
Molecular Insights into the Diagnosis of Anaplastic Large Cell Lymphoma: Beyond Morphology and Immunophenotype

 , , , , , , ,
, , , , , , , 

Abstract
1. Introduction
1.1. Mechanisms Leading to CD30 Expression
1.2. ALCL Classification and Subtypes
- Systemic ALK-positive Anaplastic Large Cell Lymphoma (ALK+ ALCL).
- Systemic ALK-negative Anaplastic Large Cell Lymphoma (ALK- ALCL).
- Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL).
- Primary cutaneous Anaplastic Large Cell Lymphoma (pcALCL).
1.3. ALCL Etiopathogenesis
2. Systemic ALK-Positive Anaplastic Large Cell Lymphoma
2.1. General and Clinical Aspects
2.2. Morphology and Immunophenotype
2.3. Cytogenetic Alterations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosomal Translocation | ALK Partner | Partner Gene Function | % of Cases | Expression Pattern |
|---|---|---|---|---|
| t(2;5)(p23;q35) | NPM1 [5,90,91,92] | Nuclear protein that shuttles between the nucleus and the cytoplasm | 80 | Nuclear and cytoplasmic |
| t(1;2)(q25;p23) | TPM3 [92,93] | Cytoskeletal protein | 13 | Cytoplasmic |
| Inv(2)(p23q53) | ATIC [92,93] | Purine biosynthesis pathway | 1 | Cytoplasmic |
| t(2;3)(p23;q21) [92,93] | TFG Xlong TFG long TFG short | Associated with ER and microtubules | <1 | Cytoplasmic |
| t(2;17)(p23;q23) | CLTC [92,93] | Component of the cytoplasmic face of intracellular organelles | <1 | Cytoplasmic |
| t(2;X)(p23;q11.12) | MSN [92,93] | Submembranous cytoskeleton | <1 | Cytoplasmic |
| t(2;19)(p23;p13.1) | TPM4 [92,93] | Cytoskeletal protein | <1 | Cytoplasmic |
| t(2;22)(p23;q11.2) | MYH9 [92,93] | Cytoskeletal (major contractile protein) | <1 | Cytoplasmic |
| t(2;9)(p23;q33–34) | TRAF1 [92,93] | TNF signaling, signaling adaptor | <1 | Cytoplasmic |
| t(2;11)(2p23;11q12.3) | EEF1G [92,93] | Translation elongation factor activity; subunit of the elongation factor-1 | <1 | Cytoplasmic |
| t(2;17)(p23;q25) | RNF213/ALO17 [92,93] | E3 ligase | <1 | Cytoplasmic |
| t(2;5)(p23;q35) | SQSTM1 [94] | Regulates activation of the NF-kB signaling pathway | <1 | Cytoplasmic |
| t(2;11)(p23;p13) | CAPRIN1 [94] | ATP binding, scaffold activity and signaling adaptor | <1 | Cytoplasmic |
2.4. Molecular Alterations
2.5. Role of Non-Coding RNAs in ALK+ ALCL
2.6. Prognosis and Prediction
2.7. Differential Diagnosis with Other ALK+ Tumors
3. Systemic ALK−Negative Anaplastic Large Cell Lymphoma
3.1. General and Clinical Aspects
3.2. Morphology and Immunophenotype
3.3. Cytogenetic Alterations
3.4. Molecular Subtypes with Prognostic Significance
3.4.1. DUSP22 Rearrangements
3.4.2. TP63 Rearrangements
3.4.3. Triple-Negative ALK- ALCL
3.4.4. New Genetic Approaches and Novel Potential Subgroups in ALK- ALCL
“Double Hit” Cases with DUSP22 and TP63 Rearrangements
JAK2 Rearrangements and Morphology Variants
ERBB4 Expression Subclass Morphology
FRK Fusions
MYC Rearrangements May Be Associated with Poor Prognosis
3.5. Signaling Alterations, Pathogenesis Mechanism and Prognostic Significance of STAT3 Activation
- Oncogenic fusion genes displaying concomitant transcriptional and kinase activities capable of sustaining the ALCL phenotype via STAT3 activation, such as NFκB2::ROS1, NCOR2::ROS1, NFκB2::TYK2, and PABPC4::TYK2 fusions [195].
3.6. Role of Non-Coding RNAs in ALK- ALCL
3.7. Mutational Landscape of ALK- ALCL
3.8. Prognosis and Prediction
3.9. Differential Diagnosis: ALK- ALCL vs. CD30+ PTCL-NOS
4. Breast Implant-Associated Anaplastic Large Cell Lymphoma
4.1. General and Clinical Aspects
4.2. Morphology and Immunophenotype
4.3. Pathogenesis of BIA-ALCL
4.4. Molecular Alterations
4.5. BIA-ALCL: Staging, Subtypes, Treatment Approaches and Prognosis
- IA: malignant cells are confined to the fluid or form a layer on the luminal side of the capsule.
- IB: early capsule infiltration is observed, but cells are confined to the internal capsule.
- IC: cell aggregates or sheets may infiltrate the capsule.
- IIA: cell infiltrates beyond the capsule.
- IIB: involvement of one regional lymph node.
- III: multiple regional lymph nodes involvement.
- IV: spread to other organs and distant sites.
- In situ BIA-ALCL (Stage I): anaplastic cell proliferation is confined to the fibrous capsule. Patients have an indolent clinical course and generally remain free of disease after capsulectomy and implant removal.
- Infiltrative BIA-ALCL (Stage II and beyond): pleomorphic cells massively infiltrate the adjacent tissue. These cases have a more aggressive clinical course that may require aggressive therapy in addition to implant removal (justifying cytotoxic chemotherapy).
4.6. Differential Diagnosis with Other Entities
5. Updated Perspectives in Systemic ALCL
6. Primary Cutaneous Anaplastic Large Cell Lymphoma (pcALCL)
6.1. General Aspects of pcALCL
6.2. LyP Morphology and Immunophenotype
6.3. pcALCL Morphology and Immunophenotype
6.4. Intralymphatic CD30+ Large T-Cell Lymphoma Morphology and Phenotype
6.5. Molecular Alterations
6.5.1. Chromosomal Rearrangements
6.5.2. Copy Number Alterations
6.5.3. Small-Nucleotide Variants
6.5.4. Role of Non-Coding miRNA
6.6. Molecular Biomarkers and Prognosis
6.7. Differential Diagnosis of pcALCL with Other CD30+ Cutaneous Entities
7. Microenvironment in ALCL
7.1. ALK+ ALCL
7.2. ALK- ALCL
7.3. BIA-ALCL
7.4. pcALCL
8. Future Direction of ALCL Diagnosis
Author Contributions
Funding
Conflicts of Interest
References
- Stansfeld, A.G.; Diebold, J.; Noel, H.; Kapanci, Y.; Rilke, F.; Kelényi, G.; Sundstrom, C.; Lennert, K.; van Unnik, J.A.; Mioduszewska, O. Updated Kiel Classification for Lymphomas. Lancet 1988, 1, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.; Jaffe, E.; Stein, H.; Banks, P.; Chan, J.; Cleary, M.; Delsol, G.; De Wolf- Peeters, C.; Falini, B.; Gatter, K. A Revised European-American Classification of Lymphoid Neoplasms: A Proposal from the International Lymphoma Study Group [See Comments]. Blood 1994, 84, 1361–1392. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, S.; Wlodarska, I.; Pulford, K.; Campo, E.; Morris, S.W.; Van den Berghe, H.; De Wolf-Peeters, C. The Monoclonal Antibody ALK1 Identifies a Distinct Morphological Subtype of Anaplastic Large Cell Lymphoma Associated with 2p23/ALK Rearrangements. Am. J. Pathol. 1997, 151, 343–351. [Google Scholar]
- Pulford, K.; Lamant, L.; Morris, S.W.; Butler, L.H.; Wood, K.M.; Stroud, D.; Delsol, G.; Mason, D.Y. Detection of Anaplastic Lymphoma Kinase (ALK) and Nucleolar Protein Nucleophosmin (NPM)-ALK Proteins in Normal and Neoplastic Cells with the Monoclonal Antibody ALK1. Blood 1997, 89, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Duyster, J.; Bai, R.Y.; Morris, S.W. Translocations Involving Anaplastic Lymphoma Kinase (ALK). Oncogene 2001, 20, 5623–5637. [Google Scholar] [CrossRef]
- Gascoyne, R.D.; Aoun, P.; Wu, D.; Chhanabhai, M.; Skinnider, B.F.; Greiner, T.C.; Morris, S.W.; Connors, J.M.; Vose, J.M.; Viswanatha, D.S.; et al. Prognostic Significance of Anaplastic Lymphoma Kinase (ALK) Protein Expression in Adults with Anaplastic Large Cell Lymphoma. Blood 1999, 93, 3913–3921. [Google Scholar] [CrossRef]
- Hernández, L.; Pinyol, M.; Hernández, S.; Beà, S.; Pulford, K.; Rosenwald, A.; Lamant, L.; Falini, B.; Ott, G.; Mason, D.Y.; et al. TRK-Fused Gene (TFG) Is a New Partner of ALK in Anaplastic Large Cell Lymphoma Producing Two Structurally Different TFG-ALK Translocations. Blood 1999, 94, 3265–3268. [Google Scholar] [CrossRef]
- Tort, F.; Pinyol, M.; Pulford, K.; Roncador, G.; Hernandez, L.; Nayach, I.; Kluin-Nelemans, H.C.; Kluin, P.; Touriol, C.; Delsol, G.; et al. Molecular Characterization of a New ALK Translocation Involving Moesin (MSN-ALK) in Anaplastic Large Cell Lymphoma. Lab. Investig. 2001, 81, 419–426. [Google Scholar] [CrossRef]
- Chan, J.K. The New World Health Organization Classification of Lymphomas: The Past, the Present and the Future. Hematol. Oncol. 2001, 19, 129–150. [Google Scholar] [CrossRef]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO Classification of Lymphoid Neoplasms and beyond: Evolving Concepts and Practical Applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef]
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H. The Expression of the Hodgkin’s Disease Associated Antigen Ki-1 in Reactive and Neoplastic Lymphoid Tissue: Evidence That Reed-Sternberg Cells and Histiocytic Malignancies Are Derived from Activated Lymphoid Cells. Blood 1985, 66, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Nakashima, M.; Uchimaru, K. CD30 Expression and Its Functions during the Disease Progression of Adult T-Cell Leukemia/Lymphoma. Int. J. Mol. Sci. 2023, 24, 8731. [Google Scholar] [CrossRef]
- Gardner, L.J.; Polski, J.M.; Evans, H.L.; Perkins, S.L.; Dunphy, C.H. CD30 Expression in Follicular Lymphoma. Arch. Pathol. Lab. Med. 2001, 125, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Parente, P.; Zanelli, M.; Sanguedolce, F.; Mastracci, L.; Graziano, P. Hodgkin Reed–Sternberg-Like Cells in Non-Hodgkin Lymphoma. Diagnostics 2020, 10, 1019. [Google Scholar] [CrossRef]
- Kojima, N.; Mori, T.; Motoi, T.; Kobayashi, E.; Yoshida, M.; Yatabe, Y.; Ichikawa, H.; Kawai, A.; Yonemori, K.; Antonescu, C.R.; et al. Frequent CD30 Expression in an Emerging Group of Mesenchymal Tumors With NTRK, BRAF, RAF1, or RET Fusions. Mod. Pathol. 2023, 36, 100083. [Google Scholar] [CrossRef]
- Iwakoshi, A.; Kikui, H.; Nakashima, R.; Goto, Y.; Ichikawa, D.; Sasaki, E.; Sekimizu, M.; Hattori, H.; Maeda, N. CD30 Expression in an Emerging Group of Mesenchymal Spindle Cell Neoplasms with ALK Fusion Detected by Flow Cytometry and Immunohistochemistry. Genes. Chromosomes Cancer 2024, 63, e23228. [Google Scholar] [CrossRef]
- Sabattini, E.; Pizzi, M.; Tabanelli, V.; Baldin, P.; Sacchetti, C.S.; Agostinelli, C.; Zinzani, P.L.; Pileri, S.A. CD30 Expression in Peripheral T-Cell Lymphomas. Haematologica 2013, 98, e81–e82. [Google Scholar] [CrossRef]
- Bossard, C.; Dobay, M.P.; Parrens, M.; Lamant, L.; Missiaglia, E.; Haioun, C.; Martin, A.; Fabiani, B.; Delarue, R.; Tournilhac, O.; et al. Immunohistochemistry as a Valuable Tool to Assess CD30 Expression in Peripheral T-Cell Lymphomas: High Correlation with mRNA Levels. Blood 2014, 124, 2983–2986. [Google Scholar] [CrossRef] [PubMed]
- Lamarque, M.; Bossard, C.; Contejean, A.; Brice, P.; Parrens, M.; Le Gouill, S.; Brière, J.; Bouabdallah, R.; Canioni, D.; Tilly, H.; et al. Brentuximab Vedotin in Refractory or Relapsed Peripheral T-Cell Lymphomas: The French Named Patient Program Experience in 56 Patients. Haematologica 2016, 101, e103–e106. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pinilla, S.M.; Domingo-Domenech, E.; Climent, F.; Sanchez, J.; Perez Seoane, C.; Lopez Jimenez, J.; Garcia-Cosio, M.; Caballero, D.; Blanco Muñez, O.J.; Carpio, C.; et al. Clinical and Pathological Characteristics of Peripheral T-Cell Lymphomas in a Spanish Population: A Retrospective Study. Br. J. Haematol. 2021, 192, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Aoki, R.; Nomura, Y.; Yamamoto, K.; Shimizu, K.; Yoshida, S.; Komatani, H.; Sugita, Y.; Ohshima, K. Usefulness of Flow Cytometry for Differential Diagnosis of Precursor and Peripheral T-Cell and NK-Cell Lymphomas: Analysis of 490 Cases. Pathol. Int. 2008, 58, 89–97. [Google Scholar] [CrossRef]
- Asano, N.; Kinoshita, T.; Tamaru, J.-I.; Ohshima, K.; Yoshino, T.; Niitsu, N.; Tsukamoto, N.; Hirabayashi, K.; Izutsu, K.; Taniwaki, M.; et al. Cytotoxic Molecule-Positive Classical Hodgkin’s Lymphoma: A Clinicopathological Comparison with Cytotoxic Molecule-Positive Peripheral T-Cell Lymphoma of Not Otherwise Specified Type. Haematologica 2011, 96, 1636–1643. [Google Scholar] [CrossRef]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK- Anaplastic Large-Cell Lymphoma Is Clinically and Immunophenotypically Different from Both ALK+ ALCL and Peripheral T-Cell Lymphoma, Not Otherwise Specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef]
- Weisenburger, D.D.; Savage, K.J.; Harris, N.L.; Gascoyne, R.D.; Jaffe, E.S.; MacLennan, K.A.; Rüdiger, T.; Pileri, S.; Nakamura, S.; Nathwani, B.; et al. Peripheral T-Cell Lymphoma, Not Otherwise Specified: A Report of 340 Cases from the International Peripheral T-Cell Lymphoma Project. Blood 2011, 117, 3402–3408. [Google Scholar] [CrossRef]
- Wang, G.-N.; Zhao, W.-G.; Li, L.; Zhang, D.-D.; Gao, X.-Z.; Zhou, J.; Zhang, L.; Fu, X.-R.; Zheng, X.-Y.; Li, Y.; et al. Prognostic Significance of CD30 Expression in Nasal Natural Killer/T-Cell Lymphoma. Oncol. Lett. 2017, 13, 1211. [Google Scholar] [CrossRef]
- Kawamoto, K.; Miyoshi, H.; Suzuki, T.; Sasaki, Y.; Yamada, K.; Yanagida, E.; Muto, R.; Kiryu, M.; Sone, H.; Seto, M.; et al. Frequent Expression of CD30 in Extranodal NK/T-Cell Lymphoma: Potential Therapeutic Target for Anti-CD30 Antibody-Based Therapy. Hematol. Oncol. 2018, 36, 166–173. [Google Scholar] [CrossRef]
- Feng, Y.; Rao, H.; Lei, Y.; Huang, Y.; Wang, F.; Zhang, Y.; Xi, S.; Wu, Q.; Shao, J. CD30 Expression in Extranodal Natural Killer/T-Cell Lymphoma, Nasal Type among 622 Cases of Mature T-Cell and Natural Killer-Cell Lymphoma at a Single Institution in South China. Chin. J. Cancer 2017, 36, 43. [Google Scholar] [CrossRef]
- Hartmann, S.; Goncharova, O.; Portyanko, A.; Sabattini, E.; Meinel, J.; Küppers, R.; Agostinelli, C.; Pileri, S.A.; Hansmann, M.-L. CD30 Expression in Neoplastic T Cells of Follicular T Cell Lymphoma Is a Helpful Diagnostic Tool in the Differential Diagnosis of Hodgkin Lymphoma. Mod. Pathol. 2019, 32, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Onaindia, A.; Martínez, N.; Montes-Moreno, S.; Almaraz, C.; Rodríguez-Pinilla, S.M.; Cereceda, L.; Revert, J.B.; Ortega, C.; Tardio, A.; González, L.; et al. CD30 Expression by B and T Cells: A Frequent Finding in Angioimmunoblastic T-Cell Lymphoma and Peripheral T-Cell Lymphoma-Not Otherwise Specified. Am. J. Surg. Pathol. 2016, 40, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wang, Y.; Xie, R.; Zhang, Q.; Xing, X.; Zhang, S.; Liu, H.; Sang, W. Clinicopathologic Features and Survival Outcomes of CD30 Expression in Extranodal Natural Killer/T-Cell Lymphoma. Am. J. Clin. Pathol. 2024, 162, 95–102. [Google Scholar] [CrossRef]
- Nakashima, M.; Utsunomiya, A.; Watanabe, T.; Horie, R.; Uchimaru, K. The Oncogenic Driving Force of CD30 Signaling-Induced Chromosomal Instability in Adult T-Cell Leukemia/Lymphoma. Cancer Sci. 2023, 114, 1556–1568. [Google Scholar] [CrossRef]
- Cerutti, A.; Schaffer, A.; Goodwin, R.G.; Shah, S.; Zan, H.; Ely, S.; Casali, P. Engagement of CD153 (CD30 Ligand) by CD30+ T Cells Inhibits Class Switch DNA Recombination and Antibody Production in Human IgD+ IgM+ B Cells. J. Immunol. 2000, 165, 786–794. [Google Scholar] [CrossRef]
- Horie, R.; Watanabe, T. CD30: Expression and Function in Health and Disease. Semin. Immunol. 1998, 10, 457–470. [Google Scholar] [CrossRef]
- Takaesu, G.; Surabhi, R.M.; Park, K.-J.; Ninomiya-Tsuji, J.; Matsumoto, K.; Gaynor, R.B. TAK1 Is Critical for IkappaB Kinase-Mediated Activation of the NF-kappaB Pathway. J. Mol. Biol. 2003, 326, 105–115. [Google Scholar] [CrossRef]
- Wright, C.W.; Rumble, J.M.; Duckett, C.S. CD30 Activates Both the Canonical and Alternative NF-κB Pathways in Anaplastic Large Cell Lymphoma Cells*. J. Biol. Chem. 2007, 282, 10252–10262. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, W.; Zhang, J.-P.; Song, Z.; Li, Y.; Xiao, W.; Liu, Y.; Zeng, M.-S.; Petrus, M.N.; Thomas, C.J.; et al. A Novel Model of Alternative NF-κB Pathway Activation in Anaplastic Large Cell Lymphoma. Leukemia 2021, 35, 1976–1989. [Google Scholar] [CrossRef]
