

Methionine Restriction Attenuates Scar Formation in Fibroblasts Derived from Patients with Post-Burn Hypertrophic Scar
 , ,
, , 
Abstract
1. Introduction
2. Results
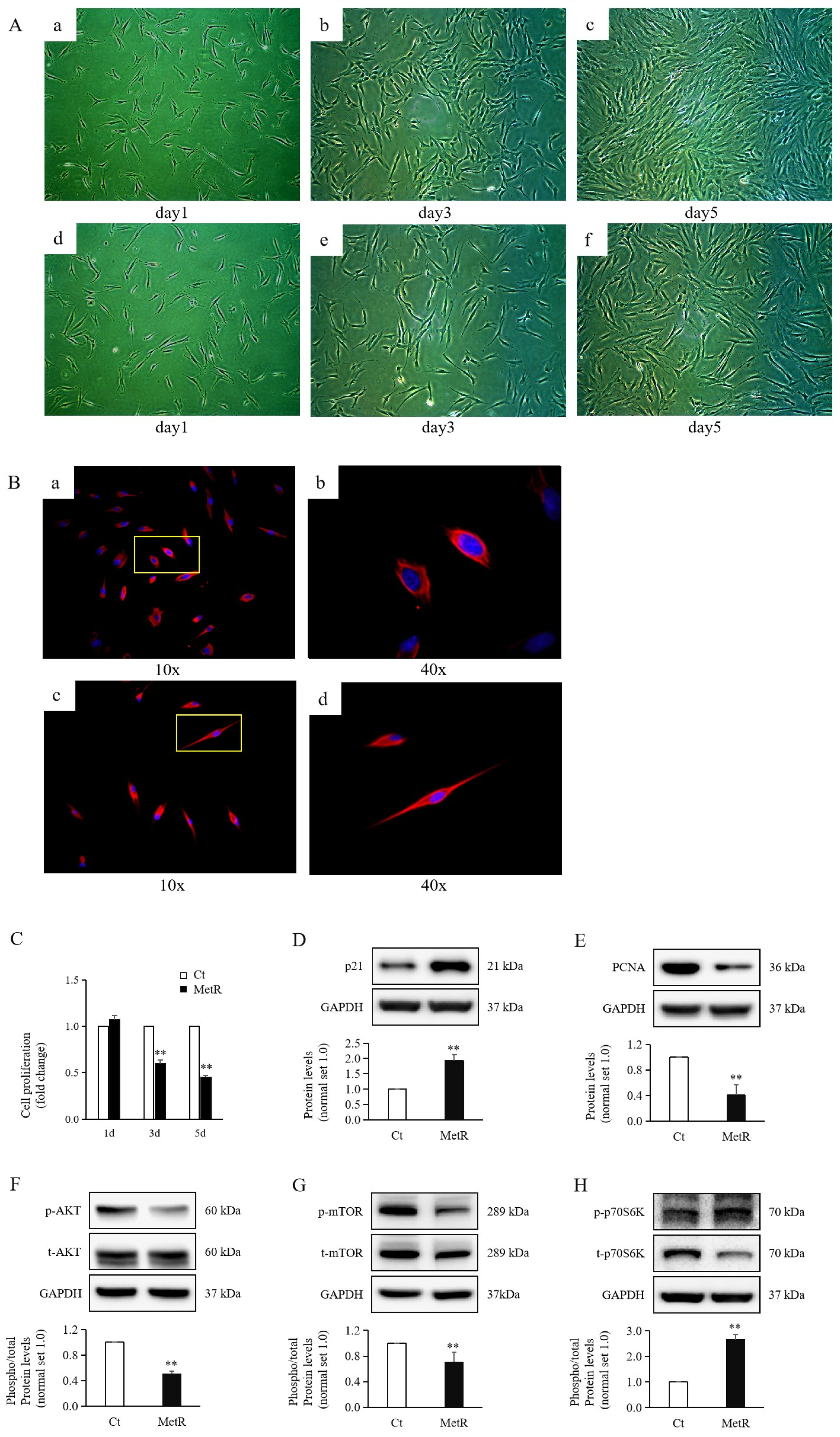
2.1. Methionine Restriction Reduced HTSF Proliferation
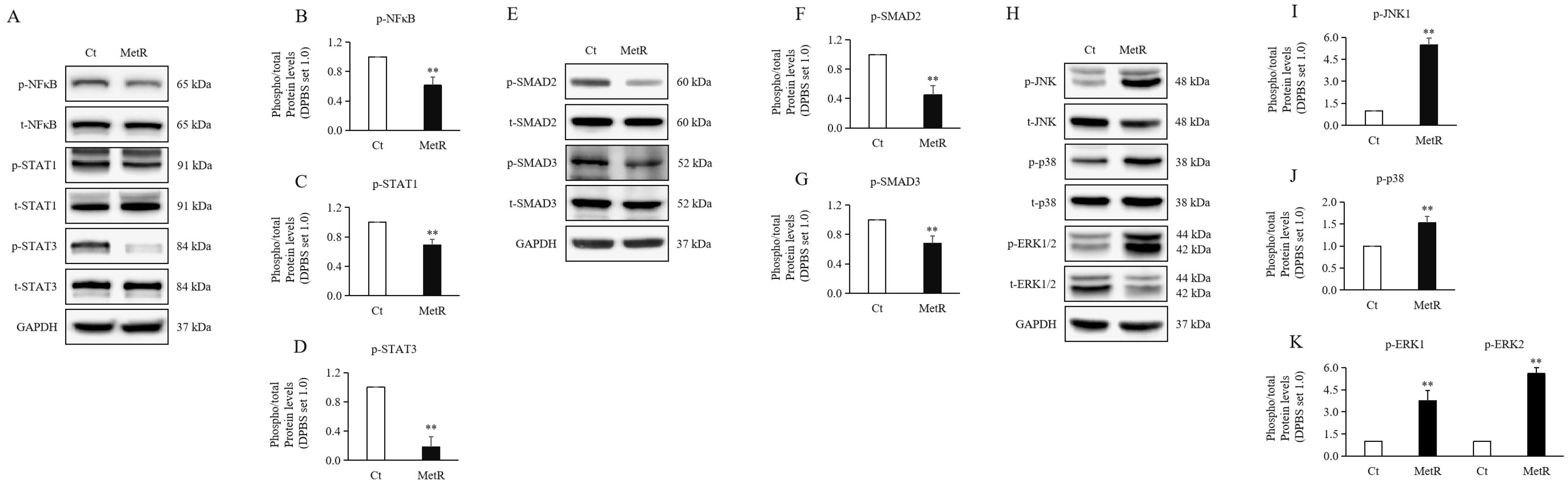
2.2. Methionine Restriction Reduced Inflammation in HTSFs
2.3. Methionine Restriction Reduced the Expression Levels of SMAD Proteins in HTSFs
2.4. Methionine Restriction Reduced MAPKs in HTSFs
2.5. Methionine Restriction Increased Apoptosis of HTSFs
2.6. Methionine Restriction Reduced the Expression Levels of Mesenchymal Markers in HTSFs
2.7. Methionine Restriction Reduced Expression of Fibrosis Markers in HTSFs
3. Discussion
4. Materials and Methods
4.1. Human Specimens
4.2. Fibroblast Isolation and Culture
4.3. Immunocytochemistry (ICC) Staining
4.4. Cell Proliferation Assay
4.5. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Western Blotting
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bharadia, S.K.; Burnett, L.; Gabriel, V. Hypertrophic Scar. Phys. Med. Rehabil. Clin. N. Am. 2023, 34, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Faour, S.; Farahat, M.; Aijaz, A.; Jeschke, M.G. Fibrosis in burns: An overview of mechanisms and therapies. Am. J. Physiol. Cell Physiol. 2023, 325, C1545–C1557. [Google Scholar] [CrossRef] [PubMed]
- Armour, A.; Scott, P.G.; Tredget, E.E. Cellular and molecular pathology of HTS: Basis for treatment. Wound Repair. Regen. 2007, 15 (Suppl. S1), S6–S17. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.G.; Dodd, C.M.; Tredget, E.E.; Ghahary, A.; Rahemtulla, F. Immunohistochemical localization of the proteoglycans decorin, biglycan and versican and transforming growth factor-beta in human post-burn hypertrophic and mature scars. Histopathology 1995, 26, 423–431. [Google Scholar] [CrossRef]
- Zhao, R.; Yan, Q.; Huang, H.; Lv, J.; Ma, W. Transdermal siRNA-TGFβ1-337 patch for hypertrophic scar treatment. Matrix Biol. 2013, 32, 265–276. [Google Scholar] [CrossRef]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-β signaling in health, disease, and therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar]
- Wang, Z.C.; Zhao, W.Y.; Cao, Y.; Liu, Y.Q.; Sun, Q.; Shi, P.; Cai, J.Q.; Shen, X.Z.; Tan, W.Q. The Roles of Inflammation in Keloid and Hypertrophic Scars. Front. Immunol. 2020, 11, 603187. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- La Russa, D.; Barberio, L.; Marrone, A.; Perri, A.; Pellegrino, D. Caloric Restriction Mitigates Kidney Fibrosis in an Aged and Obese Rat Model. Antioxidants 2023, 12, 1778. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef]
