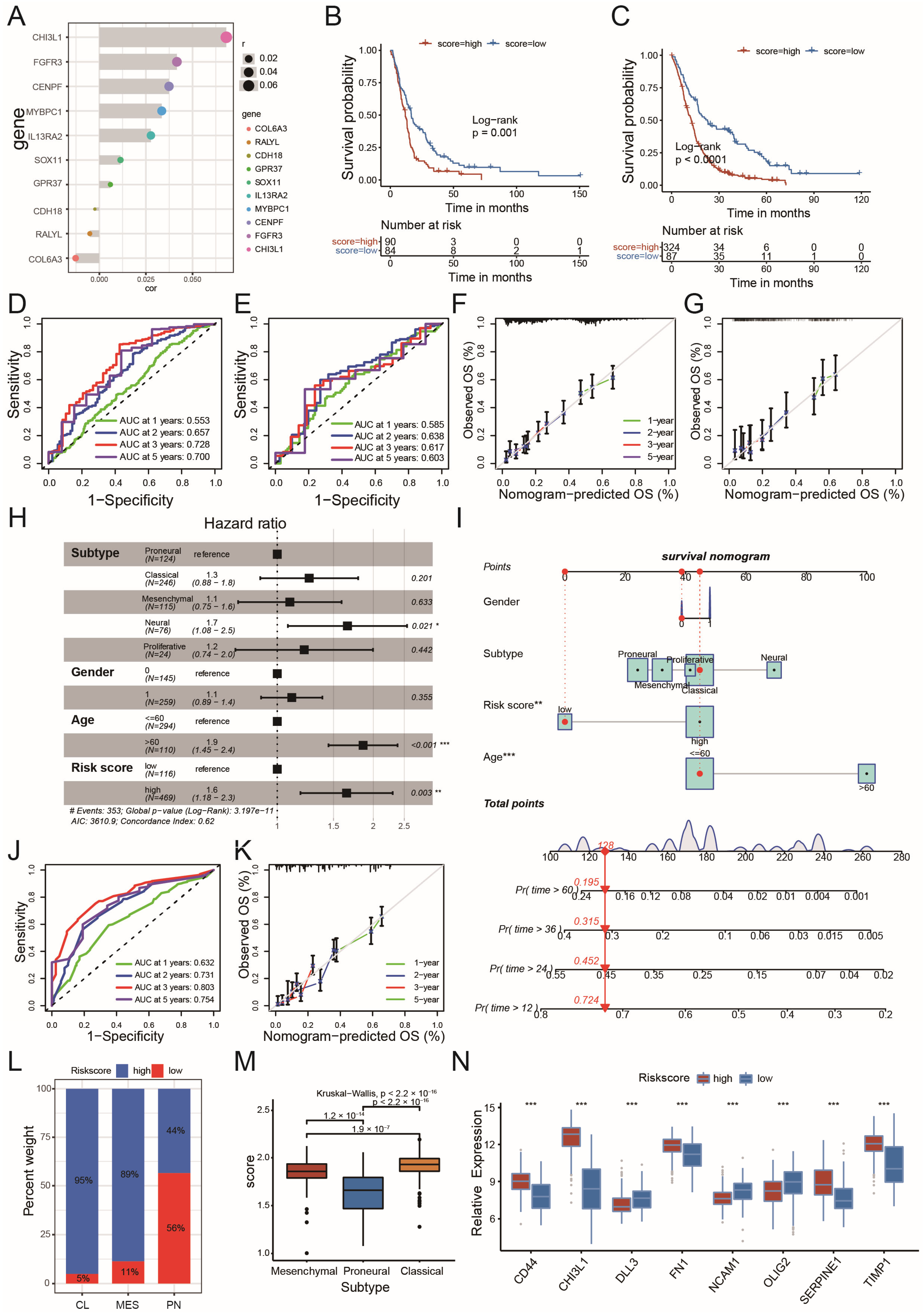
2.3. Circadian Risk Score Could Show Its Prognostic Value in GBM
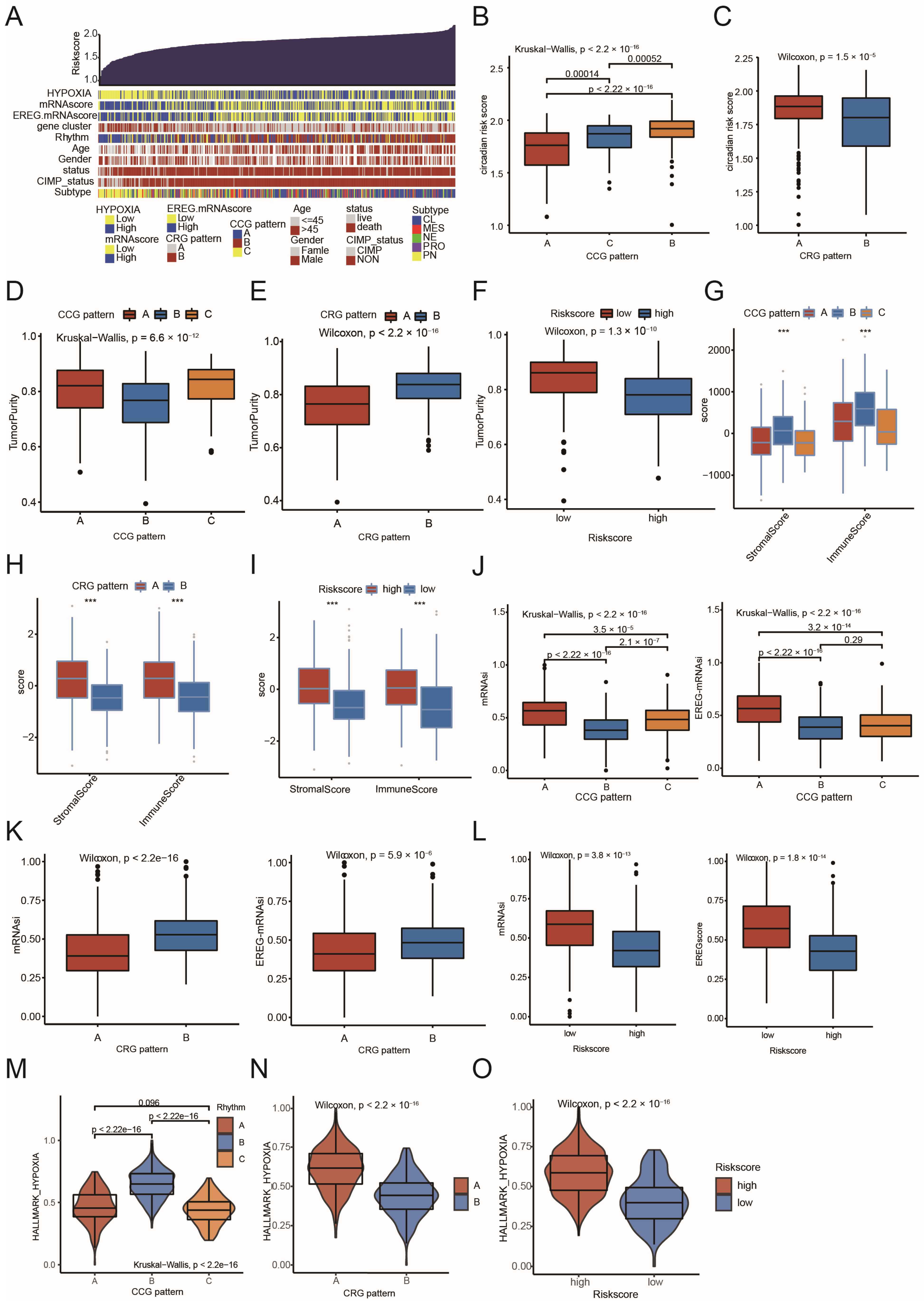
2.4. Comparison of CCG Patterns, CRG Patterns, Circadian Risk Score, and Their Association with Fundamental Characteristics of GBM
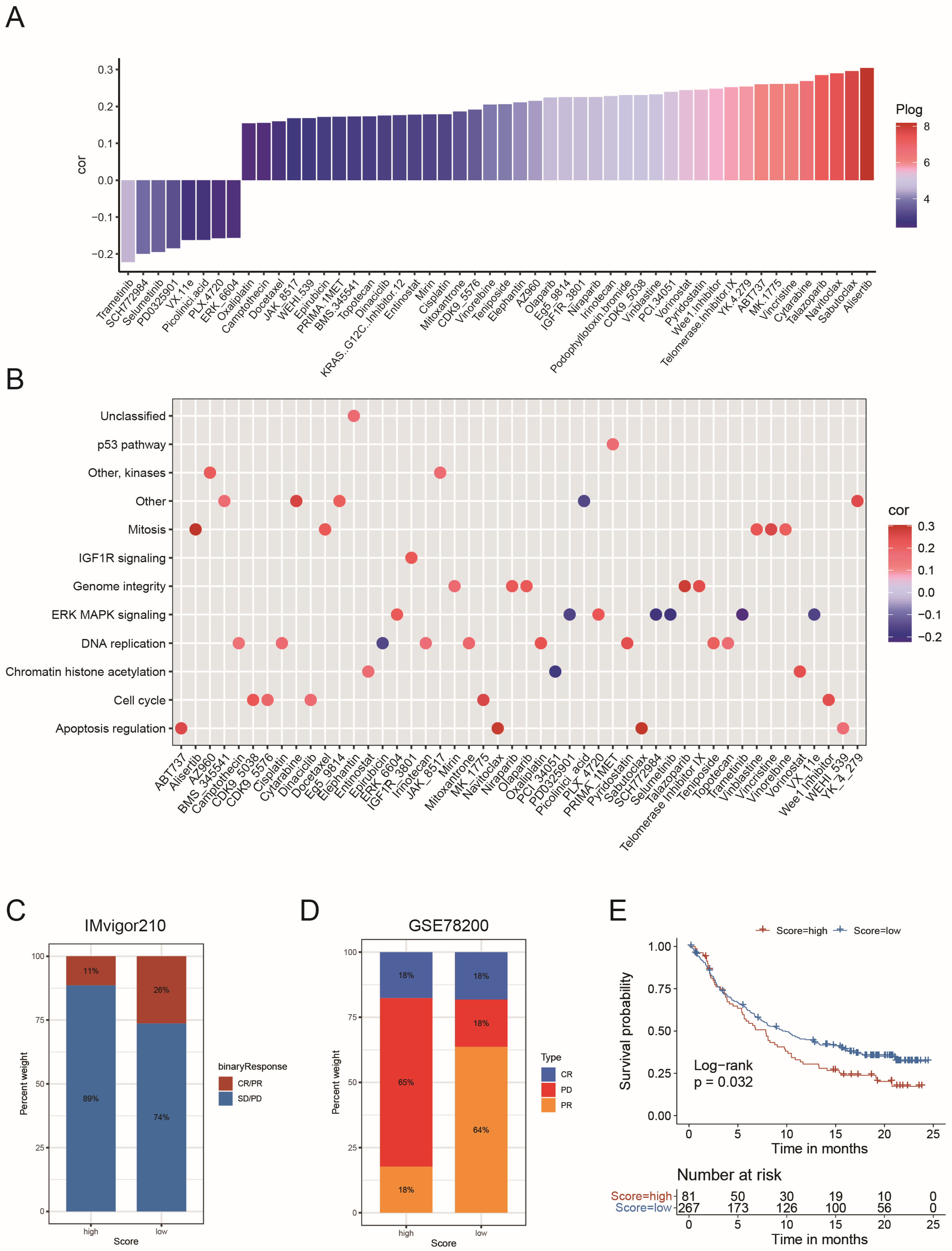
2.5. The Role of the Risk Score in the Prediction of Therapeutic Benefits
3. Discussion
4. Methods
4.1. Data Collection and Processing
4.2. Unsupervised Clustering for 17 Circadian Rhythm Genes
4.3. Single-Cell RNA Sequencing Analysis
4.4. Gene Functional Annotation Based on Gene Set Variation Analysis (GSVA)
4.5. TME Cell Infiltration and Hypoxia Estimation
4.6. Identification of DEGs Among Circadian Core-Gene Patterns
4.7. Constructing the Circadian Risk Scoring System to Evaluate Individual GBM Samples
4.8. Analyzing the Association Between Circadian Risk Score and Response to Immunotherapy
4.9. Association Analysis Between Drug Sensitivity and Circadian Risk Score
4.10. Nomogram Construction
4.11. Western Blot Analysis
4.12. Ethynyl-2′-Deoxyuridine (EdU) Cell Proliferation Assay and Trametinib Pretreatment
4.13. Cell Transfection
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APCs | Antigen-presenting cells |
| AUCs | area under the curves |
| CCLE | Cancer Cell Line Encyclopedia |
| CGGA | Chinese Glioma Genome Atlas |
| CL | Classical |
| CNS | Central Nervous System |
| CNV | copy number variation |
| DEGs | differential expressing genes |
| GBM | glioblastoma |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| GEO | Gene Expression Omnibus |
| GSVA | gene set variation analysis |
| HR | hazard ratio |
| IDH | Isocitrate dehydrogenase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LASSO | Least Absolute Shrinkage and Selection Operator regression |
| MES | Mesenchymal |
| MDSet | merged dataset |
| PCA | principal component analysis |
| PN | Proneural |