- Watanabe, M.; Ogawa, Y.; Itoh, K.; Koiwa, T.; Kadin, M.E.; Watanabe, T.; Okayasu, I.; Higashihara, M.; Horie, R. Hypomethylation of CD30 CpG Islands with Aberrant JunB Expression Drives CD30 Induction in Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma. Lab. Investig. J. Tech. Methods Pathol. 2008, 88, 48–57. [Google Scholar] [CrossRef]
- Rassidakis, G.Z.; Thomaides, A.; Atwell, C.; Ford, R.; Jones, D.; Claret, F.-X.; Medeiros, L.J. JunB Expression Is a Common Feature of CD30+ Lymphomas and Lymphomatoid Papulosis. Mod. Pathol. 2005, 18, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dörken, B.; et al. Aberrantly Expressed C-Jun and JunB Are a Hallmark of Hodgkin Lymphoma Cells, Stimulate Proliferation and Synergize with NF-Kappa B. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef]
- Drakos, E.; Leventaki, V.; Schlette, E.J.; Jones, D.; Lin, P.; Medeiros, L.J.; Rassidakis, G.Z. C-Jun Expression and Activation Are Restricted to CD30+ Lymphoproliferative Disorders. Am. J. Surg. Pathol. 2007, 31, 447–453. [Google Scholar] [CrossRef]
- Liang, H.-C.; Costanza, M.; Prutsch, N.; Zimmerman, M.W.; Gurnhofer, E.; Montes-Mojarro, I.A.; Abraham, B.J.; Prokoph, N.; Stoiber, S.; Tangermann, S.; et al. Super-Enhancer-Based Identification of a BATF3/IL-2R-Module Reveals Vulnerabilities in Anaplastic Large Cell Lymphoma. Nat. Commun. 2021, 12, 5577. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E. AP-1—The Jun Proteins: Oncogenes or Tumor Suppressors in Disguise? Cell. Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Torres, A.N.B.; Melchers, R.C.; van Grieken, L.; Out-Luiting, J.J.; Mei, H.; Agaser, C.; Kuipers, T.B.; Quint, K.D.; Willemze, R.; Vermeer, M.H.; et al. Whole-Genome Profiling of Primary Cutaneous Anaplastic Large Cell Lymphoma. Haematologica 2022, 107, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Shaffer, A.L.; Ceribelli, M.; Zhang, M.; Wright, G.; Huang, D.W.; Xiao, W.; Powell, J.; Petrus, M.N.; Yang, Y.; et al. Targeting the HTLV-I-Regulated BATF3/IRF4 Transcriptional Network in Adult T Cell Leukemia/Lymphoma. Cancer Cell 2018, 34, 286–297.e10. [Google Scholar] [CrossRef]
- Nakashima, M.; Yamochi, T.; Watanabe, M.; Uchimaru, K.; Utsunomiya, A.; Higashihara, M.; Watanabe, T.; Horie, R. CD30 Characterizes Polylobated Lymphocytes and Disease Progression in HTLV-1-Infected Individuals. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5445–5457. [Google Scholar] [CrossRef]
- Al-Hamadani, M.; Habermann, T.M.; Cerhan, J.R.; Macon, W.R.; Maurer, M.J.; Go, R.S. Non-Hodgkin Lymphoma Subtype Distribution, Geodemographic Patterns, and Survival in the US: A Longitudinal Analysis of the National Cancer Data Base from 1998 to 2011. Am. J. Hematol. 2015, 90, 790–795. [Google Scholar] [CrossRef]
- Alessandri, A.J.; Pritchard, S.L.; Schultz, K.R.; Massing, B.G. A Population-Based Study of Pediatric Anaplastic Large Cell Lymphoma. Cancer 2002, 94, 1830–1835. [Google Scholar] [CrossRef]
- Shaw, T.I.; Pounds, S.; Cao, X.; Ma, J.; Palacios, G.; Mason, J.; Perkins, S.; Wu, G.; Fan, Y.; Wang, J.; et al. Comprehensive Genomic Analysis Reveals Molecular Heterogeneity in Pediatric ALK-Positive Anaplastic Large Cell Lymphoma. Leukemia 2025, 39, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Baron, M.; Amara, N.; Haioun, C.; Dandoit, M.; Maynadié, M.; Parrens, M.; Vergier, B.; Copie-Bergman, C.; Fabiani, B.; et al. Impact of Expert Pathologic Review of Lymphoma Diagnosis: Study of Patients From the French Lymphopath Network. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2008–2017. [Google Scholar] [CrossRef]
- Vose, J.; Armitage, J.; Weisenburger, D. International T-Cell Lymphoma Project International Peripheral T-Cell and Natural Killer/T-Cell Lymphoma Study: Pathology Findings and Clinical Outcomes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [CrossRef]
- Bekkenk, M.W.; Geelen, F.A.; van Voorst Vader, P.C.; Heule, F.; Geerts, M.L.; van Vloten, W.A.; Meijer, C.J.; Willemze, R. Primary and Secondary Cutaneous CD30(+) Lymphoproliferative Disorders: A Report from the Dutch Cutaneous Lymphoma Group on the Long-Term Follow-up Data of 219 Patients and Guidelines for Diagnosis and Treatment. Blood 2000, 95, 3653–3661. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Hapgood, G.; Savage, K.J. The Biology and Management of Systemic Anaplastic Large Cell Lymphoma. Blood 2015, 126, 17–25. [Google Scholar] [CrossRef]
- Zhang, X.-R.; Chien, P.-N.; Nam, S.-Y.; Heo, C.-Y. Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment. Cancers 2022, 14, 1650. [Google Scholar] [CrossRef]
- Mussolin, L.; Le Deley, M.-C.; Carraro, E.; Damm-Welk, C.; Attarbaschi, A.; Williams, D.; Burke, A.; Horibe, K.; Nakazawa, A.; Wrobel, G.; et al. Prognostic Factors in Childhood Anaplastic Large Cell Lymphoma: Long Term Results of the International ALCL99 Trial. Cancers 2020, 12, 2747. [Google Scholar] [CrossRef]
- Sarfraz, H.; Gentille, C.; Ensor, J.; Wang, L.; Wong, S.; Ketcham, M.S.; Joshi, J.; Pingali, S.R.K. Primary Cutaneous Anaplastic Large-Cell Lymphoma: A Review of the SEER Database from 2005 to 2016. Clin. Exp. Dermatol. 2021, 46, 1420–1426. [Google Scholar] [CrossRef]
- Congras, A.; Hoareau-Aveilla, C.; Caillet, N.; Tosolini, M.; Villarese, P.; Cieslak, A.; Rodriguez, L.; Asnafi, V.; Macintyre, E.; Egger, G.; et al. ALK-Transformed Mature T Lymphocytes Restore Early Thymus Progenitor Features. J. Clin. Investig. 2020, 130, 6395–6408. [Google Scholar] [CrossRef]
- Malcolm, T.I.M.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.A.; Brugières, L.; Hughes, K.; Payet, D.; et al. Anaplastic Large Cell Lymphoma Arises in Thymocytes and Requires Transient TCR Expression for Thymic Egress. Nat. Commun. 2016, 7, 10087. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, M.H.; Dukers, D.F.; ten Berge, R.L.; Bloemena, E.; Wu, L.; Vos, W.; de Vries, E.; Tensen, C.P.; Meijer, C.J.L.M.; Willemze, R. Differential Expression of Thymus and Activation Regulated Chemokine and Its Receptor CCR4 in Nodal and Cutaneous Anaplastic Large-Cell Lymphomas and Hodgkin’s Disease. Mod. Pathol. 2002, 15, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Hassler, M.R.; Pulverer, W.; Lakshminarasimhan, R.; Redl, E.; Hacker, J.; Garland, G.D.; Merkel, O.; Schiefer, A.-I.; Simonitsch-Klupp, I.; Kenner, L.; et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-Wide DNA Methylation Profiling. Cell Rep. 2016, 17, 596–608. [Google Scholar] [CrossRef]
- Pina-Oviedo, S.; Ortiz-Hidalgo, C.; Carballo-Zarate, A.A.; Zarate-Osorno, A. ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas. Cancers 2021, 13, 4667. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Hennessey, D.; Gniadecki, R. Clonotype Pattern in T-Cell Lymphomas Map the Cell of Origin to Immature Lymphoid Precursors. Blood Adv. 2022, 6, 2334–2345. [Google Scholar] [CrossRef]
- Murthy, G.S.G.; Hamadani, M.; Bhatt, V.R.; Dhakal, I.; Mehta, P. Systemic Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma: A Population-Based Analysis of Incidence and Survival. Clin. Lymphoma Myeloma Leuk. 2017, 17, 201–206. [Google Scholar] [CrossRef]
- Gromowsky, M.J.; D’Angelo, C.R.; Lunning, M.A.; Armitage, J.O. ALK-Positive Anaplastic Large Cell Lymphoma in Adults. Fac. Rev. 2023, 12, 21. [Google Scholar] [CrossRef]
- Ferreri, A.J.M.; Govi, S.; Pileri, S.A.; Savage, K.J. Anaplastic Large Cell Lymphoma, ALK-Positive. Crit. Rev. Oncol. Hematol. 2012, 83, 293–302. [Google Scholar] [CrossRef]
- Canellas, M.C.; Bruno-Riscarolli, E.; Ferreira-Facio, C.S.; Lopes-Alves, D.V.; Botafogo, V.D.; Sutter, D.; Pontes, R.M.; Land, M.G.P.; Bedran Milito, C.; da Costa, E.S. Immunophenotypic Shifts during Minimal Residual Evaluation in a Case of Leukemic Form of Anaplastic Large Cell Lymphoma ALK+. Cancer Rep. 2021, 5, e1526. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Seo, K.; Arber, D.A. Prevalence of Bone Marrow Involvement in Systemic Anaplastic Large Cell Lymphoma: Are Immunohistochemical Studies Necessary? Hum. Pathol. 2008, 39, 1331–1340. [Google Scholar] [CrossRef]
- Benharroch, D.; Meguerian-Bedoyan, Z.; Lamant, L.; Amin, C.; Brugières, L.; Terrier-Lacombe, M.-J.; Haralambieva, E.; Pulford, K.; Pileri, S.; Morris, S.W.; et al. ALK-Positive Lymphoma: A Single Disease With a Broad Spectrum of Morphology. Blood 1998, 91, 2076–2084. [Google Scholar] [CrossRef]
- Pileri, S.; Falini, B.; Delsol, G.; Stein, H.; Baglioni, P.; Poggi, S.; Martelli, M.F.; Rivano, M.T.; Mason, D.Y.; Stansfeld, A.G. Lymphohistiocytic T-Cell Lymphoma (Anaplastic Large Cell Lymphoma CD30+/Ki-1 + with a High Content of Reactive Histiocytes). Histopathology 1990, 16, 383–391. [Google Scholar] [CrossRef]
- Stein, H.; Foss, H.D.; Dürkop, H.; Marafioti, T.; Delsol, G.; Pulford, K.; Pileri, S.; Falini, B. CD30(+) Anaplastic Large Cell Lymphoma: A Review of Its Histopathologic, Genetic, and Clinical Features. Blood 2000, 96, 3681–3695. [Google Scholar] [CrossRef] [PubMed]
- Tsuyama, N.; Sakamoto, K.; Sakata, S.; Dobashi, A.; Takeuchi, K. Anaplastic Large Cell Lymphoma: Pathology, Genetics, and Clinical Aspects. J. Clin. Exp. Hematop. JCEH 2017, 57, 120–142. [Google Scholar] [CrossRef]
- Khanlari, M.; Li, S.; Miranda, R.N.; Iyer, S.; Konoplev, S.; Lin, P.; Yin, C.C.; Tang, G.; Qiu, L.; Vega, F.; et al. Small Cell/Lymphohistiocytic Morphology Is Associated with Peripheral Blood Involvement, CD8 Positivity and Retained T-Cell Antigens, but Not Outcome in Adults with ALK+ Anaplastic Large Cell Lymphoma. Mod. Pathol. 2022, 35, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Liu, C.; Zhao, S. Transformation of Recurrent ALK-Positive Anaplastic Large-Cell Lymphoma from Common Pattern to Composite Pattern (Lymphohistiocytic and Small-Cell Pattern) with a Change in CD30 Expression. Int. J. Hematol. 2023, 118, 131–134. [Google Scholar] [CrossRef]
- Falini, B.; Bigerna, B.; Fizzotti, M.; Pulford, K.; Pileri, S.A.; Delsol, G.; Carbone, A.; Paulli, M.; Magrini, U.; Menestrina, F.; et al. ALK Expression Defines a Distinct Group of T/Null Lymphomas (“ALK Lymphomas”) with a Wide Morphological Spectrum. Am. J. Pathol. 1998, 153, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Mereu, E.; Pellegrino, E.; Scarfò, I.; Inghirami, G.; Piva, R. The Heterogeneous Landscape of ALK Negative ALCL. Oncotarget 2017, 8, 18525–18536. [Google Scholar] [CrossRef]
- Sibon, D.; Nguyen, D.-P.; Schmitz, N.; Suzuki, R.; Feldman, A.L.; Gressin, R.; Lamant, L.; Weisenburger, D.D.; Rosenwald, A.; Nakamura, S.; et al. ALK-Positive Anaplastic Large-Cell Lymphoma in Adults: An Individual Patient Data Pooled Analysis of 263 Patients. Haematologica 2019, 104, e562–e565. [Google Scholar] [CrossRef]
- Feldman, A.L.; Dasari, S.; Rimsza, L.M.; Scott, D.W.; Oishi, N.; Amador, C.; Campo, E.; Chan, W.C.; Cook, J.R.; Delabie, J.; et al. Gene Expression Profiling Reveals Two Overarching Types of Anaplastic Large Cell Lymphoma with Distinct Targetable Biology: An L.L.M.P.P. Study. Blood 2023, 142, 847. [Google Scholar] [CrossRef]
- Manso, R.; Rodríguez-Perales, S.; Torres-Ruiz, R.; Santonja, C.; Rodríguez-Pinilla, S.-M. PD-L1 Expression in Peripheral T-Cell Lymphomas Is Not Related to Either PD-L1 Gene Amplification or Rearrangements. Leuk. Lymphoma 2021, 62, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Montes-Mojarro, I.A.; Steinhilber, J.; Bonzheim, I.; Quintanilla-Martinez, L.; Fend, F. The Pathological Spectrum of Systemic Anaplastic Large Cell Lymphoma (ALCL). Cancers 2018, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Kaseb, H.; Mukkamalla, S.K.R.; Rajasurya, V. Anaplastic Large Cell Lymphoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Salaverria, I.; Beà, S.; Lopez-Guillermo, A.; Lespinet, V.; Pinyol, M.; Burkhardt, B.; Lamant, L.; Zettl, A.; Horsman, D.; Gascoyne, R.; et al. Genomic Profiling Reveals Different Genetic Aberrations in Systemic ALK-Positive and ALK-Negative Anaplastic Large Cell Lymphomas. Br. J. Haematol. 2008, 140, 516–526. [Google Scholar] [CrossRef]
- Cheng, S.H.; Ng, M.H.L.; Lau, K.M.; Liu, H.S.Y.; Chan, J.C.W.; Hui, A.B.Y.; Lo, K.W.; Jiang, H.; Hou, J.; Chu, R.W.; et al. 4q Loss Is Potentially an Important Genetic Event in MM Tumorigenesis: Identification of a Tumor Suppressor Gene Regulated by Promoter Methylation at 4q13.3, Platelet Factor 4. Blood 2007, 109, 2089–2099. [Google Scholar] [CrossRef]
- Grygalewicz, B.; Woroniecka, R.; Rymkiewicz, G.; Rygier, J.; Borkowska, K.; Kotyl, A.; Blachnio, K.; Bystydzienski, Z.; Nowakowska, B.; Pienkowska-Grela, B. The 11q-Gain/Loss Aberration Occurs Recurrently in MYC-Negative Burkitt-like Lymphoma With 11q Aberration, as Well as MYC-Positive Burkitt Lymphoma and MYC-Positive High-Grade B-Cell Lymphoma, NOS. Am. J. Clin. Pathol. 2018, 149, 17–28. [Google Scholar] [CrossRef]
- Lobello, C.; Tichy, B.; Bystry, V.; Radova, L.; Filip, D.; Mraz, M.; Montes-Mojarro, I.-A.; Prokoph, N.; Larose, H.; Liang, H.-C.; et al. STAT3 and TP53 Mutations Associate with Poor Prognosis in Anaplastic Large Cell Lymphoma. Leukemia 2021, 35, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ding, N.; Mi, L.; Shi, Y.; Liu, W.; Song, Y.; Shu, S.; Zhu, J. Correlation of Mutational Landscape and Survival Outcome of Peripheral T-Cell Lymphomas. Exp. Hematol. Oncol. 2021, 10, 9. [Google Scholar] [CrossRef]
- Cela, I.; Di Matteo, A.; Federici, L. Nucleophosmin in Its Interaction with Ligands. Int. J. Mol. Sci. 2020, 21, 4885. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a Kinase Gene, ALK, to a Nucleolar Protein Gene, NPM, in Non-Hodgkin’s Lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Wu, R.; Lim, M.S. Updates in Pathobiological Aspects of Anaplastic Large Cell Lymphoma. Front. Oncol. 2023, 13, 1241532. [Google Scholar] [CrossRef]
- Bohling, S.D.; Jenson, S.D.; Crockett, D.K.; Schumacher, J.A.; Elenitoba-Johnson, K.S.J.; Lim, M.S. Analysis of Gene Expression Profile of TPM3-ALK Positive Anaplastic Large Cell Lymphoma Reveals Overlapping and Unique Patterns with That of NPM-ALK Positive Anaplastic Large Cell Lymphoma. Leuk. Res. 2008, 32, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Kalinova, M.; Mrhalova, M.; Kabickova, E.; Svaton, M.; Skotnicova, A.; Prouzova, Z.; Krenova, Z.; Kolenova, A.; Divoka, M.; Fronkova, E.; et al. Molecular Screening in Anaplastic Lymphoma Kinase–Positive Anaplastic Large Cell Lymphoma: Anaplastic Lymphoma Kinase Analysis, Next-Generation Sequencing Fusion Gene Detection, and T-Cell Receptor Immunoprofiling. Mod. Pathol. 2024, 37, 100428. [Google Scholar] [CrossRef]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-Anaplastic Lymphoma Kinase of Large-Cell Anaplastic Lymphoma Is a Constitutively Active Tyrosine Kinase That Utilizes Phospholipase C-Gamma to Mediate Its Mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef]
- Bai, R.Y.; Ouyang, T.; Miething, C.; Morris, S.W.; Peschel, C.; Duyster, J. Nucleophosmin-Anaplastic Lymphoma Kinase Associated with Anaplastic Large-Cell Lymphoma Activates the Phosphatidylinositol 3-Kinase/Akt Antiapoptotic Signaling Pathway. Blood 2000, 96, 4319–4327. [Google Scholar] [CrossRef]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of Phosphatidylinositol 3-Kinase-Akt Pathway in Nucleophosmin/Anaplastic Lymphoma Kinase-Mediated Lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar]