- Wanders, D.; Ghosh, S.; Stone, K.P.; Van, N.T.; Gettys, T.W. Transcriptional impact of dietary methionine restriction on systemic inflammation: Relevance to biomarkers of metabolic disease during aging. Biofactors 2014, 40, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Trocha, K.; Kip, P.; MacArthur, M.R.; Mitchell, S.J.; Longchamp, A.; Treviño-Villarreal, J.H.; Tao, M.; Bredella, M.A.; De Amorim Bernstein, K.; Mitchell, J.R.; et al. Preoperative Protein or Methionine Restriction Preserves Wound Healing and Reduces Hyperglycemia. J. Surg. Res. 2019, 235, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Lin, Z.; Huang, Q.; Liu, D. USP15 Enhances the Proliferation, Migration, and Collagen Deposition of Hypertrophic Scar-Derived Fibroblasts by Deubiquitinating TGF-βR1 In Vitro. Plast. Reconstr. Surg. 2021, 148, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Kokkinakis, D.M. Methionine-stress: A pleiotropic approach in enhancing the efficacy of chemotherapy. Cancer Lett. 2006, 233, 195–207. [Google Scholar] [CrossRef]
- Kumar, A.; Pathak, R.; Palfrey, H.A.; Stone, K.P.; Gettys, T.W.; Murthy, S.N. High levels of dietary methionine improves sitagliptin-induced hepatotoxicity by attenuating oxidative stress in hypercholesterolemic rats. Nutr. Metab. 2020, 17, 2. [Google Scholar] [CrossRef]
- Schmitz, W.; Koderer, C.; El-Mesery, M.; Gubik, S.; Sampers, R.; Straub, A.; Kübler, A.C.; Seher, A. Metabolic Fingerprinting of Murine L929 Fibroblasts as a Cell-Based Tumour Suppressor Model System for Methionine Restriction. Int. J. Mol. Sci. 2021, 22, 3039. [Google Scholar] [CrossRef]
- Wang, S.Y.; Wang, W.J.; Liu, J.Q.; Song, Y.H.; Li, P.; Sun, X.F.; Cai, G.Y.; Chen, X.M. Methionine restriction delays senescence and suppresses the senescence-associated secretory phenotype in the kidney through endogenous hydrogen sulfide. Cell Cycle 2019, 18, 1573–1587. [Google Scholar] [CrossRef]
- Andreoli, A.; Ruf, M.T.; Itin, P.; Pluschke, G.; Schmid, P. Phosphorylation of the ribosomal protein S6, a marker of mTOR (mammalian target of rapamycin) pathway activation, is strongly increased in hypertrophic scars and keloids. Br. J. Dermatol. 2015, 172, 1415–1417. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, H.; Wang, H.; He, X.; Tang, J.; Qi, S.; Yang, R.; Xie, J. Inhibition of phosphatidylinositol 3-kinase catalytic subunit alpha by miR-203a-3p reduces hypertrophic scar formation via phosphatidylinositol 3-kinase/AKT/mTOR signaling pathway. Burns Trauma. 2024, 12, tkad048. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Cao, X.; Hu, Z.; Ma, D.; Guo, D.; Zhang, J.; Zhang, C.; Liu, P.; Qu, S.; Zhu, J.; et al. Pleiotropic FTY720 is a Specific and Potent Therapy for Hypertrophic Scars. J. Investig. Dermatol. 