| ROC | receiver operating characteristic |
| ssGSEA | single-sample gene set enrichment analysis |
| TCGA | The Cancer Genome Atlas |
References
- Luo, C.; Song, K.; Wu, S.; Hameed, N.F.; Kudulaiti, N.; Xu, H.; Qin, Z.Y.; Wu, J.S. The prognosis of glioblastoma: A large, multifactorial study. Br. J. Neurosurg. 2021, 35, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2018, 33, 152. [Google Scholar] [CrossRef] [PubMed]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.-H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e7. [Google Scholar] [CrossRef]
- Jesse, R.J.; Ignacio, A.; Clayton, W.L.; Carla, V.F. Circadian clock gene polymorphisms implicated in human pathologies. Trends Genet. 2024, 40, 834–852. [Google Scholar]
- Khan, S.; Liu, Y.; Siddique, R.; Nabi, G.; Xue, M.; Hou, H. Impact of chronically alternating light-dark cycles on circadian clock mediated expression of cancer (glioma)-related genes in the brain. Int. J. Biol. Sci. 2019, 15, 1816–1834. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.; Morton, A.R.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef]
- Han, S.; Kristina, C.; Harriet, E.G.; Eric, H. Hypoxia, metabolism, and the circadian clock: New links to overcome radiation resistance in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 129. [Google Scholar]
- Chen, P.; Hsu, W.H.; Chang, A.; Tan, Z.; Lan, Z.; Zhou, A.; Spring, D.J.; Lang, F.F.; Wang, Y.A.; DePinho, R.A. Circadian Regulator CLOCK Recruits Immune-Suppressive Microglia into the GBM Tumor Microenvironment. Cancer Discov. 2020, 10, 371–381. [Google Scholar] [CrossRef]
- Xuan, W.; Hsu, W.H.; Khan, F.; Dunterman, M.; Pang, L.; Wainwright, D.A.; Ahmed, A.U.; Heimberger, A.B.; Lesniak, M.S.; Chen, P. Circadian Regulator CLOCK Drives Immunosuppression in Glioblastoma. Cancer Immunol. Res. 2022, 10, 770–784. [Google Scholar] [CrossRef]