- Choudhari, R.; Minero, V.G.; Menotti, M.; Pulito, R.; Brakebusch, C.; Compagno, M.; Voena, C.; Ambrogio, C.; Chiarle, R. Redundant and Nonredundant Roles for Cdc42 and Rac1 in Lymphomas Developed in NPM-ALK Transgenic Mice. Blood 2016, 127, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Leventaki, V.; Drakos, E.; Medeiros, L.J.; Lim, M.S.; Elenitoba-Johnson, K.S.; Claret, F.X.; Rassidakis, G.Z. NPM-ALK Oncogenic Kinase Promotes Cell-Cycle Progression through Activation of JNK/cJun Signaling in Anaplastic Large-Cell Lymphoma. Blood 2007, 110, 1621–1630. [Google Scholar] [CrossRef]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel Dysregulation of STAT3 Activation in Anaplastic Lymphoma Kinase-Positive T/Null-Cell Lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.S.; Carlson, M.L.; Crockett, D.K.; Fillmore, G.C.; Abbott, D.R.; Elenitoba-Johnson, O.F.; Tripp, S.R.; Rassidakis, G.Z.; Medeiros, L.J.; Szankasi, P.; et al. The Proteomic Signature of NPM/ALK Reveals Deregulation of Multiple Cellular Pathways. Blood 2009, 114, 1585–1595. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Slupianek, A.; Xue, L.; Zhang, Q.; Raghunath, P.N.; Hoser, G.; Wasik, M.A.; Morris, S.W.; Skorski, T. Role of Signal Transducer and Activator of Transcription 5 in Nucleophosmin/Anaplastic Lymphoma Kinase-Mediated Malignant Transformation of Lymphoid Cells. Cancer Res. 2001, 61, 6517–6523. [Google Scholar]
- Ambrogio, C.; Martinengo, C.; Voena, C.; Tondat, F.; Riera, L.; di Celle, P.F.; Inghirami, G.; Chiarle, R. NPM-ALK Oncogenic Tyrosine Kinase Controls T-Cell Identity by Transcriptional Regulation and Epigenetic Silencing in Lymphoma Cells. Cancer Res. 2009, 69, 8611–8619. [Google Scholar] [CrossRef] [PubMed]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic Lymphoma Kinase (ALK) Activates Stat3 and Protects Hematopoietic Cells from Cell Death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Anastasov, N.; Bonzheim, I.; Rudelius, M.; Klier, M.; Dau, T.; Angermeier, D.; Duyster, J.; Pittaluga, S.; Fend, F.; Raffeld, M.; et al. C/EBPβ Expression in ALK-Positive Anaplastic Large Cell Lymphomas Is Required for Cell Proliferation and Is Induced by the STAT3 Signaling Pathway. Haematologica 2010, 95, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-Anaplastic Lymphoma Kinase: The Ultimate Oncogene and Therapeutic Target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef]
- Inghirami, G.; Chiarle, R.; Simmons, W.J.; Piva, R.; Schlessinger, K.; Levy, D.E. New and Old Functions of STAT3: A Pivotal Target for Individualized Treatment of Cancer. Cell Cycle Georget. Tex 2005, 4, 1131–1133. [Google Scholar] [CrossRef]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 Is Required for ALK-Mediated Lymphomagenesis and Provides a Possible Therapeutic Target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef]
- Marzec, M.; Halasa, K.; Liu, X.; Wang, H.Y.; Cheng, M.; Baldwin, D.; Tobias, J.W.; Schuster, S.J.; Woetmann, A.; Zhang, Q.; et al. Malignant Transformation of CD4+ T Lymphocytes Mediated by Oncogenic Kinase NPM/ALK Recapitulates IL-2-Induced Cell Signaling and Gene Expression Reprogramming. J. Immunol. 2013, 191, 6200–6207. [Google Scholar] [CrossRef]
- Huber, R.; Pietsch, D.; Panterodt, T.; Brand, K. Regulation of C/EBPβ and Resulting Functions in Cells of the Monocytic Lineage. Cell. Signal. 2012, 24, 1287–1296. [Google Scholar] [CrossRef]
- Bonzheim, I.; Irmler, M.; Klier-Richter, M.; Steinhilber, J.; Anastasov, N.; Schäfer, S.; Adam, P.; Beckers, J.; Raffeld, M.; Fend, F.; et al. Identification of C/EBPβ Target Genes in ALK+ Anaplastic Large Cell Lymphoma (ALCL) by Gene Expression Profiling and Chromatin Immunoprecipitation. PLoS ONE 2013, 8, e64544. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Pittaluga, S.; Miething, C.; Klier, M.; Rudelius, M.; Davies-Hill, T.; Anastasov, N.; Martinez, A.; Vivero, A.; Duyster, J.; et al. NPM-ALK-Dependent Expression of the Transcription Factor CCAAT/Enhancer Binding Protein Beta in ALK-Positive Anaplastic Large Cell Lymphoma. Blood 2006, 108, 2029–2036. [Google Scholar] [CrossRef]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential Role of IRF4 and MYC Signaling for Survival of Anaplastic Large Cell Lymphoma. Blood 2015, 125, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Lollies, A.; Hartmann, S.; Schneider, M.; Bracht, T.; Weiß, A.L.; Arnolds, J.; Klein-Hitpass, L.; Sitek, B.; Hansmann, M.-L.; Küppers, R.; et al. An Oncogenic Axis of STAT-Mediated BATF3 Upregulation Causing MYC Activity in Classical Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma. Leukemia 2018, 32, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Brandstoetter, T.; Schmoellerl, J.; Grausenburger, R.; Kollmann, S.; Doma, E.; Huuhtanen, J.; Klampfl, T.; Eder, T.; Grebien, F.; Hoermann, G.; et al. SBNO2 Is a Critical Mediator of STAT3-Driven Hematological Malignancies. Blood 2023, 141, 1831–1845. [Google Scholar] [CrossRef]
- Villa, M.; Sharma, G.G.; Malighetti, F.; Mauri, M.; Arosio, G.; Cordani, N.; Lobello, C.; Larose, H.; Pirola, A.; D’Aliberti, D.; et al. Recurrent Somatic Mutations of FAT Family Cadherins Induce an Aggressive Phenotype and Poor Prognosis in Anaplastic Large Cell Lymphoma. Br. J. Cancer 2024, 131, 1781–1795. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhong, L.; Tang, W.; Zhang, W.; Lin, T.; Zhu, W.; Chen, G.; Wang, J. TET2-Mediated 5-Hydroxymethylcytosine of TXNIP Promotes Cell Cycle Arrest in Systemic Anaplastic Large Cell Lymphoma. Clin. Epigenetics 2025, 17, 10. [Google Scholar] [CrossRef]
- Merkel, O.; Hamacher, F.; Laimer, D.; Sifft, E.; Trajanoski, Z.; Scheideler, M.; Egger, G.; Hassler, M.R.; Thallinger, C.; Schmatz, A.; et al. Identification of Differential and Functionally Active miRNAs in Both Anaplastic Lymphoma Kinase (ALK)+ and ALK- Anaplastic Large-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2010, 107, 16228–16233. [Google Scholar] [CrossRef]
- Spaccarotella, E.; Pellegrino, E.; Ferracin, M.; Ferreri, C.; Cuccuru, G.; Liu, C.; Iqbal, J.; Cantarella, D.; Taulli, R.; Provero, P.; et al. STAT3-Mediated Activation of microRNA Cluster 17~92 Promotes Proliferation and Survival of ALK-Positive Anaplastic Large Cell Lymphoma. Haematologica 2014, 99, 116–124. [Google Scholar] [CrossRef]
- Stallings, R.L. MicroRNA Involvement in the Pathogenesis of Neuroblastoma: Potential for microRNA Mediated Therapeutics. Curr. Pharm. Des. 2009, 15, 456–462. [Google Scholar] [CrossRef]
- Merkel, O.; Hamacher, F.; Sifft, E.; Kenner, L.; Greil, R. European Research Initiative on Anaplastic Large Cell Lymphoma Novel Therapeutic Options in Anaplastic Large Cell Lymphoma: Molecular Targets and Immunological Tools. Mol. Cancer Ther. 2011, 10, 1127–1136. [Google Scholar] [CrossRef]
- Steinhilber, J.; Bonin, M.; Walter, M.; Fend, F.; Bonzheim, I.; Quintanilla-Martinez, L. Next-Generation Sequencing Identifies Deregulation of microRNAs Involved in Both Innate and Adaptive Immune Response in ALK+ ALCL. PLoS ONE 2015, 10, e0117780. [Google Scholar] [CrossRef]
- Matsuyama, H.; Suzuki, H.I.; Nishimori, H.; Noguchi, M.; Yao, T.; Komatsu, N.; Mano, H.; Sugimoto, K.; Miyazono, K. miR-135b Mediates NPM-ALK-Driven Oncogenicity and Renders IL-17-Producing Immunophenotype to Anaplastic Large Cell Lymphoma. Blood 2011, 118, 6881–6892. [Google Scholar] [CrossRef] [PubMed]
- Hoareau-Aveilla, C.; Valentin, T.; Daugrois, C.; Quelen, C.; Mitou, G.; Quentin, S.; Jia, J.; Spicuglia, S.; Ferrier, P.; Ceccon, M.; et al. Reversal of microRNA-150 Silencing Disadvantages Crizotinib-Resistant NPM-ALK(+) Cell Growth. J. Clin. Invest. 2015, 125, 3505–3518. [Google Scholar] [CrossRef] [PubMed]
- Garbin, A.; Contarini, G.; Damanti, C.C.; Tosato, A.; Bortoluzzi, S.; Gaffo, E.; Pizzi, M.; Carraro, E.; Lo Nigro, L.; Vinti, L.; et al. MiR-146a-5p Enrichment in Small-Extracellular Vesicles of Relapsed Pediatric ALCL Patients Promotes Macrophages Infiltration and Differentiation. Biochem. Pharmacol. 2023, 215, 115747. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.-H.; Lu, P.-H.; Lin, Y.-H.; Tsai, M.-M.; Lin, Y.-W.; Chiu, C.-T.; Lin, K.-H. The Long Non-Coding RNA LINC01013 Enhances Invasion of Human Anaplastic Large-Cell Lymphoma. Sci. Rep. 2017, 7, 295. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Moody, S.E.; Perez, D.; Pan, T.; Sarkisian, C.J.; Portocarrero, C.P.; Sterner, C.J.; Notorfrancesco, K.L.; Cardiff, R.D.; Chodosh, L.A. The Transcriptional Repressor Snail Promotes Mammary Tumor Recurrence. Cancer Cell 2005, 8, 197–209. [Google Scholar] [CrossRef]
- Babin, L.; Piganeau, M.; Renouf, B.; Lamribet, K.; Thirant, C.; Deriano, L.; Mercher, T.; Giovannangeli, C.; Brunet, E.C. Chromosomal Translocation Formation Is Sufficient to Produce Fusion Circular RNAs Specific to Patient Tumor Cells. iScience 2018, 5, 19–29. [Google Scholar] [CrossRef]
- Dupuis-Sandoval, F.; Poirier, M.; Scott, M.S. The Emerging Landscape of Small Nucleolar RNAs in Cell Biology. Wiley Interdiscip. Rev. RNA 2015, 6, 381–397. [Google Scholar] [CrossRef]
- Valleron, W.; Ysebaert, L.; Berquet, L.; Fataccioli, V.; Quelen, C.; Martin, A.; Parrens, M.; Lamant, L.; de Leval, L.; Gisselbrecht, C.; et al. Small Nucleolar RNA Expression Profiling Identifies Potential Prognostic Markers in Peripheral T-Cell Lymphoma. Blood 2012, 120, 3997–4005. [Google Scholar] [CrossRef]
- Le Deley, M.-C.; Reiter, A.; Williams, D.; Delsol, G.; Oschlies, I.; McCarthy, K.; Zimmermann, M.; Brugières, L. European Intergroup for Childhood Non-Hodgkin Lymphoma Prognostic Factors in Childhood Anaplastic Large Cell Lymphoma: Results of a Large European Intergroup Study. Blood 2008, 111, 1560–1566. [Google Scholar] [CrossRef]
- Williams, D.; Mori, T.; Reiter, A.; Woessman, W.; Rosolen, A.; Wrobel, G.; Zsiros, J.; Uyttebroeck, A.; Marky, I.; Le Deley, M.-C.; et al. Central Nervous System Involvement in Anaplastic Large Cell Lymphoma in Childhood: Results from a Multicentre European and Japanese Study. Pediatr. Blood Cancer 2013, 60, E118–E121. [Google Scholar] [CrossRef]
- Spiegel, A.; Paillard, C.; Ducassou, S.; Perel, Y.; Plantaz, D.; Strullu, M.; Eischen, A.; Lutz, P.; Lamant, L.; Le Deley, M.-C.; et al. Paediatric Anaplastic Large Cell Lymphoma with Leukaemic Presentation in Children: A Report of Nine French Cases. Br. J. Haematol. 2014, 165, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Onciu, M.; Behm, F.G.; Raimondi, S.C.; Moore, S.; Harwood, E.L.; Pui, C.-H.; Sandlund, J.T. ALK-Positive Anaplastic Large Cell Lymphoma with Leukemic Peripheral Blood Involvement Is a Clinicopathologic Entity with an Unfavorable Prognosis. Report of Three Cases and Review of the Literature. Am. J. Clin. Pathol. 2003, 120, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Grewal, J.S.; Smith, L.B.; Winegarden, J.D.; Krauss, J.C.; Tworek, J.A.; Schnitzer, B. Highly Aggressive ALK-Positive Anaplastic Large Cell Lymphoma with a Leukemic Phase and Multi-Organ Involvement: A Report of Three Cases and a Review of the Literature. Ann. Hematol. 2007, 86, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Akahane, D.; Takeyama, K.; Sato, N.; Takayama, N.; Ando, J.; Nitta, H.; Noguchi, M.; Naganuma, K.; Momose, S.; et al. TP53 Deletion Is Associated with Poor Survival of Adult ALK-Positive ALCL Patients Receiving CHOP-Based Chemotherapy. Ann. Hematol. 2025, 104, 1801–1806. [Google Scholar] [CrossRef]
- Lamant, L.; McCarthy, K.; d’Amore, E.; Klapper, W.; Nakagawa, A.; Fraga, M.; Maldyk, J.; Simonitsch-Klupp, I.; Oschlies, I.; Delsol, G.; et al. Prognostic Impact of Morphologic and Phenotypic Features of Childhood ALK-Positive Anaplastic Large-Cell Lymphoma: Results of the ALCL99 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4669–4676. [Google Scholar] [CrossRef]
- Noguchi, K.; Ikawa, Y.; Takenaka, M.; Sakai, Y.; Fujiki, T.; Kuroda, R.; Wada, T. Characterisation of Two Tumour Cell Populations in the Small Cell Variant of Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma. Br. J. Haematol. 2022, 196, 241–243. [Google Scholar] [CrossRef]
- Damm-Welk, C.; Kutscher, N.; Zimmermann, M.; Attarbaschi, A.; Schieferstein, J.; Knörr, F.; Oschlies, I.; Klapper, W.; Woessmann, W. Quantification of Minimal Disseminated Disease by Quantitative Polymerase Chain Reaction and Digital Polymerase Chain Reaction for NPM-ALK as a Prognostic Factor in Children with Anaplastic Large Cell Lymphoma. Haematologica 2020, 105, 2141–2149. [Google Scholar] [CrossRef]
- Lowe, E.J.; Reilly, A.F.; Lim, M.S.; Gross, T.G.; Saguilig, L.; Barkauskas, D.A.; Wu, R.; Alexander, S.; Bollard, C.M. Brentuximab Vedotin in Combination with Chemotherapy for Pediatric Patients with ALK+ ALCL: Results of COG Trial ANHL12P1. Blood 2021, 137, 3595–3603. [Google Scholar] [CrossRef]
- Sukswai, N.; Lyapichev, K.; Khoury, J.D.; Medeiros, L.J. Diffuse Large B-Cell Lymphoma Variants: An Update. Pathology 2020, 52, 53–67. [Google Scholar] [CrossRef]
- Delsol, G.; Lamant, L.; Mariamé, B.; Pulford, K.; Dastugue, N.; Brousset, P.; Rigal-Huguet, F.; Al Saati, T.; Cerretti, D.P.; Morris, S.W.; et al. A New Subtype of Large B-Cell Lymphoma Expressing the ALK Kinase and Lacking the 2; 5 Translocation. Blood 1997, 89, 1483–1490. [Google Scholar] [CrossRef]
- Pan, Z.; Hu, S.; Li, M.; Zhou, Y.; Kim, Y.S.; Reddy, V.; Sanmann, J.N.; Smith, L.M.; Chen, M.; Gao, Z.; et al. ALK-Positive Large B-Cell Lymphoma: A Clinicopathologic Study of 26 Cases with Review of Additional 108 Cases in the Literature. Am. J. Surg. Pathol. 2017, 41, 25–38. [Google Scholar] [CrossRef]
- Laurent, C.; Do, C.; Gascoyne, R.D.; Lamant, L.; Ysebaert, L.; Laurent, G.; Delsol, G.; Brousset, P. Anaplastic Lymphoma Kinase–Positive Diffuse Large B-Cell Lymphoma: A Rare Clinicopathologic Entity with Poor Prognosis. J. Clin. Oncol. 2009, 27, 4211–4216. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA 2019, 322, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Siemion, K.; Reszec-Gielazyn, J.; Kisluk, J.; Roszkowiak, L.; Zak, J.; Korzynska, A. What Do We Know about Inflammatory Myofibroblastic Tumors?—A Systematic Review. Adv. Med. Sci. 2022, 67, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Kemps, P.G.; Picarsic, J.; Durham, B.H.; Hélias-Rodzewicz, Z.; Hiemcke-Jiwa, L.; van den Bos, C.; van de Wetering, M.D.; van Noesel, C.J.M.; van Laar, J.A.M.; Verdijk, R.M.; et al. ALK-Positive Histiocytosis: A New Clinicopathologic Spectrum Highlighting Neurologic Involvement and Responses to ALK Inhibition. Blood 2022, 139, 256–280. [Google Scholar] [CrossRef]
- Veija, T.; Koljonen, V.; Bohling, T.; Kero, M.; Knuutila, S.; Sarhadi, V.K. Aberrant Expression of ALK and EZH2 in Merkel Cell Carcinoma. BMC Cancer 2017, 17, 236. [Google Scholar] [CrossRef]
- Kobayashi, T.; Uehara, Y.; Watanabe, K.; Hishima, T.; Hosomi, Y. Successful Treatment of ALK-Positive Large-Cell Neuroendocrine Carcinoma of the Lung with Sequential ALK Inhibitors: A Case Report. JTO Clin. Res. Rep. 2023, 4, 100538. [Google Scholar] [CrossRef]
- Busam, K.J.; Vilain, R.E.; Lum, T.; Busam, J.A.; Hollmann, T.J.; Saw, R.P.M.; Coit, D.C.; Scolyer, R.A.; Wiesner, T. Primary and Metastatic Cutaneous Melanomas Express ALK Through Alternative Transcriptional Initiation. Am. J. Surg. Pathol. 2016, 40, 786–795. [Google Scholar] [CrossRef]
- Yeh, I.; de la Fouchardiere, A.; Pissaloux, D.; Mully, T.W.; Garrido, M.C.; Vemula, S.S.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C. Clinical, Histopathologic and Genomic Features of Spitz Tumors with ALK Fusions. Am. J. Surg. Pathol. 2015, 39, 581–591. [Google Scholar] [CrossRef]
- Kuroda, N.; Trpkov, K.; Gao, Y.; Tretiakova, M.; Liu, Y.J.; Ulamec, M.; Takeuchi, K.; Agaimy, A.; Przybycin, C.; Magi-Galluzzi, C.; et al. ALK Rearranged Renal Cell Carcinoma (ALK-RCC): A Multi-Institutional Study of Twelve Cases with Identification of Novel Partner Genes CLIP1, KIF5B and KIAA1217. Mod. Pathol. 2020, 33, 2564–2579. [Google Scholar] [CrossRef] [PubMed]
- Fadl, A.; Feldman, A.L. Epithelioid Inflammatory Myofibroblastic Sarcoma: A Pitfall in the Differential Diagnosis of ALK-Positive Anaplastic Large Cell Lymphoma. J. Hematop. 2023, 16, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Duan, C.; Wang, S.; Fu, L.; Yang, P.; Yu, T.; Deel, M.D.; Lau, L.M.S.; Ma, X.; Ni, X.; et al. Pediatric Spindle Cell/Sclerosing Rhabdomyosarcoma with FUS–TFCP2 Fusion: A Case Report and Literature Review. Transl. Pediatr. 2024, 13, 178–191. [Google Scholar] [CrossRef]