2017, 137, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, S.F.; de la Vega, M.B.; Calzetta, N.L.; Siri, S.O.; Gottifredi, V. CDK-Independent and PCNA-Dependent Functions of p21 in DNA Replication. Genes. 2020, 11, 593. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, E.; Yang, Y.; Han, F.; Han, S.; Wu, G.; Zhang, M.; Zhang, J.; Han, J.; Su, L.; et al. The Akt/FoxO/p27(Kip1) axis contributes to the anti-proliferation of pentoxifylline in hypertrophic scars. J. Cell Mol. Med. 2019, 23, 6164–6172. [Google Scholar] [CrossRef]
- Nakaoka, H.; Miyauchi, S.; Miki, Y. Proliferating activity of dermal fibroblasts in keloids and hypertrophic scars. Acta Derm. Venereol. 1995, 75, 102–104. [Google Scholar] [CrossRef]
- Rong, Y.; Darnell, A.M.; Sapp, K.M.; Vander Heiden, M.G.; Spencer, S.L. Cells use multiple mechanisms for cell-cycle arrest upon withdrawal of individual amino acids. Cell Rep. 2023, 42, 113539. [Google Scholar] [CrossRef]
- Stone, K.P.; Ghosh, S.; Kovalik, J.P.; Orgeron, M.; Wanders, D.; Sims, L.C.; Gettys, T.W. The acute transcriptional responses to dietary methionine restriction are triggered by inhibition of ternary complex formation and linked to Erk1/2, mTOR, and ATF4. Sci. Rep. 2021, 11, 3765. [Google Scholar] [CrossRef]
- Bárcena, C.; Quirós, P.M.; Durand, S.; Mayoral, P.; Rodríguez, F.; Caravia, X.M.; Mariño, G.; Garabaya, C.; Fernández-García, M.T.; Kroemer, G.; et al. Methionine Restriction Extends Lifespan in Progeroid Mice and Alters Lipid and Bile Acid Metabolism. Cell Rep. 2018, 24, 2392–2403. [Google Scholar] [CrossRef]
- Booher, K.; Lin, D.W.; Borrego, S.L.; Kaiser, P. Downregulation of Cdc6 and pre-replication complexes in response to methionine stress in breast cancer cells. Cell Cycle 2012, 11, 4414–4423. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Z.; Ye, J. Phosphorylation and degradation of S6K1 (p70S6K1) in response to persistent JNK1 Activation. Biochim. Biophys. Acta 2013, 1832, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Ma, S. Resveratrol-laden mesoporous silica nanoparticles regulate the autophagy and apoptosis via ROS-mediated p38-MAPK/HIF-1a/p53 signaling in hypertrophic scar fibroblasts. Heliyon 2024, 10, e24985. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Shi, S.; Xie, W.; Zhao, M.; Li, Y.; Zhang, J.; Li, N.; Bai, X.; Cai, W.; Hu, X.; et al. IL-10 alleviates lipopolysaccharide-induced skin scarring via IL-10R/STAT3 axis regulating TLR4/NF-κB pathway in dermal fibroblasts. J. Cell Mol. Med. 2021, 25, 1554–1567. [Google Scholar] [CrossRef] [PubMed]
- Jere, S.W.; Abrahamse, H.; Houreld, N.N. The JAK/STAT signaling pathway and photobiomodulation in chronic wound healing. Cytokine Growth Factor Rev. 2017, 38, 73–79. [Google Scholar] [CrossRef]