- Xiao, K.; Tan, J.; Yuan, J.; Peng, G.; Long, W.; Su, J.; Xiao, Y.; Xiao, Q.; Wu, C.; Qin, C.; et al. Prognostic value and immune cell infiltration of hypoxic phenotype-related gene signatures in glioblastoma microenvironment. J. Cell. Mol. Med. 2020, 24, 13235–13247. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.C.; Felipe, G.-C.; Jiali, L.; Steven, M.P.; Sergio, A.Q.; Yinyin, Y. The tumour ecology of quiescence: Niches across scales of complexity. Semin Cancer Biol. 2023, 92, 139–149. [Google Scholar]
- Wagner, P.M.; Monjes, N.M.; Guido, M.E. Chemotherapeutic Effect of SR9009, a REV-ERB Agonist, on the Human Glioblastoma T98G Cells. ASN Neuro 2019, 11, 1759091419892713. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef]
- Wang, Z.; Su, G.; Dai, Z.; Meng, M.; Zhang, H.; Fan, F.; Liu, Z.; Zhang, L.; Weygant, N.; He, F.; et al. Circadian clock genes promote glioma progression by affecting tumour immune infiltration and tumour cell proliferation. Cell Prolif. 2021, 54, e12988. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Yang, T.; Xing, H.; Wang, Y.; Gao, L.; Guo, X.; Xing, B.; Wang, Y.; Ma, W. Machine learning revealed stemness features and a novel stemness-based classification with appealing implications in discriminating the prognosis, immunotherapy and temozolomide responses of 906 glioblastoma patients. Brief. Bioinform. 2021, 22, bbab032. [Google Scholar] [CrossRef]
- Du, J.; Hou, K.; Mi, S.; Ji, H.; Ma, S.; Ba, Y.; Hu, S.; Xie, R.; Chen, L. Malignant Evaluation and Clinical Prognostic Values of m6A RNA Methylation Regulators in Glioblastoma. Front. Oncol. 2020, 10, 208. [Google Scholar] [CrossRef]
- Sun, Z.; Zhao, Y.; Wei, Y.; Ding, X.; Tan, C.; Wang, C. Identification and validation of an anoikis-associated gene signature to predict clinical character, stemness, IDH mutation, and immune filtration in glioblastoma. Front. Immunol. 2022, 13, 939523. [Google Scholar] [CrossRef]