- Mano, H. ALKoma: A Cancer Subtype with a Shared Target. Cancer Discov. 2012, 2, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic Insight into ALK Receptor Tyrosine Kinase in Human Cancer Biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef]
- Ferreri, A.J.M.; Govi, S.; Pileri, S.A.; Savage, K.J. Anaplastic Large Cell Lymphoma, ALK-Negative. Crit. Rev. Oncol. Hematol. 2013, 85, 206–215. [Google Scholar] [CrossRef]
- Harb, M.; Abrassart, T.; Dewispeleare, L.; Sidon, P.; Dirckx, N.; Trepant, A.-L.; Castiaux, J.; Heimann, P.; Emile, J.-F.; Farhat, H. Synchronous Clonally Related Anaplastic Large Cell Lymphoma and Malignant Histiocytosis. Diagn. Pathol. 2025, 20, 6. [Google Scholar] [CrossRef]
- Bonzheim, I.; Geissinger, E.; Roth, S.; Zettl, A.; Marx, A.; Rosenwald, A.; Müller-Hermelink, H.K.; Rüdiger, T. Anaplastic Large Cell Lymphomas Lack the Expression of T-Cell Receptor Molecules or Molecules of Proximal T-Cell Receptor Signaling. Blood 2004, 104, 3358–3360. [Google Scholar] [CrossRef]
- Krenacs, L.; Wellmann, A.; Sorbara, L.; Himmelmann, A.W.; Bagdi, E.; Jaffe, E.S.; Raffeld, M. Cytotoxic Cell Antigen Expression in Anaplastic Large Cell Lymphomas of T- and Null-Cell Type and Hodgkin’s Disease: Evidence for Distinct Cellular Origin. Blood 1997, 89, 980–989. [Google Scholar] [CrossRef]
- Foss, H.-D.; Anagnostopoulos, l.; Araujo, I.; Assaf, C.; Demel, G.; Kummer, J.A.; Hummel, M.; Stein, H. Anaplastic Large-Cell Lymphomas of T-Cell and Null-Cell Phenotype Express Cytotoxic Molecules. Blood 1996, 88, 4005–4011. [Google Scholar] [CrossRef]
- Parrilla Castellar, E.R.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-Negative Anaplastic Large Cell Lymphoma Is a Genetically Heterogeneous Disease with Widely Disparate Clinical Outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Medeiros, L.J.; Li, S.; Wang, S.A.; Lin, P.; Khanlari, M.; Iyer, S.P.; Yin, C.C.; Tang, G.; Jorgensen, J.L.; et al. CD8 Expression in Anaplastic Large Cell Lymphoma Correlates with Noncommon Morphologic Variants and T-Cell Antigen Expression Suggesting Biological Differences with CD8-Negative Anaplastic Large Cell Lymphoma. Hum. Pathol. 2020, 98, 1–9. [Google Scholar] [CrossRef]
- Ma, L.; Katz, Y.; Sharan, K.P.; Schwarting, R.; Kim, A.S. Epstein-Barr Virus Positive Anaplastic Large Cell Lymphoma: Myth or Reality? Int. J. Clin. Exp. Pathol. 2011, 4, 100–110. [Google Scholar]
- Kim, Y.C.; Yang, W.I.; Lee, M.-G.; Kim, S.N.; Cho, K.H.; Lee, S.J.; Lee, M.W.; Koh, J.K. Epstein–Barr Virus in CD30+ Anaplastic Large Cell Lymphoma Involving the Skin and Lymphomatoid Papulosis in South Korea. Int. J. Dermatol. 2006, 45, 1312–1316. [Google Scholar] [CrossRef]
- Arun, I.; Roy, P.; Arora, N.; Bhave, S.J.; Nair, R.; Chandy, M. PAX-5 Positivity in Anaplastic Lymphoma Kinase-Negative Anaplastic Large Cell Lymphoma: A Case Report and Review of Literature. Int. J. Surg. Pathol. 2017, 25, 333–338. [Google Scholar] [CrossRef]
- Feldman, A.L.; Law, M.E.; Inwards, D.J.; Dogan, A.; McClure, R.F.; Macon, W.R. PAX5-Positive T-Cell Anaplastic Large Cell Lymphomas Associated with Extra Copies of the PAX5 Gene Locus. Mod. Pathol. 2010, 23, 593–602. [Google Scholar] [CrossRef]
- Ong, D.M.; Cummins, K.D.; Pham, A.; Grigoriadis, G. PAX5-Expressing ALK-Negative Anaplastic Large Cell Lymphoma with Extensive Extranodal and Nodal Involvement. BMJ Case Rep. 2015, 2015, bcr2015211159. [Google Scholar] [CrossRef] [PubMed]
- Salyana, M.A.; Khan, S.; Zhang, X. ALK-Negative Anaplastic Large Cell Lymphoma, Null Type with Aberrant Expression of PAX5 and CD138: A Diagnostic Pitfall. Diagn. Cytopathol. 2021, 49, E395–E399. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.L.; Oishi, N.; Ketterling, R.P.; Ansell, S.M.; Shi, M.; Dasari, S. Immunohistochemical Approach to Genetic Subtyping of Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2022, 46, 1490–1499. [Google Scholar] [CrossRef]
- Zettl, A.; Rüdiger, T.; Konrad, M.-A.; Chott, A.; Simonitsch-Klupp, I.; Sonnen, R.; Müller-Hermelink, H.K.; Ott, G. Genomic Profiling of Peripheral T-Cell Lymphoma, Unspecified, and Anaplastic Large T-Cell Lymphoma Delineates Novel Recurrent Chromosomal Alterations. Am. J. Pathol. 2004, 164, 1837–1848. [Google Scholar] [CrossRef]
- Boi, M.; Rinaldi, A.; Kwee, I.; Bonetti, P.; Todaro, M.; Tabbò, F.; Piva, R.; Rancoita, P.M.V.; Matolcsy, A.; Timar, B.; et al. PRDM1/BLIMP1 Is Commonly Inactivated in Anaplastic Large T-Cell Lymphoma. Blood 2013, 122, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Sait, S.N.J.; Block, A.W.; Ward, P.M.; Kelly, F.L.R.; Cheney, R.T.; Czuczman, M.; Brecher, M.L.; Barcos, M. Extra Copies of Chromosome 2 Are a Recurring Aberration in ALK-Negative Lymphomas with Anaplastic Morphology. Mod. Pathol. 2005, 18, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Dasari, S.; Oishi, N.; Pedersen, M.B.; Hu, G.; Rech, K.L.; Ketterling, R.P.; Sidhu, J.; Wang, X.; Katoh, R.; et al. Molecular Profiling Reveals Immunogenic Cues in Anaplastic Large Cell Lymphomas with DUSP22 Rearrangements. Blood 2018, 132, 1386–1398. [Google Scholar] [CrossRef]
- Feldman, A.L.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Özsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of Recurrent t(6;7)(P25.3;Q32.3) Translocations in ALK-Negative Anaplastic Large Cell Lymphomas by Massively Parallel Genomic Sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef]
- Onaindia, A.; de Villambrosía, S.G.; Prieto-Torres, L.; Rodríguez-Pinilla, S.M.; Montes-Moreno, S.; González-Vela, C.; Piris, M.A. DUSP22-Rearranged Anaplastic Lymphomas Are Characterized by Specific Morphological Features and a Lack of Cytotoxic and JAK/STAT Surrogate Markers. Haematologica 2019, 104, e158–e162. [Google Scholar] [CrossRef]
- King, R.L.; Dao, L.N.; McPhail, E.D.; Jaffe, E.S.; Said, J.; Swerdlow, S.H.; Sattler, C.A.; Ketterling, R.P.; Sidhu, J.S.; Hsi, E.D.; et al. Morphologic Features of ALK-Negative Anaplastic Large Cell Lymphomas with DUSP22 Rearrangements. Am. J. Surg. Pathol. 2016, 40, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, A.; Feldman, A.L.; Ketterling, R.P.; Dasari, S.; Rech, K.L.; McPhail, E.D.; Kurtin, P.J.; Shi, M. Striking Association of Lymphoid Enhancing Factor (LEF1) Overexpression and DUSP22 Rearrangements in Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2021, 45, 550–557. [Google Scholar] [CrossRef]
- Díaz de la Pinta, F.J.; Rodríguez Moreno, M.; Salgado, R.N.; Carvajal García, N.; Santonja, C.; Pérez Buira, S.; Piris, M.A.; Requena, L.; Manso, R.; Rodríguez-Pinilla, S.M. Anaplastic Large Cell Lymphomas with the 6p25.3 Rearrangement Are a Heterogeneous Group of Tumours with a Diverse Molecular Background. Hum. Pathol. 2023, 137, 71–78. [Google Scholar] [CrossRef]
- Fadl, A.; Oishi, N.; Shi, M.; Dasari, S.; Ansell, S.M.; Ketterling, R.P.; Feldman, A.L. Anaplastic Large Cell Lymphomas with Equivocal DUSP22 FISH Results: Recommendations for Clinical Reporting and Diagnostic Evaluation. Hum. Pathol. 2023, 141, 6–14. [Google Scholar] [CrossRef]
- Wang, X.; Boddicker, R.L.; Dasari, S.; Sidhu, J.S.; Kadin, M.E.; Macon, W.R.; Ansell, S.M.; Ketterling, R.P.; Rech, K.L.; Feldman, A.L. Expression of P63 Protein in Anaplastic Large Cell Lymphoma: Implications for Genetic Subtyping. Hum. Pathol. 2017, 64, 19–27. [Google Scholar] [CrossRef]
- Vasmatzis, G.; Johnson, S.H.; Knudson, R.A.; Ketterling, R.P.; Braggio, E.; Fonseca, R.; Viswanatha, D.S.; Law, M.E.; Kip, N.S.; Ozsan, N.; et al. Genome-Wide Analysis Reveals Recurrent Structural Abnormalities of TP63 and Other P53-Related Genes in Peripheral T-Cell Lymphomas. Blood 2012, 120, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Woodstock, D.L.; Sammons, M.A.; Fischer, M. P63 and P53: Collaborative Partners or Dueling Rivals? Front. Cell Dev. Biol. 2021, 9, 701986. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xiong, Q.; Han, J.; Zhu, Q. The Dual Role of P63 in Cancer. Front. Oncol. 2023, 13, 1116061. [Google Scholar] [CrossRef]
- Wu, G.; Yoshida, N.; Liu, J.; Zhang, X.; Xiong, Y.; Heavican-Foral, T.B.; Mandato, E.; Liu, H.; Nelson, G.M.; Yang, L.; et al. TP63 Fusions Drive Multicomplex Enhancer Rewiring, Lymphomagenesis, and EZH2 Dependence. Sci. Transl. Med. 2023, 15, eadi7244. [Google Scholar] [CrossRef] [PubMed]
- Hapgood, G.; Ben-Neriah, S.; Mottok, A.; Lee, D.G.; Robert, K.; Villa, D.; Sehn, L.H.; Connors, J.M.; Gascoyne, R.D.; Feldman, A.L.; et al. Identification of High-Risk DUSP22-Rearranged ALK-Negative Anaplastic Large Cell Lymphoma. Br. J. Haematol. 2019, 186, e28–e31. [Google Scholar] [CrossRef]
- Wang, J.-C.; Zhong, L.-H.; Lin, W.-Q.; Zhang, W.-F.; Xi, Y.-F.; Liu, Y.-P.; Zhu, Q.; Liu, W.; Zhu, W.-F.; Chen, Y.-P.; et al. JAK/STAT3 Signaling Activation Related to Distinct Clinicopathologic Features in Systemic ALK-Anaplastic Large Cell Lymphomas: New Insights into Their Heterogeneity. Am. J. Surg. Pathol. 2023, 47, 55–64. [Google Scholar] [CrossRef]
- Karube, K.; Feldman, A.L. “Double-Hit” of DUSP22 and TP63 Rearrangements in Anaplastic Large Cell Lymphoma, ALK-Negative. Blood 2020, 135, 700. [Google Scholar] [CrossRef] [PubMed]
- Klairmont, M.M.; Ward, N. Co-Occurring Rearrangements of DUSP22 and TP63 Define a Rare Genetic Subset of ALK-Negative Anaplastic Large Cell Lymphoma with Inferior Survival Outcomes. Leuk. Lymphoma 2022, 63, 506–508. [Google Scholar] [CrossRef]
- Fitzpatrick, M.J.; Massoth, L.R.; Marcus, C.; Vergilio, J.-A.; Severson, E.; Duncan, D.; Ramkissoon, S.H.; Hasserjian, R.P.; Kim, A.S.; Sohani, A.R.; et al. JAK2 Rearrangements Are a Recurrent Alteration in CD30+ Systemic T-Cell Lymphomas with Anaplastic Morphology. Am. J. Surg. Pathol. 2021, 45, 895–904. [Google Scholar] [CrossRef]
- Scarfò, I.; Pellegrino, E.; Mereu, E.; Kwee, I.; Agnelli, L.; Bergaggio, E.; Garaffo, G.; Vitale, N.; Caputo, M.; Machiorlatti, R.; et al. Identification of a New Subclass of ALK-Negative ALCL Expressing Aberrant Levels of ERBB4 Transcripts. Blood 2016, 127, 221–232. [Google Scholar] [CrossRef]
- Hu, G.; Dasari, S.; Asmann, Y.; Greipp, P.; Knudson, R.; Benson, H.; Li, Y.; Eckloff, B.; Jen, J.; Link, B.; et al. Targetable Fusions of the FRK Tyrosine Kinase in ALK-Negative Anaplastic Large Cell Lymphoma. Leukemia 2018, 32, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Khanlari, M.; Tang, G.; Hao, S.; Gong, Y.; Li, S.; Miranda, R.N.; Lin, P.; Iyer, S.; Yin, C.C.; Xie, W.; et al. Anaplastic Lymphoma Kinase (ALK)-Negative Anaplastic Large Cell Lymphoma with MYC Rearrangement. Br. J. Haematol. 2021, 192, e17–e21. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo’, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent Mutations and Kinase Fusions Lead to Oncogenic STAT3 Activation in Anaplastic Large Cell Lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.V.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic Mutations Activating STAT3 in Human Inflammatory Hepatocellular Adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Rajala, H.; Gómez-Seguí, I.; Olson, T.; McGraw, K.; Przychodzen, B.; Kulasekararaj, A.; Afable, M.; et al. STAT3 Mutations Indicate the Presence of Subclinical T-Cell Clones in a Subset of Aplastic Anemia and Myelodysplastic Syndrome Patients. Blood 2013, 122, 2453–2459. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Petrus, M.N.; Xiao, W.; Nicolae, A.; Raffeld, M.; Pittaluga, S.; Bamford, R.N.; Nakagawa, M.; Ouyang, S.T.; et al. Cytokine Receptor Signaling Is Required for the Survival of ALK- Anaplastic Large Cell Lymphoma, Even in the Presence of JAK1/STAT3 Mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 3975–3980. [Google Scholar] [CrossRef]
- Fragliasso, V.; Verma, A.; Bareja, R.; Heavican, T.; Iqbal, J.; Chan, W.C.; Merli, F.; Ciarrocchi, A.; Elemento, O.; Inghirami, G. Novel Long Non Coding RNA Blackmamba Is Associated to ALK- Anaplastic Large Cell Lymphoma. Blood 2016, 128, 461. [Google Scholar] [CrossRef]
- Mularoni, V.; Donati, B.; Tameni, A.; Manicardi, V.; Reggiani, F.; Sauta, E.; Zanelli, M.; Tigano, M.; Vitale, E.; Torricelli, F.; et al. Long Non-Coding RNA Mitophagy and ALK-Negative Anaplastic Lymphoma-Associated Transcript: A Novel Regulator of Mitophagy in T-Cell Lymphoma. Haematologica 2023, 108, 3333–3346. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-S.; Chung, I.-H.; Lin, Y.-H.; Lin, T.-K.; Chen, W.-J.; Lin, K.-H. The Long Non-Coding RNA MIR503HG Enhances Proliferation of Human ALK-Negative Anaplastic Large-Cell Lymphoma. Int. J. Mol. Sci. 2018, 19, 1463. [Google Scholar] [CrossRef]
- Luchtel, R.A.; Zimmermann, M.T.; Hu, G.; Dasari, S.; Jiang, M.; Oishi, N.; Jacobs, H.K.; Zeng, Y.; Hundal, T.; Rech, K.L.; et al. Recurrent MSC E116K Mutations in ALK-Negative Anaplastic Large Cell Lymphoma. Blood 2019, 133, 2776–2789. [Google Scholar] [CrossRef]
- Rassidakis, G.Z.; Thomaides, A.; Wang, S.; Jiang, Y.; Fourtouna, A.; Lai, R.; Medeiros, L.J. P53 Gene Mutations Are Uncommon but P53 Is Commonly Expressed in Anaplastic Large-Cell Lymphoma. Leukemia 2005, 19, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Cannella, S.; Santoro, A.; Bruno, G.; Pillon, M.; Mussolin, L.; Mangili, G.; Rosolen, A.; Aricò, M. Germline Mutations of the Perforin Gene Are a Frequent Occurrence in Childhood Anaplastic Large Cell Lymphoma. Cancer 2007, 109, 2566–2571. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Higashi, M.; Kawano, R.; Momose, S.; Tokuhira, M.; Kizaki, M.; Tamaru, J. Anaplastic Large Cell Lymphoma with TP63 Rearrangement: A Dismal Prognosis. Pathol. Int. 2019, 69, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.B.; Relander, T.; Lauritzsen, G.F.; Ellin, F.; Leppä, S.; Mannisto, S.; Jantunen, E.; Ketterling, R.P.; Bedroske, P.; Luoma, I.; et al. The Impact of Upfront Autologous Transplant on the Survival of Adult Patients with ALCL and PTCL-NOS According to Their ALK, DUSP22 and TP63 Gene Rearrangement Status-a Joined Nordic Lymphoma Group and Mayo Clinic Analysis. Blood 2017, 130, 822. [Google Scholar] [CrossRef]
- d’Amore, F.; Relander, T.; Lauritzsen, G.F.; Jantunen, E.; Hagberg, H.; Anderson, H.; Holte, H.; Österborg, A.; Merup, M.; Brown, P.; et al. Up-Front Autologous Stem-Cell Transplantation in Peripheral T-Cell Lymphoma: NLG-T-01. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 3093–3099. [Google Scholar] [CrossRef]
- Pedersen, M.B.; Hamilton-Dutoit, S.J.; Bendix, K.; Ketterling, R.P.; Bedroske, P.P.; Luoma, I.M.; Sattler, C.A.; Boddicker, R.L.; Bennani, N.N.; Nørgaard, P.; et al. DUSP22 and TP63 Rearrangements Predict Outcome of ALK-Negative Anaplastic Large Cell Lymphoma: A Danish Cohort Study. Blood 2017, 130, 554–557. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Ansell, S.M.; Ai, W.Z.; Barnes, J.; Barta, S.K.; Choi, M.; Clemens, M.W.; Dogan, A.; Greer, J.P.; Halwani, A.; et al. NCCN Guidelines Insights: T-Cell Lymphomas, Version 2.2018. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 123–135. [Google Scholar] [CrossRef]
- Qiu, L.; Tang, G.; Li, S.; Vega, F.; Lin, P.; Wang, S.A.; Wang, W.; Iyer, S.P.; Malpica, L.; Miranda, R.N.; et al. DUSP22 Rearrangement Is Associated with a Distinctive Immunophenotype but Not Outcome in Patients with Systemic ALK-Negative Anaplastic Large Cell Lymphoma. Haematologica 2022, 108, 1604–1615. [Google Scholar] [CrossRef]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Gene Expression Signatures Delineate Biological and Prognostic Subgroups in Peripheral T-Cell Lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef]