- Ray, S.; Ju, X.; Sun, H.; Finnerty, C.C.; Herndon, D.N.; Brasier, A.R. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J. Investig. Dermatol. 2013, 133, 1212–1220. [Google Scholar] [CrossRef]
- Cui, H.S.; Joo, S.Y.; Cho, Y.S.; Kim, J.B.; Seo, C.H. CPEB1 or CPEB4 knockdown suppresses the TAK1 and Smad signalings in THP-1 macrophage-like cells and dermal fibroblasts. Arch. Biochem. Biophys. 2020, 683, 108322. [Google Scholar] [CrossRef]
- Zou, Y.; Yuan, Y.; Zhou, Q.; Yue, Z.; Liu, J.; Fan, L.; Xu, H.; Xin, L. The Role of Methionine Restriction in Gastric Cancer: A Summary of Mechanisms and a Discussion on Tumor Heterogeneity. Biomolecules 2024, 14, 161. [Google Scholar] [CrossRef]
- Maebashi, M.; Miyake, K.; Yamamoto, J.; Sahara, K.; Akiyama, T.; Kimura, Y.; Endo, I. Methionine restriction inhibits pancreatic cancer proliferation while suppressing JAK2/STAT3 pathway. Pancreatology 2025, 25, 108–117. [Google Scholar] [CrossRef]
- Wang, L.; Ren, B.; Hui, Y.; Chu, C.; Zhao, Z.; Zhang, Y.; Zhao, B.; Shi, R.; Ren, J.; Dai, X.; et al. Methionine Restriction Regulates Cognitive Function in High-Fat Diet-Fed Mice: Roles of Diurnal Rhythms of SCFAs Producing- and Inflammation-Related Microbes. Mol. Nutr. Food Res. 2020, 64, e2000190. [Google Scholar] [CrossRef]
- Xin, L.; Cao, W.X.; Fei, X.F.; Wang, Y.; Liu, W.T.; Liu, B.Y.; Zhu, Z.G. Applying proteomic methodologies to analyze the effect of methionine restriction on proliferation of human gastric cancer SGC7901 cells. Clin. Chim. Acta 2007, 377, 206–212. [Google Scholar] [CrossRef]
- Lu, S.; Hoestje, S.M.; Choo, E.M.; Epner, D.E. Methionine restriction induces apoptosis of prostate cancer cells via the c-Jun N-terminal kinase-mediated signaling pathway. Cancer Lett. 2002, 179, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Merkt, W.; Zhou, Y.; Han, H.; Lagares, D. Myofibroblast fate plasticity in tissue repair and fibrosis: Deactivation, apoptosis, senescence and reprogramming. Wound Repair. Regen. 2021, 29, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Guo, Z.; Gao, W. miR-181b-5p promotes proliferation and inhibits apoptosis of hypertrophic scar fibroblasts through regulating the MEK/ERK/p21 pathway. Exp. Ther. Med. 2019, 17, 1537–1544. [Google Scholar] [CrossRef]
- Lu, S.; Hoestje, S.M.; Choo, E.; Epner, D.E. Induction of caspase-dependent and -independent apoptosis in response to methionine restriction. Int. J. Oncol. 2003, 22, 415–420. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef]
- Danial, N.N. BCL-2 family proteins: Critical checkpoints of apoptotic cell death. Clin. Cancer Res. 2007, 13, 7254–7263. [Google Scholar] [CrossRef]