- Lok, R.; Qian, J.; Chellappa, S.L. Sex differences in sleep, circadian rhythms, and metabolism: Implications for precision medicine. Sleep Med. Rev. 2024, 75, 101926. [Google Scholar] [CrossRef]
- Okyar, A.; Kumar, S.A.; Filipski, E.; Piccolo, E.; Ozturk, N.; Xandri-Monje, H.; Pala, Z.; Abraham, K.; Gomes, A.R.; Orman, M.N.; et al. Sex-, feeding-, and circadian time-dependency of P-glycoprotein expression and activity—Implications for mechanistic pharmacokinetics modeling. Sci. Rep. 2019, 9, 10505. [Google Scholar] [CrossRef]
- Diamantopoulou, Z.; Castro-Giner, F.; Schwab, F.D.; Foerster, C.; Saini, M.; Budinjas, S.; Strittmatter, K.; Krol, I.; Seifert, B.; Heinzelmann-Schwarz, V.; et al. The metastatic spread of breast cancer accelerates during sleep. Nature 2022, 607, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Furtado, A.; Costa, D.; Lemos, M.C.; Cavaco, J.E.; Santos, C.R.; Quintela, T. The impact of biological clock and sex hormones on the risk of disease. Adv. Protein Chem. Struct. Biol. 2023, 137, 39–81. [Google Scholar] [PubMed]
- Jarabo, P.; de Pablo, C.; González-Blanco, A.; Casas-Tintó, S. Circadian Gene Controls Tumorigenesis through Modulation of Myc Accumulation in Glioblastoma Cells. Int. J. Mol. Sci. 2022, 23, 2043. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.; Rich, J.N.; Kay, S.A. Watching the clock in glioblastoma. Neuro Oncol. 2023, 25, 1932–1946. [Google Scholar] [CrossRef]
- Jia, J.L.; Alshamsan, B.; Ng, T.L. Temozolomide Chronotherapy in Glioma: A Systematic Review. Curr Oncol. 2023, 30, 1893–1902. [Google Scholar] [CrossRef]
- Damato, A.R.; Katumba, R.G.; Luo, J.; Atluri, H.; Talcott, G.R.; Govindan, A.; Slat, E.A.; Weilbaecher, K.N.; Tao, Y.; Huang, J.; et al. A randomized feasibility study evaluating temozolomide circadian medicine in patients with glioma. Neurooncol. Pract. 2022, 9, 193–200. [Google Scholar] [CrossRef]