- Agnelli, L.; Mereu, E.; Pellegrino, E.; Limongi, T.; Kwee, I.; Bergaggio, E.; Ponzoni, M.; Zamò, A.; Iqbal, J.; Piccaluga, P.P.; et al. Identification of a 3-Gene Model as a Powerful Diagnostic Tool for the Recognition of ALK-Negative Anaplastic Large-Cell Lymphoma. Blood 2012, 120, 1274–1281. [Google Scholar] [CrossRef]
- Lamant, L.; de Reyniès, A.; Duplantier, M.-M.; Rickman, D.S.; Sabourdy, F.; Giuriato, S.; Brugières, L.; Gaulard, P.; Espinos, E.; Delsol, G. Gene-Expression Profiling of Systemic Anaplastic Large-Cell Lymphoma Reveals Differences Based on ALK Status and Two Distinct Morphologic ALK+ Subtypes. Blood 2007, 109, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Wu, W.; Fan, M.; Wang, Z.; Feng, X.; Liu, C.; Liu, J.; Liu, G.; Xia, L.; Si, H.; et al. Phosphorylated STAT3 as a Potential Diagnostic and Predictive Biomarker in ALK- ALCL vs. CD30high PTCL, NOS. Front. Immunol. 2023, 14, 1132834. [Google Scholar] [CrossRef]
- Shauly, O.; Gould, D.J.; Siddiqi, I.; Patel, K.M.; Carey, J. The First Reported Case of Gluteal Implant-Associated Anaplastic Large Cell Lymphoma (ALCL). Aesthet. Surg. J. 2019, 39, NP253–NP258. [Google Scholar] [CrossRef]
- Kolasiński, J.; Sorotos, M.; Firmani, G.; Panagiotakos, D.; Płonka, J.; Kolenda, M.; Santanelli di Pompeo, F. BIA-ALCL Epidemiology in an Aesthetic Breast Surgery Cohort of 1501 Patients. Aesthet. Surg. J. 2023, 43, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Santanelli Di Pompeo, F.; Panagiotakos, D.; Firmani, G.; Sorotos, M. BIA-ALCL Epidemiological Findings From a Retrospective Study of 248 Cases Extracted from Relevant Case Reports and Series: A Systematic Review. Aesthet. Surg. J. 2023, 43, 545–555. [Google Scholar] [CrossRef]
- Brody, G.S.; Deapen, D.; Taylor, C.R.; Pinter-Brown, L.; House-Lightner, S.R.; Andersen, J.S.; Carlson, G.; Lechner, M.G.; Epstein, A.L. Anaplastic Large Cell Lymphoma Occurring in Women with Breast Implants: Analysis of 173 Cases. Plast. Reconstr. Surg. 2015, 135, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Delas, A.; Gaulard, P.; Haioun, C.; Moreau, A.; Xerri, L.; Traverse-Glehen, A.; Rousset, T.; Quintin-Roue, I.; Petrella, T.; et al. Breast Implant-Associated Anaplastic Large Cell Lymphoma: Two Distinct Clinicopathological Variants with Different Outcomes. Ann. Oncol. 2016, 27, 306–314. [Google Scholar] [CrossRef]
- Ionescu, P.; Vibert, F.; Amé, S.; Mathelin, C. New Data on the Epidemiology of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Eur. J. Breast Health 2021, 17, 302–307. [Google Scholar] [CrossRef]
- Ebner, P.J.; Liu, A.; Gould, D.J.; Patel, K.M. Breast Implant-Associated Anaplastic Large Cell Lymphoma, a Systematic Review and in-Depth Evaluation of the Current Understanding. J. Surg. Oncol. 2019, 120, 573–577. [Google Scholar] [CrossRef]
- Kricheldorff, J.; Fallenberg, E.M.; Solbach, C.; Gerber-Schäfer, C.; Rancsó, C.; von Fritschen, U. Breast Implant-Associated Lymphoma. Dtsch. Arzteblatt Int. 2018, 115, 628–635. [Google Scholar] [CrossRef]
- Nelson, J.A.; Dabic, S.; Mehrara, B.J.; Cordeiro, P.G.; Disa, J.J.; Pusic, A.L.; Matros, E.; Dayan, J.H.; Allen, R.J.; Coriddi, M.; et al. Breast Implant-Associated Anaplastic Large Cell Lymphoma Incidence: Determining an Accurate Risk. Ann. Surg. 2020, 272, 403–409. [Google Scholar] [CrossRef]
- Quesada, A.E.; Medeiros, L.J.; Clemens, M.W.; Ferrufino-Schmidt, M.C.; Pina-Oviedo, S.; Miranda, R.N. Breast Implant-Associated Anaplastic Large Cell Lymphoma: A Review. Mod. Pathol. 2019, 32, 166–188. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, A.A.; Wirtz, E.C.; Ollila, D.W.; Bhatt, N. An Unusual Case of BIA-ALCL Associated with Prolonged/Complicated Biocell-Textured Expander, Followed by Smooth Round Breast Implant Exposure, and Concurrent Use of Adalimumab. Plast. Reconstr. Surg. 2021, 148, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, A.; Pepe, G.; Giarnieri, E.; Cippitelli, C.; Bonifacino, A.; Mattei, M.; Martelli, M.; Falasca, C.; Cox, M.C.; Santino, I.; et al. Cytological Diagnostic Features of Late Breast Implant Seromas: From Reactive to Anaplastic Large Cell Lymphoma. PLoS ONE 2017, 12, e0181097. [Google Scholar] [CrossRef]
- Tardío, J.C.; Granados, R. Axillary Lymphadenopathy: An Outstanding Presentation for Breast Implant-Associated ALK-Negative Anaplastic Large Cell Lymphoma. Int. J. Surg. Pathol. 2015, 23, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Ferrufino-Schmidt, M.C.; Medeiros, L.J.; Liu, H.; Clemens, M.W.; Hunt, K.K.; Laurent, C.; Lofts, J.; Amin, M.B.; Ming Chai, S.; Morine, A.; et al. Clinicopathologic Features and Prognostic Impact of Lymph Node Involvement in Patients With Breast Implant-Associated Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2018, 42, 293–305. [Google Scholar] [CrossRef]
- Longo, B.; Di Napoli, A.; Curigliano, G.; Veronesi, P.; Pileri, S.; Martelli, M.; De Vita, R.; Felici, N.; Cirillo, P.; Bernardi, C.; et al. Clinical Recommendations for Diagnosis and Treatment According to Current Updated Knowledge on BIA-ALCL. Breast Edinb. Scotl. 2022, 66, 332–341. [Google Scholar] [CrossRef]
- Ghione, P.; Cordeiro, P.G. Current Updates on Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Rev. Senol. Patol. Mamar. 2019, 32, 79–80. [Google Scholar] [CrossRef]
- Premji, S.; Barbieri, A.; Roth, C.; Rohren, E.M.; Rivero, G.; Teegavarapu, S.P. An Unusual Case of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Case Rep. Hematol. 2022, 2022, 4700787. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Ashar, B.S.; Clemens, M.W.; Feldman, A.L.; Gaulard, P.; Miranda, R.N.; Sohani, A.R.; Stenzel, T.; Yoon, S.W. Best Practices Guideline for the Pathologic Diagnosis of Breast Implant–Associated Anaplastic Large-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 1102–1111. [Google Scholar] [CrossRef]
- Barbé, E.; de Boer, M.; de Jong, D. A Practical Cytological Approach to the Diagnosis of Breast-Implant Associated Anaplastic Large Cell Lymphoma. Cytopathol. Off. J. Br. Soc. Clin. Cytol. 2019, 30, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.E.; Zhang, Y.; Ptashkin, R.; Ho, C.; Horwitz, S.; Benayed, R.; Dogan, A.; Arcila, M.E. Next Generation Sequencing of Breast Implant-Associated Anaplastic Large Cell Lymphomas Reveals a Novel STAT3-JAK2 Fusion among Other Activating Genetic Alterations within the JAK-STAT Pathway. Breast J. 2021, 27, 314–321. [Google Scholar] [CrossRef]
- De Azambuja, A.P.; Gevert, F.; Oliveira, R.M.; Sebastião, A.P.; Groth, A.K. Use of Flow Cytometry and Cytology to Differentiate Breast Implant-Associated Anaplastic Large Cell Lymphoma from Reactive Seromas in Brazilian Patients. Cytometry B Clin. Cytom. 2022, 102, 312–316. [Google Scholar] [CrossRef]
- Ezekwudo, D.E.; Ifabiyi, T.; Gbadamosi, B.; Haberichter, K.; Yu, Z.; Amin, M.; Shaheen, K.; Stender, M.; Jaiyesimi, I. Breast Implant–Associated Anaplastic Large Cell Lymphoma: A Case Report and Review of the Literature. Case Rep. Oncol. Med. 2017, 2017, 6478467. [Google Scholar] [CrossRef] [PubMed]
- de Leval, L. Approach to Nodal-Based T-Cell Lymphomas. Pathology 2020, 52, 78–99. [Google Scholar] [CrossRef]
- Dashevsky, B.Z.; Gallagher, K.M.; Grabenstetter, A.; Cordeiro, P.G.; Dogan, A.; Morris, E.A.; Horwitz, S.M.; Sutton, E.J. Breast Implant-Associated Anaplastic Large Cell Lymphoma: Clinical and Imaging Findings at a Large US Cancer Center. Breast J. 2019, 25, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kadin, M.E.; Morgan, J.; Kouttab, N.; Xu, H.; Adams, W.P.; Glicksman, C.; McGuire, P.; Sieber, D.; Epstein, A.L.; Miranda, R.N.; et al. Comparative Analysis of Cytokines of Tumor Cell Lines, Malignant and Benign Effusions Around Breast Implants. Aesthet. Surg. J. 2020, 40, 630–637. [Google Scholar] [CrossRef]
- Di Napoli, A.; Greco, D.; Scafetta, G.; Ascenzi, F.; Gulino, A.; Aurisicchio, L.; Santanelli Di Pompeo, F.; Bonifacino, A.; Giarnieri, E.; Morgan, J.; et al. IL-10, IL-13, Eotaxin and IL-10/IL-6 Ratio Distinguish Breast Implant-Associated Anaplastic Large-Cell Lymphoma from All Types of Benign Late Seromas. Cancer Immunol. Immunother. 2021, 70, 1379–1392. [Google Scholar] [CrossRef]
- Hu, H.; Shklovskaya, E.; Deva, A.; Xu, H.; Fan, K.; Brosamer, K.; Willson, R.; Khan, I.; Sinha, M.; Kadin, M. Diagnosis of Breast Implant Associated Anaplastic Large Cell Lymphoma by Analysis of Cytokines in Peri-implant Seromas. Am. J. Hematol. 2023, 98, E312–E314. [Google Scholar] [CrossRef]
- Kadin, M.E. What Cytokines Can Tell Us About the Pathogenesis of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Aesthet. Surg. J. 2019, 39, S28–S35. [Google Scholar] [CrossRef]
- Alessandri-Bonetti, M.; Jeong, T.; Vaienti, L.; De La Cruz, C.; Gimbel, M.L.; Nguyen, V.T.; Egro, F.M. The Role of Microorganisms in the Development of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Pathogens 2023, 12, 313. [Google Scholar] [CrossRef]
- Kadin, M.E.; Deva, A.; Xu, H.; Morgan, J.; Khare, P.; MacLeod, R.A.F.; Van Natta, B.W.; Adams, W.P.; Brody, G.S.; Epstein, A.L. Biomarkers Provide Clues to Early Events in the Pathogenesis of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Aesthet. Surg. J. 2016, 36, 773–781. [Google Scholar] [CrossRef]
- Oishi, N.; Hundal, T.; Phillips, J.L.; Dasari, S.; Hu, G.; Viswanatha, D.S.; He, R.; Mai, M.; Jacobs, H.K.; Ahmed, N.H.; et al. Molecular Profiling Reveals a Hypoxia Signature in Breast Implant-Associated Anaplastic Large Cell Lymphoma. Haematologica 2021, 106, 1714–1724. [Google Scholar] [CrossRef]
- Carbonaro, R.; Accardo, G.; Mazzocconi, L.; Pileri, S.; Derenzini, E.; Veronesi, P.; Caldarella, P.; De Lorenzi, F. BIA-ALCL in Patients with Genetic Predisposition for Breast Cancer: Our Experience and a Review of the Literature. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. ECP 2023, 32, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Nicolae, A.; Laurent, C.; Le Bras, F.; Haioun, C.; Fataccioli, V.; Amara, N.; Adélaïde, J.; Guille, A.; Schiano, J.-M.; et al. Gene Alterations in Epigenetic Modifiers and JAK-STAT Signaling Are Frequent in Breast Implant–Associated ALCL. Blood 2020, 135, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Thompson, E.; Ryland, G.L.; Joyce, R.; Byrne, D.J.; Khoo, C.; Lade, S.; Hertzberg, M.; Hapgood, G.; Marlton, P.; et al. Frequent Activating STAT3 Mutations and Novel Recurrent Genomic Abnormalities Detected in Breast Implant-Associated Anaplastic Large Cell Lymphoma. Oncotarget 2018, 9, 36126–36136. [Google Scholar] [CrossRef] [PubMed]
- Harrop, S.; Mehta-Shah, N.; Dsouza, C.; Thompson, E.; Deva, A.; Prince, H.M. An Update on the Current Genomic Landscape of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Cancers 2021, 13, 4921. [Google Scholar] [CrossRef]
- Le Bras, F.; Gaulard, P.; Andre, M.; Haioun, C.; Bosc, R.; Laurent, C.; Tortelano, L.; Dao, T.-H.; Itti, E.; Malhaire, C.; et al. Breast Implant Associated-Anaplastic Large Cell Lymphoma (BIA-ALCL): The Lymphoma Study Association (LYSA) Registry Data. Blood 2019, 134, 4021. [Google Scholar] [CrossRef]
- Deva, A.K.; Turner, S.D.; Kadin, M.E.; Magnusson, M.R.; Prince, H.M.; Miranda, R.N.; Inghirami, G.G.; Adams, W.P. Etiology of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL): Current Directions in Research. Cancers 2020, 12, 3861. [Google Scholar] [CrossRef]
- Tabanelli, V.; Corsini, C.; Fiori, S.; Agostinelli, C.; Calleri, A.; Orecchioni, S.; Melle, F.; Motta, G.; Rotili, A.; Napoli, A.; et al. Recurrent PDL1 Expression and PDL1 (CD274) Copy Number Alterations in Breast Implant-Associated Anaplastic Large-Cell Lymphomas. Hum. Pathol. 2019, 90, 60–69. [Google Scholar] [CrossRef]
- Los-de Vries, G.T.; de Boer, M.; van Dijk, E.; Stathi, P.; Hijmering, N.J.; Roemer, M.G.M.; Mendeville, M.; Miedema, D.M.; de Boer, J.P.; Rakhorst, H.A.; et al. Chromosome 20 Loss Is Characteristic of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Blood 2020, 136, 2927–2932. [Google Scholar] [CrossRef]
- Kadin, M.E.; Morgan, J.; Wei, W.; Song, Z.; Yang, Y. CD30 Regulation of IL-13-STAT6 Pathway in Breast Implant-Associated Anaplastic Large Cell Lymphoma. Aesthet. Surg. J. 2023, 43, 137–146. [Google Scholar] [CrossRef]
- Miranda, R.N.; Aladily, T.N.; Prince, H.M.; Kanagal-Shamanna, R.; de Jong, D.; Fayad, L.E.; Amin, M.B.; Haideri, N.; Bhagat, G.; Brooks, G.S.; et al. Breast Implant-Associated Anaplastic Large-Cell Lymphoma: Long-Term Follow-up of 60 Patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Barrington, S.F.; Mikhaeel, N.G.; Kostakoglu, L.; Meignan, M.; Hutchings, M.; Müeller, S.P.; Schwartz, L.H.; Zucca, E.; Fisher, R.I.; Trotman, J.; et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 3048–3058. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A. Recommendations for Initial Evaluation, Staging, and Response Assessment of Hodgkin and Non-Hodgkin Lymphoma: The Lugano Classification. J. Clin. Oncol. 2014, 32, 3059–3067. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.W.; Jacobsen, E.D.; Horwitz, S.M. 2019 NCCN Consensus Guidelines on the Diagnosis and Treatment of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Aesthet. Surg. J. 2019, 39, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- St Cyr, T.L.; Pockaj, B.A.; Northfelt, D.W.; Craig, F.E.; Clemens, M.W.; Mahabir, R.C. Breast Implant-Associated Anaplastic Large-Cell Lymphoma: Current Understanding and Recommendations for Management. Plast. Surg. Oakv. Ont 2020, 28, 117–126. [Google Scholar] [CrossRef]
- Clemens, M.W.; Nava, M.B.; Rocco, N.; Miranda, R.N. Understanding Rare Adverse Sequelae of Breast Implants: Anaplastic Large-Cell Lymphoma, Late Seromas, and Double Capsules. Gland Surg. 2017, 6, 169–184. [Google Scholar] [CrossRef]
- Clemens, M.W.; Medeiros, L.J.; Butler, C.E.; Hunt, K.K.; Fanale, M.A.; Horwitz, S.; Weisenburger, D.D.; Liu, J.; Morgan, E.A.; Kanagal-Shamanna, R.; et al. Complete Surgical Excision Is Essential for the Management of Patients with Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 160–168. [Google Scholar] [CrossRef]
- McKernan, C. Treating Breast Implant-Associated Anaplastic Large Cell Lymphoma. JAAPA Off. J. Am. Acad. Physician Assist. 2021, 34, 47–51. [Google Scholar] [CrossRef]
- Vets, J.; Marcelis, L.; Schepers, C.; Dorreman, Y.; Verbeek, S.; Vanwalleghem, L.; Gieraerts, K.; Meylaerts, L.; Lesaffer, J.; Devos, H.; et al. Breast Implant Associated EBV-Positive Diffuse Large B-Cell Lymphoma: An Underrecognized Entity? Diagn. Pathol. 2023, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Mescam, L.; Camus, V.; Schiano, J.-M.; Adélaïde, J.; Picquenot, J.-M.; Guille, A.; Bannier, M.; Ruminy, P.; Viailly, P.-J.; Jardin, F.; et al. EBV+ Diffuse Large B-Cell Lymphoma Associated with Chronic Inflammation Expands the Spectrum of Breast Implant-Related Lymphomas. Blood 2020, 135, 2004–2009. [Google Scholar] [CrossRef]
- Malata, C.M.; Madada-Nyakauru, R.N.; Follows, G.; Wright, P. Epstein-Barr Virus-Associated Diffuse Large B-Cell Lymphoma Identified in a Breast Implant Capsule: A New Breast Implant-Associated Lymphoma? Ann. Plast. Surg. 2021, 86, 383–386. [Google Scholar] [CrossRef] [PubMed]
- D’Orsi, G.; Giacalone, M.; Calicchia, A.; Gagliano, E.; Vannucchi, L.; Vanni, G.; Buonomo, O.C.; Cervelli, V.; Longo, B. BIA-ALCL and BIA-SCC: Updates on Clinical Features and Genetic Mutations for Latest Recommendations. Medicina 2024, 60, 793. [Google Scholar] [CrossRef]
- Thursday, S. 8 ASPS Statement on Breast Implant Associated-Squamous Cell Carcinoma (BIA-SCC). Available online: https://www.plasticsurgery.org/for-medical-professionals/publications/psn-extra/news/asps-statement-on-breast-implant-associated-squamous-cell-carcinoma (accessed on 12 March 2025).