- Kocab, A.J.; Duckett, C.S. Inhibitor of apoptosis proteins as intracellular signaling intermediates. Febs. J. 2016, 283, 221–231. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, R.; Yoo, H.; Hong, K.; Song, H. Nonylphenol Induces Apoptosis through ROS/JNK Signaling in a Spermatogonia Cell Line. Int. J. Mol. Sci. 2020, 22, 307. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, D.; Ning, S.; Jia, H.; Yu, S. Botulinum toxin type A prevents the phenotypic transformation of fibroblasts induced by TGF-β1 via the PTEN/PI3K/Akt signaling pathway. Int. J. Mol. Med. 2019, 44, 661–671. [Google Scholar] [CrossRef]
- Feng, Y.; Wu, J.J.; Sun, Z.L.; Liu, S.Y.; Zou, M.L.; Yuan, Z.D.; Yu, S.; Lv, G.Z.; Yuan, F.L. Targeted apoptosis of myofibroblasts by elesclomol inhibits hypertrophic scar formation. EBioMedicine 2020, 54, 102715. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes. Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska-Podhorodecka, Z.; Ding, I.; Norouzi, M.; McCulloch, C.A. Impact of Vimentin on Regulation of Cell Signaling and Matrix Remodeling. Front. Cell Dev. Biol. 2022, 10, 869069. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef]
- Phan, S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Cui, H.S.; Hong, A.R.; Kim, J.B.; Yu, J.H.; Cho, Y.S.; Joo, S.Y.; Seo, C.H. Extracorporeal Shock Wave Therapy Alters the Expression of Fibrosis-Related Molecules in Fibroblast Derived from Human Hypertrophic Scar. Int. J. Mol. Sci. 2018, 19, 124. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Sivagurunathan, S.; Vahabikashi, A.; Yang, H.; Zhang, J.; Vazquez, K.; Rajasundaram, D.; Politanska, Y.; Abdala-Valencia, H.; Notbohm, J.; Guo, M.; et al. Expression of vimentin alters cell mechanics, cell-cell adhesion, and gene expression profiles suggesting the induction of a hybrid EMT in human mammary epithelial cells. Front. Cell Dev. Biol. 2022, 10, 929495. [Google Scholar] [CrossRef]
- Schuster, R.; Younesi, F.; Ezzo, M.; Hinz, B. The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harb. Perspect. Biol. 2023, 15, a041231. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Yoo, J.D.; Choi, S.Y.; Kwon, O.S. The expression and secretion of vimentin in the progression of non-alcoholic steatohepatitis. BMB Rep. 2014, 47, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Cooke, D.; Ouattara, A.; Ables, G.P. Dietary methionine restriction modulates renal response and attenuates kidney injury in mice. FASEB J. 2018, 32, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Development of recombinant methioninase to target the general cancer-specific metabolic defect of methionine dependence: A 40-year odyssey. Expert. Opin. Biol. Ther. 2015, 15, 21–31. [Google Scholar] [CrossRef]