- Sapienza, L.G.; Nasra, K.; Berry, R.; Danesh, L.; Little, T.; Abu-Isa, E. Clinical effects of morning and afternoon radiotherapy on high-grade gliomas. Chronobiol. Int. 2021, 38, 732–741. [Google Scholar] [CrossRef]
- Slat, E.A.; Sponagel, J.; Marpegan, L.; Simon, T.; Kfoury, N.; Kim, A.; Binz, A.; Herzog, E.D.; Rubin, J.B. Cell-intrinsic, Bmal1-dependent Circadian Regulation of Temozolomide Sensitivity in Glioblastoma. J. Biol. Rhythms 2017, 32, 121–129. [Google Scholar] [CrossRef]
- Buhr, E.D.; Takahashi, J.S. Molecular components of the Mammalian circadian clock. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–27. [Google Scholar]
- Tahira, K.; Ueno, T.; Fukuda, N.; Aoyama, T.; Tsunemi, A.; Matsumoto, S.; Nagura, C.; Matsumoto, T.; Soma, M.; Shimba, S.; et al. Obesity alters the expression profile of clock genes in peripheral blood mononuclear cells. Arch. Med. Sci. AMS 2011, 7, 933–940. [Google Scholar] [CrossRef]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Karen, D.W.; Richard, J.G. To infinium, and beyond! Cancer Cell 2010, 17, 419–420. [Google Scholar]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Chen, H.; Yao, J.; Bao, R.; Dong, Y.; Zhang, T.; Du, Y.; Wang, G.; Ni, D.; Xun, Z.; Niu, X.; et al. Cross-talk of four types of RNA modification writers defines tumor microenvironment and pharmacogenomic landscape in colorectal cancer. Mol. Cancer 2021, 20, 29. [Google Scholar] [CrossRef]
- Madhavan, S.; Zenklusen, J.C.; Kotliarov, Y.; Sahni, H.; Fine, H.A.; Buetow, K. Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 2009, 7, 157–167. [Google Scholar] [CrossRef]
- Gravendeel, L.A.; Kouwenhoven, M.C.; Gevaert, O.; de Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; Daemen, A.; Bleeker, F.E.; Bralten, L.B.; Kloosterhof, N.K.; et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.F.; De Tribolet, N.; Regli, L.; Wick, W.; Kouwenhoven, M.C.; et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef]