- Niraula, S.; Katel, A.; Barua, A.; Weiss, A.; Strawderman, M.S.; Zhang, H.; Manrique, O.; O’Connell, A.; Pandey, S.R.; Dhakal, A. A Systematic Review of Breast Implant-Associated Squamous Cell Carcinoma. Cancers 2023, 15, 4516. [Google Scholar] [CrossRef]
- Kempf, W. A New Era for Cutaneous CD30-Positive T-Cell Lymphoproliferative Disorders. Semin. Diagn. Pathol. 2017, 34, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Benner, M.F.; Willemze, R. Applicability and Prognostic Value of the New TNM Classification System in 135 Patients with Primary Cutaneous Anaplastic Large Cell Lymphoma. Arch. Dermatol. 2009, 145, 1399–1404. [Google Scholar] [CrossRef]
- Sciallis, A.P.; Law, M.E.; Inwards, D.J.; McClure, R.F.; Macon, W.R.; Kurtin, P.J.; Dogan, A.; Feldman, A.L. Mucosal CD30-Positive T-Cell Lymphoproliferations of the Head and Neck Show a Clinicopathologic Spectrum Similar to Cutaneous CD30-Positive T-Cell Lymphoproliferative Disorders. Mod. Pathol. 2012, 25, 983–992. [Google Scholar] [CrossRef]
- Kempf, W.; Mitteldorf, C.; Karai, L.J.; Robson, A. Lymphomatoid Papulosis-Making Sense of the Alphabet Soup: A Proposal to Simplify Terminology. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. JDDG 2017, 15, 390–394. [Google Scholar] [CrossRef]
- Saleh, J.S.; Subtil, A.; Hristov, A.C. Primary Cutaneous T-Cell Lymphoma: A Review of the Most Common Entities with Focus on Recent Updates. Hum. Pathol. 2023, 138, 75–100. [Google Scholar] [CrossRef]
- Rodríguez-Pinilla, S.M.; Ortiz-Romero, P.L.; Monsalvez, V.; Tomás, I.E.; Almagro, M.; Sevilla, A.; Camacho, G.; Longo, M.I.; Pulpillo, Á.; Diaz-Pérez, J.A.; et al. TCR-γ Expression in Primary Cutaneous T-Cell Lymphomas. Am. J. Surg. Pathol. 2013, 37, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Meawad, H.; Song, J.Y.; Ulrickson, M.L.; Weisenburger, D.D. Primary Cutaneous Gamma-Delta T-Cell Lymphoma Mimicking Anaplastic Lymphoma Kinase-1-Negative Anaplastic Large Cell Lymphoma: A Case Report. Am. J. Dermatopathol. 2022, 44, 62–65. [Google Scholar] [CrossRef]
- Tran, T.A. Does a Subset of Localized Chronic Fibrosing Vasculitis Represent Cutaneous Manifestation of IgG4-Related Disease/a Histologic Pattern of IgG4-Related Skin Disease? A Reappraisal of an Enigmatic Pathologic Entity. Am. J. Dermatopathol. 2020, 42, 683–688. [Google Scholar] [CrossRef]
- Amador, C.; Feldman, A.L. How I Diagnose Anaplastic Large Cell Lymphoma. Am. J. Clin. Pathol. 2021, 155, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, M.; Biskup, E.; Gniadecki, R. Notch Signalling in Primary Cutaneous CD30+ Lymphoproliferative Disorders: A New Therapeutic Approach? Br. J. Dermatol. 2010, 163, 781–788. [Google Scholar] [CrossRef]
- Kong, Y.-Y.; Dai, B.; Kong, J.-C.; Lu, H.-F.; Shi, D.-R. Neutrophil/Eosinophil-Rich Type of Primary Cutaneous Anaplastic Large Cell Lymphoma: A Clinicopathological, Immunophenotypic and Molecular Study of Nine Cases. Histopathology 2009, 55, 189–196. [Google Scholar] [CrossRef]
- Kempf, W.; Kazakov, D.V.; Schärer, L.; Rütten, A.; Mentzel, T.; Paredes, B.E.; Palmedo, G.; Panizzon, R.G.; Kutzner, H. Angioinvasive Lymphomatoid Papulosis: A New Variant Simulating Aggressive Lymphomas. Am. J. Surg. Pathol. 2013, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Hidalgo, C.; Pina-Oviedo, S. Primary Cutaneous Anaplastic Large Cell Lymphoma—A Review of Clinical, Morphological, Immunohistochemical, and Molecular Features. Cancers 2023, 15, 4098. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Cavicchini, S.; Corradin, M.T. Hypopigmented Atypical Spitzoid Neoplasms (Atypical Spitz Nevi, Atypical Spitz Tumors, Spitzoid Melanoma): A Clinicopathological Update. Dermatol. Pract. Concept. 2015, 5, 45–52. [Google Scholar] [CrossRef]
- Samols, M.A.; Su, A.; Ra, S.; Cappel, M.A.; Louissant, A.J.; Knudson, R.A.; Ketterling, R.P.; Said, J.; Binder, S.; Harris, N.L.; et al. Intralymphatic Cutaneous Anaplastic Large Cell Lymphoma/Lymphomatoid Papulosis: Expanding the Spectrum of CD30-Positive Lymphoproliferative Disorders. Am. J. Surg. Pathol. 2014, 38, 1203. [Google Scholar] [CrossRef]
- Vilas Boas, P.; Cerroni, L.; Requena, L. Intravascular Cutaneous Disorders. A Clinicopathologic Review. Am. J. Dermatopathol. 2021, 43, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, M.; Parente, P.; Sanguedolce, F.; Zizzo, M.; Palicelli, A.; Bisagni, A.; Carosi, I.; Trombetta, D.; Mastracci, L.; Ricci, L.; et al. Intravascular NK/T-Cell Lymphoma: What We Know about This Diagnostically Challenging, Aggressive Disease. Cancers 2022, 14, 5458. [Google Scholar] [CrossRef] [PubMed]
- Wada, D.A.; Law, M.E.; Hsi, E.D.; DiCaudo, D.J.; Ma, L.; Lim, M.S.; de Souza, A.; Comfere, N.I.; Weenig, R.H.; Macon, W.R.; et al. Specificity of IRF4 Translocations for Primary Cutaneous Anaplastic Large Cell Lymphoma: A Multicenter Study of 204 Skin Biopsies. Mod. Pathol. 2011, 24, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, F.; Pope, E.; Ngan, B.; Abla, O. Primary Cutaneous Lymphomas in Children and Adolescents. Pediatr. Blood Cancer 2016, 63, 1886–1894. [Google Scholar] [CrossRef]
- Massone, C.; El-Shabrawi-Caelen, L.; Kerl, H.; Cerroni, L. The Morphologic Spectrum of Primary Cutaneous Anaplastic Large T-Cell Lymphoma: A Histopathologic Study on 66 Biopsy Specimens from 47 Patients with Report of Rare Variants. J. Cutan. Pathol. 2008, 35, 46–53. [Google Scholar] [CrossRef]
- Greisser, J.; Palmedo, G.; Sander, C.; Kutzner, H.; Kazakov, D.V.; Roos, M.; Burg, G.; Kempf, W. Detection of Clonal Rearrangement of T-Cell Receptor Genes in the Diagnosis of Primary Cutaneous CD30 Lymphoproliferative Disorders. J. Cutan. Pathol. 2006, 33, 711–715. [Google Scholar] [CrossRef]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A Novel Recurrent NPM1-TYK2 Gene Fusion in Cutaneous CD30-Positive Lymphoproliferative Disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef]
- Melchers, R.C.; Willemze, R.; van de Loo, M.; van Doorn, R.; Jansen, P.M.; Cleven, A.H.G.; Solleveld, N.; Bekkenk, M.W.; van Kester, M.S.; Diercks, G.F.H.; et al. Clinical, Histologic, and Molecular Characteristics of Anaplastic Lymphoma Kinase-Positive Primary Cutaneous Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2020, 44, 776–781. [Google Scholar] [CrossRef]
- Schrader, A.M.R.; Chung, Y.-Y.; Jansen, P.M.; Szuhai, K.; Bastidas Torres, A.N.; Tensen, C.P.; Willemze, R. No TP63 Rearrangements in a Selected Group of Primary Cutaneous CD30+ Lymphoproliferative Disorders with Aggressive Clinical Course. Blood 2016, 128, 141–143. [Google Scholar] [CrossRef]
- Di Napoli, A.; Vacca, D.; Bertolazzi, G.; Lopez, G.; Piane, M.; Germani, A.; Rogges, E.; Pepe, G.; Santanelli Di Pompeo, F.; Salgarello, M.; et al. RNA Sequencing of Primary Cutaneous and Breast-Implant Associated Anaplastic Large Cell Lymphomas Reveals Infrequent Fusion Transcripts and Upregulation of PI3K/AKT Signaling via Neurotrophin Pathway Genes. Cancers 2021, 13, 6174. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S. Genetic Alterations in Primary Cutaneous CD30+ Anaplastic Large Cell Lymphoma. Genes. Chromosomes Cancer 2003, 37, 176–185. [Google Scholar] [CrossRef]
- Abdulla, F.R.; Zhang, W.; Wu, X.; Honda, K.; Qin, H.; Cho, H.; Querfeld, C.; Zain, J.; Rosen, S.T.; Chan, W.C.; et al. Genomic Analysis of Cutaneous CD30-Positive Lymphoproliferative Disorders. JID Innov. Skin Sci. Mol. Popul. Health 2022, 2, 100068. [Google Scholar] [CrossRef] [PubMed]
- Laharanne, E.; Oumouhou, N.; Bonnet, F.; Carlotti, M.; Gentil, C.; Chevret, E.; Jouary, T.; Longy, M.; Vergier, B.; Beylot-Barry, M.; et al. Genome-Wide Analysis of Cutaneous T-Cell Lymphomas Identifies Three Clinically Relevant Classes. J. Invest. Dermatol. 2010, 130, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Nicolae-Cristea, A.R.; Benner, M.F.; Zoutman, W.H.; van Eijk, R.; Jansen, P.M.; Tensen, C.P.; Willemze, R. Diagnostic and Prognostic Significance of CDKN2A/CDKN2B Deletions in Patients with Transformed Mycosis Fungoides and Primary Cutaneous CD30-Positive Lymphoproliferative Disease. Br. J. Dermatol. 2015, 172, 784–788. [Google Scholar] [CrossRef]
- Chen, B.-J.; Hsieh, S.-M.; Hsieh, T.-H.; Jhuang, J.-Y.; Kao, Y.-C. DUSP22-Rearranged Primary Cutaneous CD30-Positive T-Cell Lymphoproliferative Disorders and Adult T-Cell Leukemia/Lymphoma Frequently Share the LEF1+/TIA1- Immunophenotype. Hum. Pathol. 2024, 150, 58–66. [Google Scholar] [CrossRef]
- Gallardo, F.; Pujol, R.M. Genetics Abnormalities with Clinical Impact in Primary Cutaneous Lymphomas. Cancers 2022, 14, 4972. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xu, Y.; Jing, Z.; Wang, X.; Zha, X.; Zeng, C.; Chen, S.; Yang, L.; Luo, G.; Li, B.; et al. Altered Expression Pattern of miR-29a, miR-29b and the Target Genes in Myeloid Leukemia. Exp. Hematol. Oncol. 2014, 3, 17. [Google Scholar] [CrossRef]
- Sandoval, J.; Díaz-Lagares, A.; Salgado, R.; Servitje, O.; Climent, F.; Ortiz-Romero, P.L.; Pérez-Ferriols, A.; Garcia-Muret, M.P.; Estrach, T.; Garcia, M.; et al. MicroRNA Expression Profiling and DNA Methylation Signature for Deregulated microRNA in Cutaneous T-Cell Lymphoma. J. Invest. Dermatol. 2015, 135, 1128–1137. [Google Scholar] [CrossRef]
- Gallardo, F.; Sandoval, J.; Díaz-Lagares, A.; Garcia, R.; D’Altri, T.; González, J.; Alegre, V.; Servitje, O.; Crujeiras, A.-B.; Stefánsson, Ó.-A.; et al. Notch1 Pathway Activation Results from the Epigenetic Abrogation of Notch-Related MicroRNAs in Mycosis Fungoides. J. Investig. Dermatol. 2015, 135, 3144–3152. [Google Scholar] [CrossRef]
- Hara, N.; Sawada, Y. Epigenetics of Cutaneous T-Cell Lymphomas. Int. J. Mol. Sci. 2022, 23, 3538. [Google Scholar] [CrossRef]
- Yi, S.; Sun, J.; Qiu, L.; Fu, W.; Wang, A.; Liu, X.; Yang, Y.; Kadin, M.E.; Tu, P.; Wang, Y. Dual Role of EZH2 in Cutaneous Anaplastic Large Cell Lymphoma: Promoting Tumor Cell Survival and Regulating Tumor Microenvironment. J. Investig. Dermatol. 2018, 138, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Joosten, M.; Seitz, V.; Zimmermann, K.; Sommerfeld, A.; Berg, E.; Lenze, D.; Leser, U.; Stein, H.; Hummel, M. Histone Acetylation and DNA Demethylation of T Cells Result in an Anaplastic Large Cell Lymphoma-like Phenotype. Haematologica 2013, 98, 247–254. [Google Scholar] [CrossRef]
- Kempf, W.; Pfaltz, K.; Vermeer, M.H.; Cozzio, A.; Ortiz-Romero, P.L.; Bagot, M.; Olsen, E.; Kim, Y.H.; Dummer, R.; Pimpinelli, N.; et al. EORTC, ISCL, and USCLC Consensus Recommendations for the Treatment of Primary Cutaneous CD30-Positive Lymphoproliferative Disorders: Lymphomatoid Papulosis and Primary Cutaneous Anaplastic Large-Cell Lymphoma. Blood 2011, 118, 4024–4035. [Google Scholar] [CrossRef]
- Blanchard, M.; Morren, M.-A.; Busschots, A.-M.; Hauben, E.; Alberti-Violetti, S.; Berti, E.; Avallone, G.; Tavoletti, G.; Panzone, M.; Quaglino, P.; et al. Paediatric-Onset Lymphomatoid Papulosis: Results of a Multicentre Retrospective Cohort Study on Behalf of the EORTC Cutaneous Lymphoma Tumours Group (CLTG). Br. J. Dermatol. 2024, 191, 233–242. [Google Scholar] [CrossRef]
- Ferenczi, K.; Makkar, H.S. Cutaneous Lymphoma: Kids Are Not Just Little People. Clin. Dermatol. 2016, 34, 749–759. [Google Scholar] [CrossRef]
- Gleason, L.; Afifi, L.; Banner, L.; Talasila, S.; Joffe, D.; Bhatti, S.; Alpdogan, O.; Porcu, P.; Nikbakht, N. Challenges in Utilizing ALK Expression to Distinguish Primary Cutaneous from Systemic Anaplastic Large Cell Lymphoma. Mol. Clin. Oncol. 2024, 20, 35. [Google Scholar] [CrossRef] [PubMed]
- Swallow, M.A.; Micevic, G.; Zhou, A.; Carlson, K.R.; Foss, F.M.; Girardi, M. Clinical and Histologic Variants of CD8+ Cutaneous T-Cell Lymphomas. Cancers 2024, 16, 3087. [Google Scholar] [CrossRef] [PubMed]
- Melchers, R.C.; Willemze, R.; Bekkenk, M.W.; de Haas, E.R.M.; Horvath, B.; van Rossum, M.M.; Sanders, C.J.G.; Veraart, J.C.J.M.; Putter, H.; Jansen, P.M.; et al. Frequency and Prognosis of Associated Malignancies in 504 Patients with Lymphomatoid Papulosis. J. Eur. Acad. Dermatol. Venereol. JEADV 2020, 34, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cabriales, S.A.; Walsh, S.; Sade, S.; Shear, N.H. Lymphomatoid Papulosis: An Update and Review. J. Eur. Acad. Dermatol. Venereol. JEADV 2020, 34, 59–73. [Google Scholar] [CrossRef]
- Badje, E.D.; Tejasvi, T.; Hristov, A. Γδ Lymphomatoid Papulosis Type D: A Histologic Mimic of Primary Cutaneous Γδ T-Cell Lymphoma. JAAD Case Rep. 2019, 5, 264–266. [Google Scholar] [CrossRef]
- Xing, X.; Feldman, A.L. Anaplastic Large Cell Lymphomas: ALK Positive, ALK Negative, and Primary Cutaneous. Adv. Anat. Pathol. 2015, 22, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Kempf, W.; Kazakov, D.V.; Kerl, K. Cutaneous Lymphomas: An Update. Part 1: T-Cell and Natural Killer/t-Cell Lymphomas and Related Conditions. Am. J. Dermatopathol. 2014, 36, 105–123. [Google Scholar] [CrossRef]
- Tokura, Y.; Sugita, K.; Yagi, H.; Shimauchi, T.; Kabashima, K.; Takigawa, M. Primary Cutaneous Anaplastic Large Cell Lymphoma with Fatal Leukemic Outcome in Association with CLA and CCR4-Negative Conversion. J. Am. Acad. Dermatol. 2007, 57, S92–S96. [Google Scholar] [CrossRef] [PubMed]
- Fauconneau, A.; Pham-Ledard, A.; Cappellen, D.; Frison, E.; Prochazkova-Carlotti, M.; Parrens, M.; Dalle, S.; Joly, P.; Viraben, R.; Franck, F.; et al. Assessment of Diagnostic Criteria between Primary Cutaneous Anaplastic Large-Cell Lymphoma and CD30-Rich Transformed Mycosis Fungoides; a Study of 66 Cases. Br. J. Dermatol. 2015, 172, 1547–1554. [Google Scholar] [CrossRef]
- Collins, K.; Gu, J.; Aung, P.P.; Nagarajan, P.; Curry, J.L.; Huen, A.; Ivan, D.; Prieto, V.G.; Tetzlaff, M.T.; Duvic, M.; et al. Is Immunohistochemical Expression of GATA3 Helpful in the Differential Diagnosis of Transformed Mycosis Fungoides and Primary Cutaneous CD30-Positive T Cell Lymphoproliferative Disorders? Virchows Arch. Int. J. Pathol. 2021, 479, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Mitteldorf, C.; Robson, A.; Tronnier, M.; Pfaltz, M.C.; Kempf, W. Galectin-3 Expression in Primary Cutaneous CD30-Positive Lymphoproliferative Disorders and Transformed Mycosis Fungoides. Dermatology 2015, 231, 164–170. [Google Scholar] [CrossRef]
- Lai, P.; Liu, F.; Liu, X.; Sun, J.; Wang, Y. Differential Molecular Programs of Cutaneous Anaplastic Large Cell Lymphoma and CD30-Positive Transformed Mycosis Fungoides. Front. Immunol. 2023, 14, 1270365. [Google Scholar] [CrossRef]
- Runge, J.S.; Novice, M.L.; Briones, N.; Williams, K.; Lowe, L.; Boyer, D.F.; Wilcox, R.A.; Tejasvi, T.; Hristov, A.C. Patch/Plaque Mycosis-Fungoides-like Presentations of DUSP22-Translocated T-Cell Lymphomas. J. Cutan. Pathol. 2022, 49, 299–305. [Google Scholar] [CrossRef]
- Diaz de la Pinta, F.J.; Machan, S.; Manso Alonso, R.; Carvajal, N.; Nieves Salgado, R.; Piris, M.A.; Rodriguez-Pinilla, S.M. DUSP22 Rearrangement in Primary Cutaneous T Cell Lymphoma with Features Intermediate between Mycosis Fungoides, Anaplastic Large-Cell Lymphoma and Lymphomatoid Papulosis. Histopathology 2022, 80, 446–449. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Forchhammer, S.; Otto, F.; Fend, F.; Frauenfeld, L. Primary Cutaneous Anaplastic Large Cell Lymphoma with DUSP22-Rearrangement Presenting as a Mimicker of Mycosis Fungoides: A Case Report and Review of the Literature. Leuk. Lymphoma 2024, 65, 265–269. [Google Scholar] [CrossRef]
- Cieza-Díaz, D.E.; Prieto-Torres, L.; Rodríguez-Pinilla, S.M.; Córdoba Mascuñano, R.; Manso Alonso, R.; Machan, S.; Piris Pinilla, M.Á.; Requena Caballero, L. Mycosis Fungoides Associated With Lesions in the Spectrum of Primary Cutaneous CD30+ Lymphoproliferative Disorders: The Same Process or 3 Coexisting Lymphomas? Am. J. Dermatopathol. 2019, 41, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Bisig, B.; Cairoli, A.; Gaide, O.; Somja, J.; Bregnard, C.; Gaulard, P.; Xerri, L.; Lefort, K.; Missiaglia, E.; Gilliet, M.; et al. Cutaneous presentation of enteropathy-associated T-cell lymphoma masquerading as a DUSP22-rearranged CD30+ lymphoproliferation. Virchows. Arch. 2022, 481, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Pileri, A.; Cavicchi, M.; Bertuzzi, C.; Righi, S.; Zengarini, C.; Sabattini, E.; Roncador, G.; Agostinelli, C. TOX Expression in Mycosis Fungoides and Sezary Syndrome. Diagnostics 2022, 12, 1582. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Yu, R.; Huang, Y.; Su, M.; Xiao, C.; Martinka, M.; Dutz, J.P.; Zhang, X.; Zheng, Z.; et al. Molecular Markers of Early-Stage Mycosis Fungoides. J. Invest. Dermatol. 2012, 132, 1698–1706. [Google Scholar] [CrossRef]
- Cerroni, L. Mycosis Fungoides-Clinical and Histopathologic Features, Differential Diagnosis, and Treatment. Semin. Cutan. Med. Surg. 2018, 37, 2–10. [Google Scholar] [CrossRef]
- Hematopathology of the Skin. Available online: https://shop.lww.com/Hematopathology-of-the-Skin/p/9781975158552 (accessed on 8 October 2024).