- Cavuoto, P.; Fenech, M.F. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat. Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef]
- Navik, U.; Sheth, V.G.; Khurana, A.; Jawalekar, S.S.; Allawadhi, P.; Gaddam, R.R.; Bhatti, J.S.; Tikoo, K. Methionine as a double-edged sword in health and disease: Current perspective and future challenges. Ageing Res. Rev. 2021, 72, 101500. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Location of Specimens | Age (Years) | Sex | Months Post-Burn |
|---|---|---|---|---|
| 1 | Foot | 37 | Male | 9 |
| 2 | Hand | 30 | Male | 12 |
| 3 | Foot | 34 | Male | 11 |
| 4 | Hand | 47 | Male | 10 |
| Gene | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| CYCS | AAGGGAGGCAAGCACAAGACTG | CTCCATCAGTGTATCCTCTCCC |
| BCL2 | TGCGGCCTCTGTTTGATTT | AGGCATGTTGACTTCACTTGT |
| BAD | CCAACCTCTGGGCAGCACAGC | TTTGCCGCATCTGCGTTGCTGT |
| BID | TGGGACACTGTGAACCAGGAGT | GAGGAAGCCAAACACCAGTAGG |
| BAX | CCTTTTGCTTCAGGGTTTCA | CCATGTTACTGTCCAGTTCG |
| ACTA2 | CCGACCGAATGCAGAAGGA | ACAGAGTATTTGCGCTCCGAA |
| COL1A1 | ATGTTCAGCTTTGTGGACCTC | CTGTACGCAGGTGATTGGTG |
| COL3A1 | CACTGGGGAATGGAGCAAAAC | ATCAGGACCACCAATGTCATAGG |
| FN1 | CCAGTCCACAGCTATTCCTG | ACAACCACGGATGAGCTG |
| GAPDH | CATGAGAAGTATGACAACAGCCT | AGTCCTTCCACGATACCAAAGT |
| Target | Host | Dilution | Company (Cat. No.) |
|---|---|---|---|
| GAPDH | Rabbit | 1:1000 | Cell Signaling Technology (2118S), Danvers, MA, USA |
| GAPDH | Mouse | 1:1000 | Santa Cruz Technology (sc-47724), Dallas, TX, USA |
| α-SMA | Mouse | 1:500 | Abcam (ab7817), Cambridge, UK |
| Fibronectin | Rabbit | 1:2000 | Abcam (ab6328) |
| Collagen I | Rabbit | 1:1000 | Abcam (ab34710) |
| Collagen III | Rabbit | 1:1000 | Abcam (ab7778) |
| Vimentin | Mouse | 1:3000 | Abcam (ab92547) |
| E-Cadherin | Rabbit | 1:1000 | Cell Signaling Technology (3195S) |
| N-Cadherin | Mouse | 1:1000 | Thermo Fisher Scientific (333900), Waltham, MA, USA |
| Snail1 | Rabbit | 1:1000 | Millipore (ABD38), Billerica, MA, USA |
| Slug | Rabbit | 1:1000 | Cell Signaling Technology (9585S) |
| Twist1 | Mouse | 1:300 | Santa Cruz Biotechnology (sc-81417) |
| Phospho-SMAD2 | Rabbit | 1:1000 | Cell Signaling Technology (3108S) |
| SMAD2 | Rabbit | 1:1000 | Abcam (ab33875) |
| Phospho-SMAD3 | Rabbit | 1:1000 | Invitrogen (MA5-14936), Carlsbad, CA, USA |
| SMAD3 | Rabbit | 1:1000 | Cell Signaling Technology (9523S) |
| Phospho-STAT1 | Rabbit | 1:1000 | Cusabio Technology (CSB-PA050162), Wuhan, Hubei, China |
| STAT1 | Rabbit | 1:1000 | Cusabio Technology (CSB-PA825331) |
| Phospho-STAT3 | Rabbit | 1:1000 | Cell Signaling Technology (9134S) |
| STAT3 | Rabbit | 1:1000 | Cusabio Technology (CSB-PA004173) |
| Phospho-NF-κB | Rabbit | 1:1000 | Cell Signaling Technology (3033S) |
| NF-κB | Rabbit | 1:1000 | Abcam (ab32536) |