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y.L.; Park, W.Y.; Kong, D.S.; et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Tang, X.R.; Li, Y.Q.; Liang, S.B.; Jiang, W.; Liu, F.; Ge, W.X.; Tang, L.L.; Mao, Y.P.; He, Q.M.; Yang, X.J.; et al. Development and validation of a gene expression-based signature to predict distant metastasis in locoregionally advanced nasopharyngeal carcinoma: A retrospective, multicentre, cohort study. Lancet Oncol. 2018, 19, 382–393. [Google Scholar] [CrossRef]
- Du, J.; Ji, H.; Ma, S.; Jin, J.; Mi, S.; Hou, K.; Dong, J.; Wang, F.; Zhang, C.; Li, Y.; et al. m6A regulator-mediated methylation modification patterns and characteristics of immunity and stemness in low-grade glioma. Brief. Bioinform. 2021, 22, bbab013. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, F.; Zhang, Z.; Zhang, J.; Hu, J.; Hu, W.; Gao, J.; Fu, M.; Feng, Y.; Kong, L. The Circadian Rhythm Gene Network Could Distinguish Molecular Profile and Prognosis for Glioblastoma. Int. J. Mol. Sci. 2025, 26, 5873. https://doi.org/10.3390/ijms26125873
Wan F, Zhang Z, Zhang J, Hu J, Hu W, Gao J, Fu M, Feng Y, Kong L. The Circadian Rhythm Gene Network Could Distinguish Molecular Profile and Prognosis for Glioblastoma. International Journal of Molecular Sciences. 2025; 26(12):5873. https://doi.org/10.3390/ijms26125873
Chicago/Turabian StyleWan, Fangzhu, Zongpu Zhang, Jinsen Zhang, Jiyi Hu, Weixu Hu, Jing Gao, Minjie Fu, Yuan Feng, and Lin Kong. 2025. "The Circadian Rhythm Gene Network Could Distinguish Molecular Profile and Prognosis for Glioblastoma" International Journal of Molecular Sciences 26, no. 12: 5873. https://doi.org/10.3390/ijms26125873
APA StyleWan, F., Zhang, Z., Zhang, J., Hu, J., Hu, W., Gao, J., Fu, M., Feng, Y., & Kong, L. (2025). The Circadian Rhythm Gene Network Could Distinguish Molecular Profile and Prognosis for Glioblastoma. International Journal of Molecular Sciences, 26(12), 5873. https://doi.org/10.3390/ijms26125873