- Lopez, A.; Abrisqueta, P. Plasmablastic Lymphoma: Current Perspectives. Blood Lymphat. Cancer Targets Ther. 2018, 8, 63–70. [Google Scholar] [CrossRef]
- Pielasinski, U.; Santonja, C.; Rodríguez-Pinilla, S.M.; Requena, L. Extracavitary Primary Effusion Lymphoma Presenting as a Cutaneous Tumor: A Case Report and Literature Review. J. Cutan. Pathol. 2014, 41, 745–753. [Google Scholar] [CrossRef]
- Burg, G.; Kempf, W.; Kazakov, D.V.; Dummer, R.; Frosch, P.J.; Lange-Ionescu, S.; Nishikawa, T.; Kadin, M.E. Pyogenic Lymphoma of the Skin: A Peculiar Variant of Primary Cutaneous Neutrophil-Rich CD30+ Anaplastic Large-Cell Lymphoma. Clinicopathological Study of Four Cases and Review of the Literature. Br. J. Dermatol. 2003, 148, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Chioureas, D.; Beck, J.; Baltatzis, G.; Vardaki, I.; Fonseca, P.; Tsesmetzis, N.; Vega, F.; Leventaki, V.; Eliopoulos, A.G.; Drakos, E.; et al. ALK+ Anaplastic Large Cell Lymphoma (ALCL)-Derived Exosomes Carry ALK Signaling Proteins and Interact with Tumor Microenvironment. Cancers 2022, 14, 2939. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Manohar, V.; Zhao, S.; Bangs, C.D.; Cherry, A.; Azevedo, R.S.; Lage, L.A.P.C.; Pereira, J.; Zerbini, M.C.N.; Gratzinger, D.; et al. Genetic Subtypes of Systemic Anaplastic Large Cell Lymphoma Show Distinct Differences in PD-L1 Expression and Regulatory and Cytotoxic T Cells in the Tumor Microenvironment. Appl. Immunohistochem. Mol. Morphol. AIMM 2020, 28, 10–16. [Google Scholar] [CrossRef]
- Lovisa, F.; Di Battista, P.; Gaffo, E.; Damanti, C.C.; Garbin, A.; Gallingani, I.; Carraro, E.; Pillon, M.; Biffi, A.; Bortoluzzi, S.; et al. RNY4 in Circulating Exosomes of Patients with Pediatric Anaplastic Large Cell Lymphoma: An Active Player? Front. Oncol. 2020, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Zhao, N.; Zeng, Z.; Zhou, X.; Chang, C.-C.; Zu, Y. Interleukin-2 Functions in Anaplastic Large Cell Lymphoma Cells through Augmentation of Extracellular Signal-Regulated Kinases 1/2 Activation. Int. J. Biomed. Sci. IJBS 2011, 7, 181–190. [Google Scholar] [PubMed]
- Li, Y.; Yue, S.; Cao, J.; Zhu, C.; Wang, Y.; Hai, X.; Song, W.; Bi, S. pH-Responsive DNA Nanomicelles for Chemo-Gene Synergetic Therapy of Anaplastic Large Cell Lymphoma. Theranostics 2020, 10, 8250–8263. [Google Scholar] [CrossRef] [PubMed]
- Drieux, F.; Ruminy, P.; Abdel-Sater, A.; Lemonnier, F.; Viailly, P.-J.; Fataccioli, V.; Marchand, V.; Bisig, B.; Letourneau, A.; Parrens, M.; et al. Defining Signatures of Peripheral T-Cell Lymphoma with a Targeted 20-Marker Gene Expression Profiling Assay. Haematologica 2020, 105, 1582–1592. [Google Scholar] [CrossRef]
- Sibon, D.; Bisig, B.; Bonnet, C.; Poullot, E.; Bachy, E.; Cavalieri, D.; Fataccioli, V.; Bregnard, C.; Drieux, F.; Bruneau, J.; et al. ALK-Negative Anaplastic Large Cell Lymphoma with DUSP22 Rearrangement Has Distinctive Disease Characteristics with Better Progression-Free Survival: A LYSA Study. Haematologica 2023, 108, 1590–1603. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Dasari, S.; Feldman, A.L. PD-L1 Expression in Anaplastic Large Cell Lymphoma. Mod. Pathol. 2020, 33, 1232–1233. [Google Scholar] [CrossRef]
- Shen, J.; Li, S.; Medeiros, L.J.; Lin, P.; Wang, S.A.; Tang, G.; Yin, C.C.; You, M.J.; Khoury, J.D.; Iyer, S.P.; et al. PD-L1 Expression Is Associated with ALK Positivity and STAT3 Activation, but Not Outcome in Patients with Systemic Anaplastic Large Cell Lymphoma. Mod. Pathol. 2020, 33, 324–333. [Google Scholar] [CrossRef]
- Holst, J.M.; Ludvigsen, M.; Hamilton-Dutoit, S.J.; Bendix, K.; Plesner, T.L.; Nørgaard, P.; Møller, M.B.; Steiniche, T.; Rabinovich, G.A.; d’Amore, F.; et al. High Intratumoural Galectin-1 Expression Predicts Adverse Outcome in ALK- ALCL and CD30+ PTCL-NOS. Hematol. Oncol. 2020, 38, 59–66. [Google Scholar] [CrossRef]
- DeCoster, R.C.; Clemens, M.W.; Di Napoli, A.; Lynch, E.B.; Bonaroti, A.R.; Rinker, B.D.; Butterfield, T.A.; Vasconez, H.C. Cellular and Molecular Mechanisms of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Plast. Reconstr. Surg. 2021, 147, 30e–41e. [Google Scholar] [CrossRef]
- Orciani, M.; Sorgentoni, G.; Torresetti, M.; Di Primio, R.; Di Benedetto, G. MSCs and Inflammation: New Insights into the Potential Association between ALCL and Breast Implants. Breast Cancer Res. Treat. 2016, 156, 65–72. [Google Scholar] [CrossRef]
- Kleinhans, M.; Tun-Kyi, A.; Gilliet, M.; Kadin, M.E.; Dummer, R.; Burg, G.; Nestle, F.O. Functional Expression of the Eotaxin Receptor CCR3 in CD30+ Cutaneous T-Cell Lymphoma. Blood 2003, 101, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.; Tinguely, M.; Burghart, D.R.; Berisha, A.; Mertz, K.D.; Kempf, W. Characterization of the Tumor Microenvironment in Primary Cutaneous CD30-Positive Lymphoproliferative Disorders: A Predominance of CD163-Positive M2 Macrophages. J. Cutan. Pathol. 2016, 43, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.-X.; Bai, B.; Cai, Q.-C.; Cai, Q.-Q.; Wang, X.-X.; Wu, X.-Y.; Huang, H.-Q. High Numbers of Tumor-Associated Macrophages Correlate with Poor Prognosis in Patients with Mature T- and Natural Killer Cell Lymphomas. Med. Oncol. 2012, 29, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Vaid, T.; Gunning, T.S.; Cohen, R.; Della Pia, A.; Voss, J.; Weber, M.; Pecora, A.L.; Leslie, L.A.; Feldman, T.; Goy, A.H.; et al. Next-Generation Sequencing Guides Diagnosis and Treatment in a Complex Presentation of ALK-Positive Anaplastic Large-Cell Lymphoma: A Case Report. Front. Oncol. 2025, 15, 1502782. [Google Scholar] [CrossRef]
- Hsieh, W.-C.; Budiarto, B.R.; Wang, Y.-F.; Lin, C.-Y.; Gwo, M.-C.; So, D.K.; Tzeng, Y.-S.; Chen, S.-Y. Spatial Multi-Omics Analyses of the Tumor Immune Microenvironment. J. Biomed. Sci. 2022, 29, 96. [Google Scholar] [CrossRef]
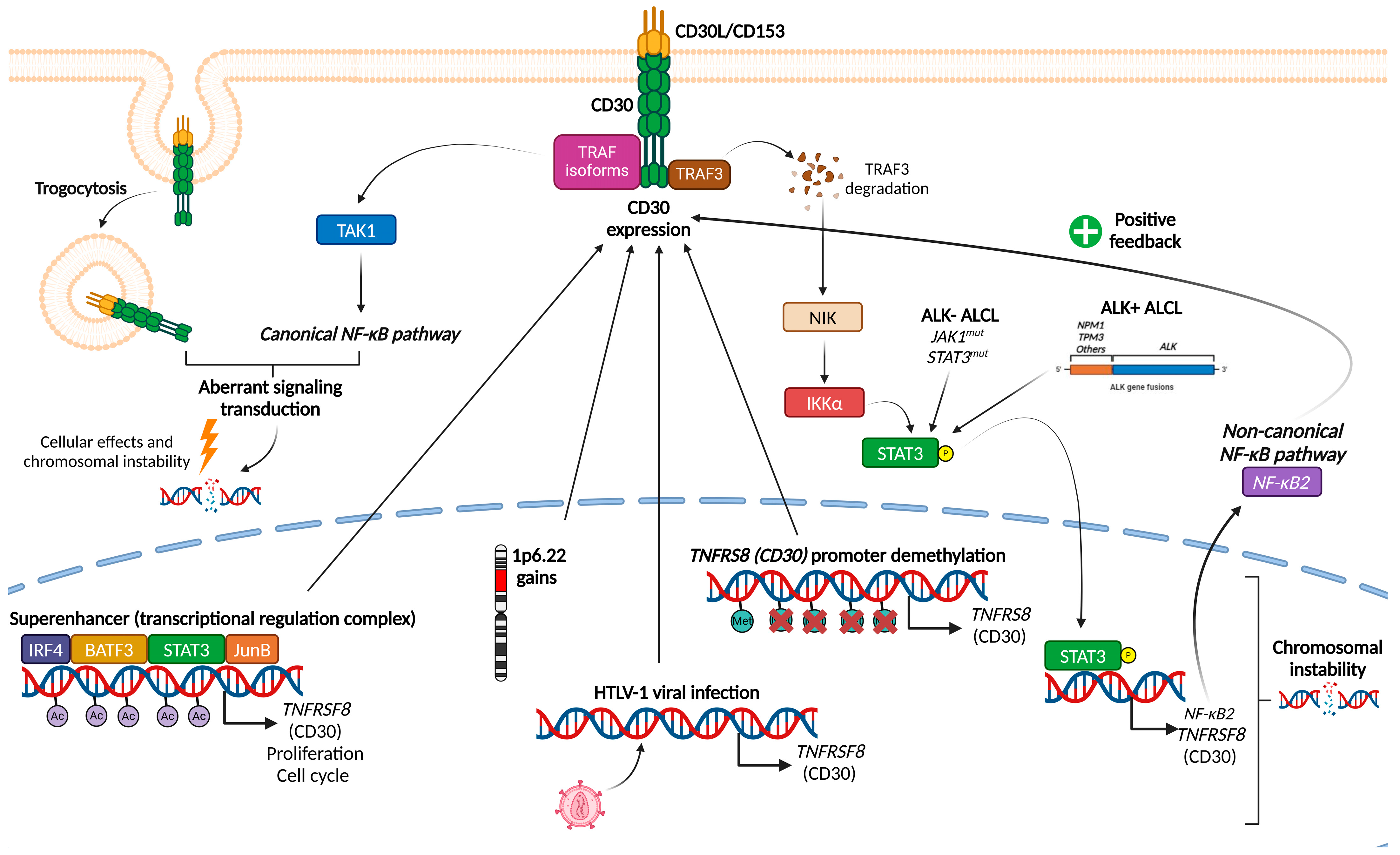
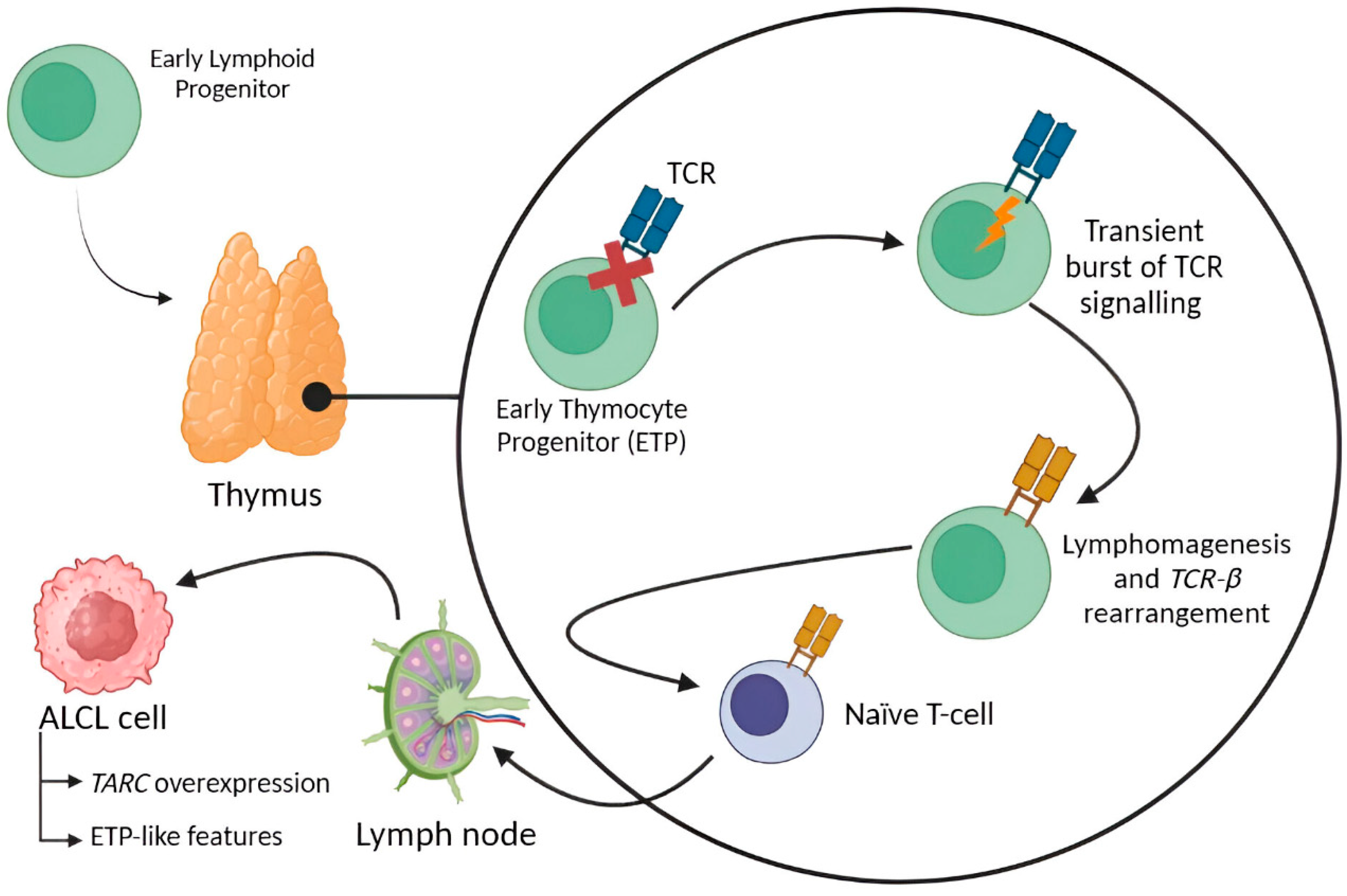
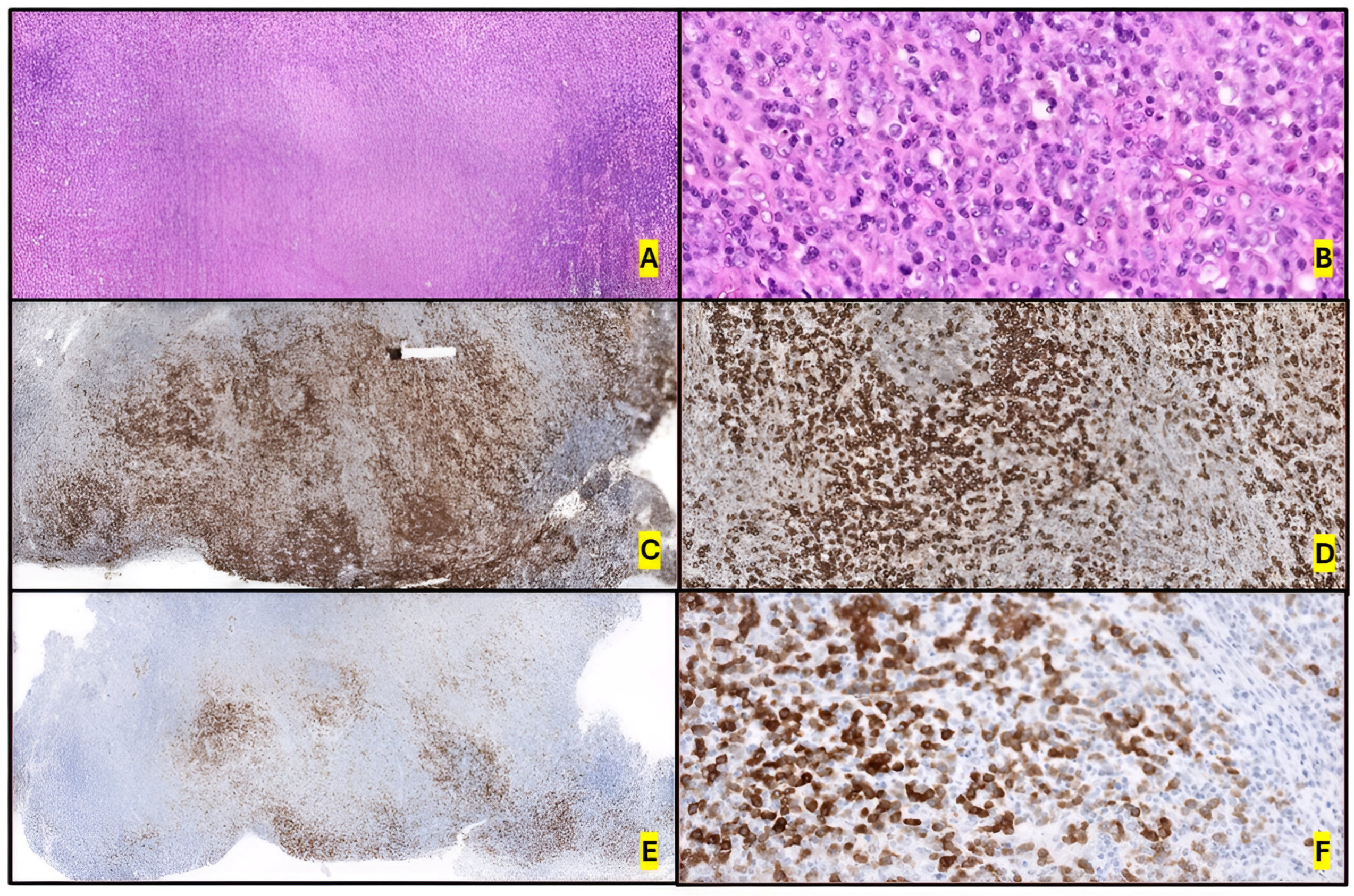
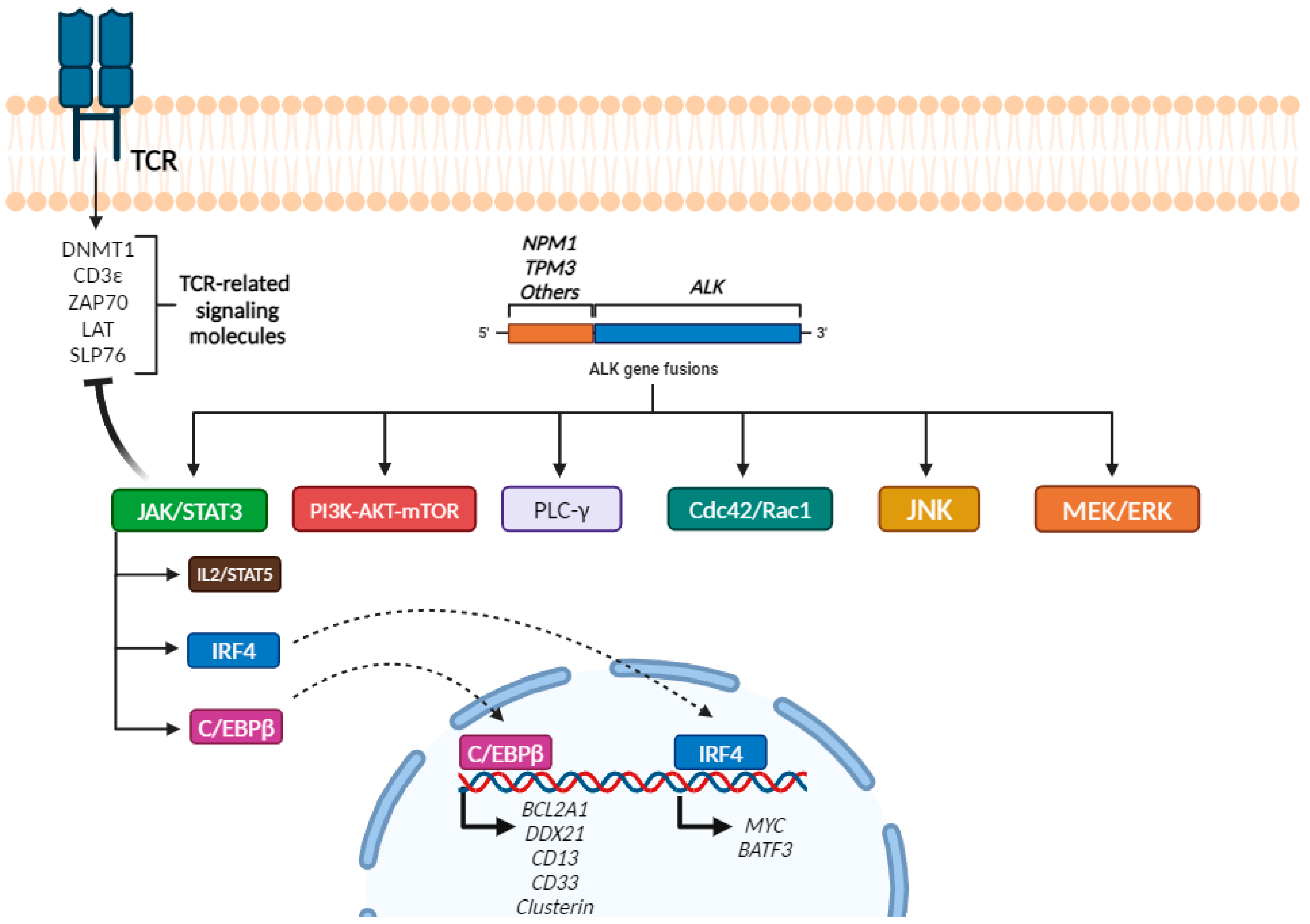


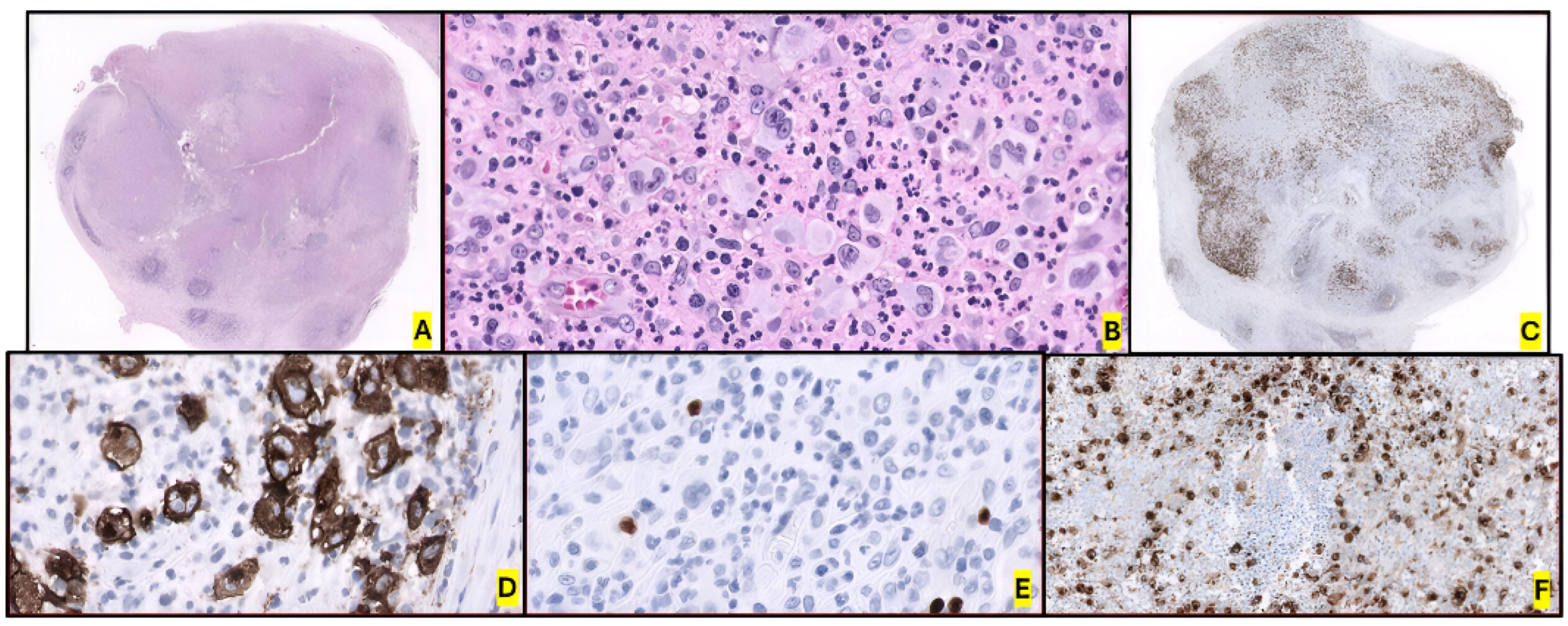




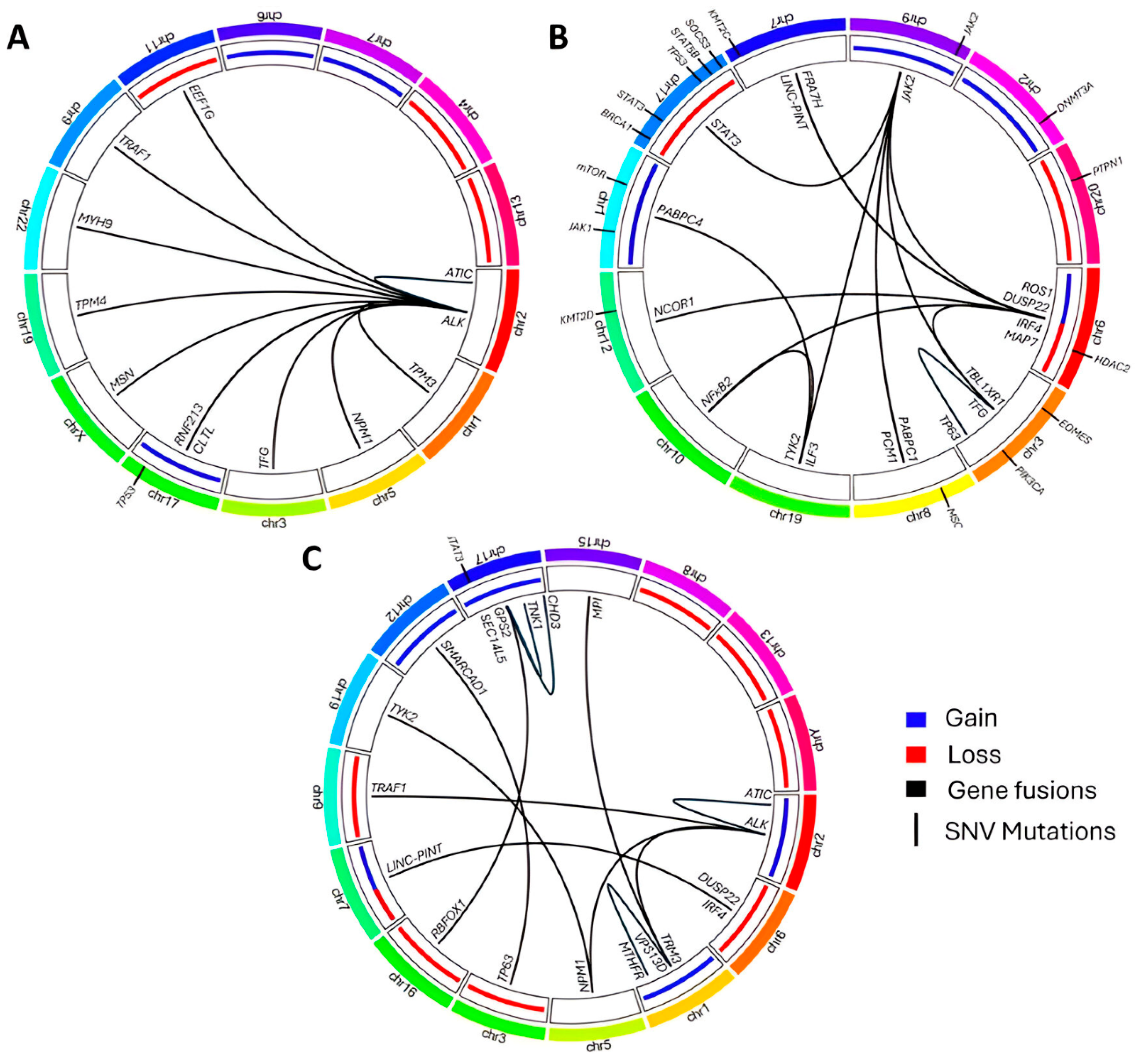
| Study Reference | CD30+ Cutoff | PTCL-NOS | CTCL/MF | ALK- ALCL | ALK+ ALCL | AITL | ATLL | ENKTL | EATL | F-PTCL |
|---|---|---|---|---|---|---|---|---|---|---|
| Sabattini et al., 2013 [21] (N = 192) | 0: No staining 1: 0 to <25% 2: 25 to 50% 3: >50 to 75% 4: >75% | 36% 13% 21% 13% 18% | 41% 47% 6% 0% 6% | NA | NA | 51% 21% 12% 10% 0% | NA | 20% 10% 30% 10% 30% | 0% 0% 22% 0% 78% | NA |
| Bossard et al., 2014 [22] (N = 376) | 0: No staining 1: 0 to <25% 2: 25 to 50% 3: >50 to 75% 4: >75% | 42% 26% 9% 10% 13% | NA | 0% 0% 0% 0% 100% | 0% 0% 5% 2% 93% | 37% 47% 10% 5% 0% | 44% 11% 33% 11% 0% | 54% 7% 11% 14% 14% | 50% 0% 0% 7% 43% | NA |
| Lamarque et al., 2016 [23] (N = 56) | 0: <5% 1: 5 to 24% 2: 25 to 49% 3: 50–75% 4: >75% | 10% 10% 30% 30% 20% | 14% 0% 0% 14% 71% | 0% 0% 0% 0% 100% | 0 0 20 20 60 | 0% 100% 0 0 0 | 100% 0% 0% 0% 0% | NA | 0% 100% 0% 0% 0% | NA |
| Rodriguez-Pinilla et al., 2021 [24] (N = 175) | Mean (SD) of % stained cells in each group | 25.0% (34.8) | NA | 74.5% (32) | 97.3% (6.5) | 18.5% (21) | NA | 53.1% (41) | 31.7% (46.6) | NA |
| Karube et al., 2008 [25] (N = 319) * | >70% 20 to 70% | 5% 11% | 9% 9% | 58% 35% | NA | 0% 32% | 15% 24% | 0% 64% | NA | NA |
| Asano et al., 2011 [26] (N = 47) | >30% | 51% | NA | NA | NA | NA | NA | NA | NA | NA |
| Savage et al., 2008 [27] (N = 490) | >0% ≥80% | 32% 5% | NA | 100% 100% | 100% 100% | NA | NA | NA | NA | NA |
| Weisenburger et al., 2011 [28] (N = 217) | >20% | 32% | NA | NA | NA | NA | NA | NA | NA | NA |
| Wang et al., 2017 [29] (N = 122) | 0: No staining 1: 0 to <25% 2: 25 to 50% 3: >50 to 75% 4: >75% | NA | NA | NA | NA | NA | NA | 30% 38% 18% 10% 5% | NA | NA |
| Kawamoto et al., 2018 [30] (N = 97) | 0: ≥1% 1: ≥10% 2: ≥20% | NA | NA | NA | NA | NA | NA | 57% 55% 44% | NA | NA |
| Feng et al., 2017 [31] (N = 622) | ≥20% | NA | NA | NA | NA | NA | NA | 47.3% | NA | NA |
| Hartmann et al., 2019 [32] (N = 16) | >60% | NA | NA | NA | NA | NA | NA | NA | NA | 75% |
| Onaindia et al., 2016 [33] (N = 51) | 0: No staining 1: 0 to <25% 2: 25 to 50% 3: >50 | 25% 0% 37.5% 37.5% | NA | NA | NA | 7% 83.7% 9.3% 0% | NA | NA | NA | NA |