| Phospho-JNK | Rabbit | 1:1000 | Cell Signaling Technology (9251S) |
| JNK | Rabbit | 1:1000 | Cell Signaling Technology (9252S) |
| Phospho-ERK | Rabbit | 1:1000 | Cell Signaling Technology (4370S) |
| ERK | Mouse | 1:1000 | Cell Signaling Technology (4696S) |
| Phospho-p38 | Mouse | 1:1000 | Cell Signaling Technology (9216S) |
| p38 | Rabbit | 1:1000 | Cell Signaling Technology (8690S) |
| Phospho-AKT | Rabbit | 1:1000 | Cell Signaling Technology (4060S) |
| AKT | Rabbit | 1:1000 | Cell Signaling Technology (4671S) |
| Phospho-mTOR | Rabbit | 1:1000 | Cusabio Technology (CSB-PA000576) |
| mTOR | Rabbit | 1:1000 | Cusabio Technology (CSB-PA003333) |
| Phospho-p70S6K | Rabbit | 1:1000 | Cell Signaling Technology (9205S) |
| p70S6K | Rabbit | 1:1000 | Cell Signaling Technology (9202S) |
| p21 | Rabbit | 1:1000 | Abcam (ab109199) |
| PCNA | Rabbit | 1:1000 | Cell Signaling Technology (2586S) |
| Cytochrome C | Rabbit | 1:1000 | Cell Signaling Technology (4272S) |
| c-IAP1 | Mouse | 1:300 | Santa Cruz Biotechnology (sc-271419) |
| c-IAP2 | Rabbit | 1:1000 | Cell Signaling Technology (3130S) |
| Cleaved caspase3 | Rabbit | 1:1000 | Cell Signaling Technology (9661S) |
| Cleaved caspase9 | Rabbit | 1:1000 | Cell Signaling Technology (9507S) |
| Bcl-2 | Rabbit | 1:1000 | Abcam (ab196495) |
| Bad | Rabbit | 1:1000 | Cell Signaling Technology (9292S) |
| Bid | Rabbit | 1:1000 | Cell Signaling Technology (8762S) |
| Bax | Rabbit | 1:1000 | Abcam (ab199677) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, H.S.; Zheng, Y.X.; Cho, Y.S.; Ro, Y.M.; Kwak, I.S.; Joo, S.Y.; Seo, C.H. Methionine Restriction Attenuates Scar Formation in Fibroblasts Derived from Patients with Post-Burn Hypertrophic Scar. Int. J. Mol. Sci. 2025, 26, 5876. https://doi.org/10.3390/ijms26125876
Cui HS, Zheng YX, Cho YS, Ro YM, Kwak IS, Joo SY, Seo CH. Methionine Restriction Attenuates Scar Formation in Fibroblasts Derived from Patients with Post-Burn Hypertrophic Scar. International Journal of Molecular Sciences. 2025; 26(12):5876. https://doi.org/10.3390/ijms26125876
Chicago/Turabian StyleCui, Hui Song, Ya Xin Zheng, Yoon Soo Cho, Yu Mi Ro, In Suk Kwak, So Young Joo, and Cheong Hoon Seo. 2025. "Methionine Restriction Attenuates Scar Formation in Fibroblasts Derived from Patients with Post-Burn Hypertrophic Scar" International Journal of Molecular Sciences 26, no. 12: 5876. https://doi.org/10.3390/ijms26125876
APA StyleCui, H. S., Zheng, Y. X., Cho, Y. S., Ro, Y. M., Kwak, I. S., Joo, S. Y., & Seo, C. H. (2025). Methionine Restriction Attenuates Scar Formation in Fibroblasts Derived from Patients with Post-Burn Hypertrophic Scar. International Journal of Molecular Sciences, 26(12), 5876. https://doi.org/10.3390/ijms26125876