| Shen et al., 2024 [34] (N = 82) | 1: <40% 2: ≥40% | NA | NA | NA | NA | NA | NA | 18.3% 81.7% | NA | NA |
| Non-Coding RNA Type | Transcript | Regulation Status | Effect |
|---|---|---|---|
| miRNA | miR-17-92 | Overexpressed | Activation of STAT3 |
| miR-101 | Downregulated | Activation of mTOR pathway | |
| miR-155 | Downregulated | Create immunosuppressive microenvironment, favoring Th2 differentiation due to low levels of IFN-γ | |
| miR135b | Overexpressed | Activation of NPM–ALK–STAT3 axis. Favor immunosuppressive microenvironment due to IL-17 secretion (Th17 signature), activating the transcription of STAT6 and GATA3 | |
| miR-150 | Downregulated | Protumoral properties | |
| miR-146a-5p | Overexpressed | M2 macrophage infiltration, improving tumor aggressiveness and dissemination | |
| lncRNA | LINC01013 | Overexpressed | Induction of Snai1 (activating EMT) |
| snoRNA | All | Downregulated | Unknown |
| U3 | Overexpressed | Unknown, but serves as tool for diagnostic procedures | |
| circRNA | All | Overexpressed | Unknown. Formation of these non-coding RNAs between the breakpoint of NPM and ALK |
| DUSP22R | TP63R | TN pSTAT3+ | TN pSTAT3- | “Double Hit” DUSP22R and TP63R | |
|---|---|---|---|---|---|
| Neoplastic cells | Hallmark cells Monomorphism Doughnut cells | Pleomorphic cells Mitosis | Sheet-like neoplastic cells, large pleomorphic cells | Pleomorphic | NA (absence of DUSP22R morphology) |
| Inflammatory background | Scattered | >lymphocytes | Lymphocyte-rich background | Monomorphic | NA (abscence of DUSP22R morphology) |
| IHC | Non-cytotoxic phenotype (TIA1 < 20%) P63 variable LEF1 > 75% | Cytotoxic phenotype P63 > 30% | Cytotoxic phenotype | Non-cytotoxic phenotype | Non-cytotoxic phenotype |
| DUSP22R | + | - | - | - | + |
| TP63R | - | + | - | - | + |
| JAK/STAT3 pathway activation | - | + | + | - | NA |
| pSTAT3 expression | - | + | + | - | NA |
| TP53 mutations | - | +/- | -/+ | +/- | NA |
| 5-year OS | 40–90% | 20% | 50% | 20% | NA |
| Characteristic | ALK- ALCL | CD30+ PTCL-NOS |
|---|---|---|
| Immunophenotype | Strong CD30, EMA+; CD3 may be absent or weak | CD30 variable, CD3 strong, other T markers present (CD4, CD8) |
| DUSP22 rearrangement | Common (~30% of cases) | Rare |
| TP63 rearrangement | Less common (~8% of cases) | Rare |
| TNFRSF8 (CD30), BATF3, TMOD3 gene expression | Highly expressed; differentiates with 97% accuracy | Less common expression |
| Loss of 5q/9p | Rare | Common |
| STAT3 phosphorylation (pSTAT3-S727) | Overexpressed; strong marker for ALK- ALCL | Rarely overexpressed |
| Cytotoxic phenotype markers (TIA-1, granzyme B) | Frequently positive | Infrequent or absent |
| Epigenetic mutations (e.g., TET2, DNMT3A) | Rare | Frequently mutated |
| Recurrent Molecular Alterations | Frequency (%) |
|---|---|
| JAK/STAT signaling | |
| STAT3 | 11–64 |
| JAK1 | 7–44 |
| SOCS1 | 3–20 |
| SOCS3 | 6 |
| PTPN1 | 3–9 |
| Epigenetic modifiers | |
| KMT2C | 11–26 |
| CHD2 | 15 |
| CREBBP | 15 |
| KMT2D | 9 |
| DNMT3A | 6–20 |
| HDAC2 | 6 |
| TET2 | 3 |
| Cell cycle/apoptosis | |
| TP53 | 11–20 |
| Chromosomal alterations | |
| Loss of 20q13.13 | 66 |
| Focal amplification of 9p24.1 | 33 |
| Marker | ALK+ ALCL | ALK- ALCL: DUSP22-R | ALK- ALCL: TP63-R | ALK- ALCL: Triple Negative | BIA-ALCL | Primary Cutaneous ALCL |
|---|---|---|---|---|---|---|
| Lymphoid origin | ||||||
| CD45 | - | + | + | + | + | + |
| CD3 | 25% | +/- | +/- | +/- | +/- | +/- |
| CD4 | 40% | 70% | + | + | + | + |
| CD8 | - | - | - | - | - | +/- |
| CD2 | 30% | +/- | + | + | + | + |
| CD5 | 30% | +/- | +/- | +/- | - | +/- |
| CD7 | +/- | +/- | +/- | +/- | +/- | +/- |
| ALK (Anaplastic Lymphoma Kinase) | + | - | - | - | - | - |
| Cytotoxic markers (TIA1, granzyme B, and perforin) | 75–90% | - | + | +/- | + | + |
| CD30 | +++ | +++ | +++ | +++ | +++ | +++ |
| CD20 | - | - | - | - | - | - |
| EBER | - | - | - | - | - | - |
| Epithelial origin (EMA and/or Cytokeratins) | 30% | - | - | - | Only EMA in excepctional cases | - |
| Ki-67 | High (>70%) | Moderate–High (50–70%) | High (>80%) | Variable (40–70%) | High (>70%) | Variable (40–70%) |
| Others | CD43 (30%) | LEF1 (>75%) | P63 (35%) | CD43 | LEF1 and PSTAT3 in DUSP22-R cases | |
| CD56 (10%) | TCRαβ (<20%) | pSTAT3 (rarely) | CD25 | Negative for cytotoxic markers in DUSP22-R cases | ||
| pSTAT3 (75%) | pSTAT3 (<20%) | TCRαβ or TCRγδ (30%) | PD-1 and/or ICOS (rarely) | |||
| CD15 (rarely) | PAX5 (rarely) | CD15 (45%) | ||||
| PAX5 (rarely) | CD138 (rarely) | CD56 (rarely) | ||||
| BCL2 negative | TCRγδ (rarely) |
| Entity | Abnormality Type | List of Alterations |
|---|---|---|
| ALK+ ALCL | Chromosomal translocation (ALK partner) | t(2;5)(p23;q35) (NPM1), t(1;2)(q25;p23) (TPM3), Inv(2)(p23q53) (ATIC), t(2;3)(p23;q21) (TFG), t(2;17)(p23;q23) (CLTL), t(2;X)(p23;q11.12) (MSN), t(2;19)(p23;p13.1) (TPM4), t(2;22)(p23;q11.2) (MYH9), t(2;9)(p23;q33–34) (TRAF1), t(2;11)(2p23;11q12.3) (EEF1G), t(2;17)(p23;q25) (RNF213/ALO17) |
| Chromosomal alterations | 6q (gain), 7p (gain), 17p/17q24 (gain), 4q13-q21/11q14 (loss), 13q (loss) | |
| Mutations | TP53 | |
| ALK- ALCL | Gene fusions and chromosomal translocations | DUSP22::FRA7H t(6;7), DUSP22/IRF4::LINC-PINT t(6;7), TBL1XR1::TP63 inv(3)(q26q28), DUSP22::TBL1XR1 t(6;3), PABPC1::JAK2 t(8;9), TFG::JAK2 t(3;9), ILF3::JAK2 t(19;9), MAP7::JAK2 t(6;9), PCM1::JAK2 t(8;9), NFκB2::ROS1 t(10;6), NCOR2::ROS1 t(12;6), NFκB2::TYK2 t(10;19), PABPC4::TYK2 t(1;19), FRK::PABPC1 t(6;8), FRK::MAPK9 t(6;5), FRK::CAPRIN1 t(6;11), MYC::Unknown t(8;?) |
| Chromosomal alterations | 1q (gain), 6p21 (gain), 6q21 (loss), 17p13 (loss), 2 Trisomy | |
| Mutations | JAK1G1097D/S, JAK1L910P, STAT3Y640F, STAT3N647I, STAT3D661Y, STAT3A662V, TP53, MSCE116K, PRDM1, BANK1, FAS, STIM2, LRP1B, EPHA5, KMT2D | |
| BIA-ALCL | Gene fusions | STAT3::JAK2 t(17;9) |
| Chromosomal alterations | 9p24.1 (gain), 20q13.12–13.2 (loss) | |
| Mutations | TP53, BRCA1/2, JAK1, JAK2, STAT3, STAT5B, SOCS1, SOCS3, PTPN1, KMT2C, KMT2D, CHD2, CREBBP, DNMT3A, TET2, HDAC2, EOMES, PI3K/AKT/mTOR | |
| pcALCL | Gene fusions and chromosomal translocations | ALK::NPM1 t(2;5), DUSP22/IRF4::LINC-PINT t(6;7), ALK::TRM3 t(1;2), ALK::ATIC t(2;2), ALK::TRAF1 t(2;9), NPM1::TYK2 t(5;19), TP63::SMARCAD1 t(3;12), GPS2::TNK1 inv(17), GPS2::CHD3 inv(17), RBFOX1::SEC14L5 t(16;17), VPS13D::MPI t(1;15), VPS13D::MTHFR inv(1) |
| Chromosomal alterations | Gain of 1p/q, 2p/q, 7q31, 12q, 17q and loss of 3p, 6q, 7p/q, 8p, 9p21, 13q, 16q, Y | |
| Mutations | STAT3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frutos Díaz-Alejo, J.; Prieto-Potín, I.; Manso, R.; Rodríguez, M.; Rebollo-González, M.; Díaz de la Pinta, F.J.; Morales-Gallego, M.; Rodríguez-Pinilla, S.M.; Onaindia, A. Molecular Insights into the Diagnosis of Anaplastic Large Cell Lymphoma: Beyond Morphology and Immunophenotype. Int. J. Mol. Sci. 2025, 26, 5871. https://doi.org/10.3390/ijms26125871
Frutos Díaz-Alejo J, Prieto-Potín I, Manso R, Rodríguez M, Rebollo-González M, Díaz de la Pinta FJ, Morales-Gallego M, Rodríguez-Pinilla SM, Onaindia A. Molecular Insights into the Diagnosis of Anaplastic Large Cell Lymphoma: Beyond Morphology and Immunophenotype. International Journal of Molecular Sciences. 2025; 26(12):5871. https://doi.org/10.3390/ijms26125871
Chicago/Turabian StyleFrutos Díaz-Alejo, Jesús, Iván Prieto-Potín, Rebeca Manso, Marta Rodríguez, Marcos Rebollo-González, Francisco Javier Díaz de la Pinta, Miriam Morales-Gallego, Socorro María Rodríguez-Pinilla, and Arantza Onaindia. 2025. "Molecular Insights into the Diagnosis of Anaplastic Large Cell Lymphoma: Beyond Morphology and Immunophenotype" International Journal of Molecular Sciences 26, no. 12: 5871. https://doi.org/10.3390/ijms26125871
APA StyleFrutos Díaz-Alejo, J., Prieto-Potín, I., Manso, R., Rodríguez, M., Rebollo-González, M., Díaz de la Pinta, F. J., Morales-Gallego, M., Rodríguez-Pinilla, S. M., & Onaindia, A. (2025). Molecular Insights into the Diagnosis of Anaplastic Large Cell Lymphoma: Beyond Morphology and Immunophenotype. International Journal of Molecular Sciences, 26(12), 5871. https://doi.org/10.3390/ijms26125871