
Transcriptome and Metabolome Analyses of Short-Term Responses of Populus talassica × Populus euphratica to Leaf Damage



Abstract
1. Introduction
2. Results
2.1. Transcriptome Analysis
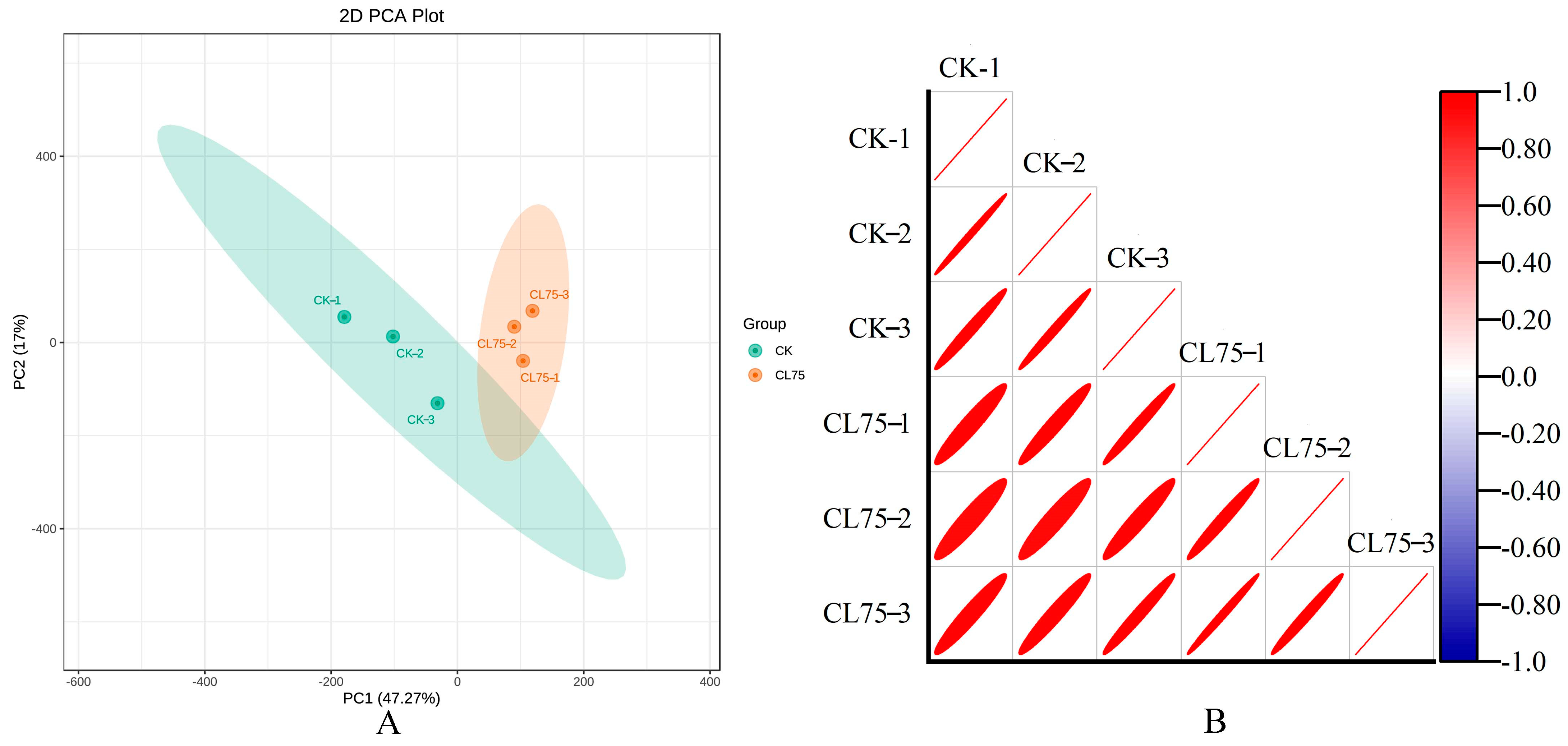
2.1.1. Quality Control Analysis and Transcriptome Data Annotation
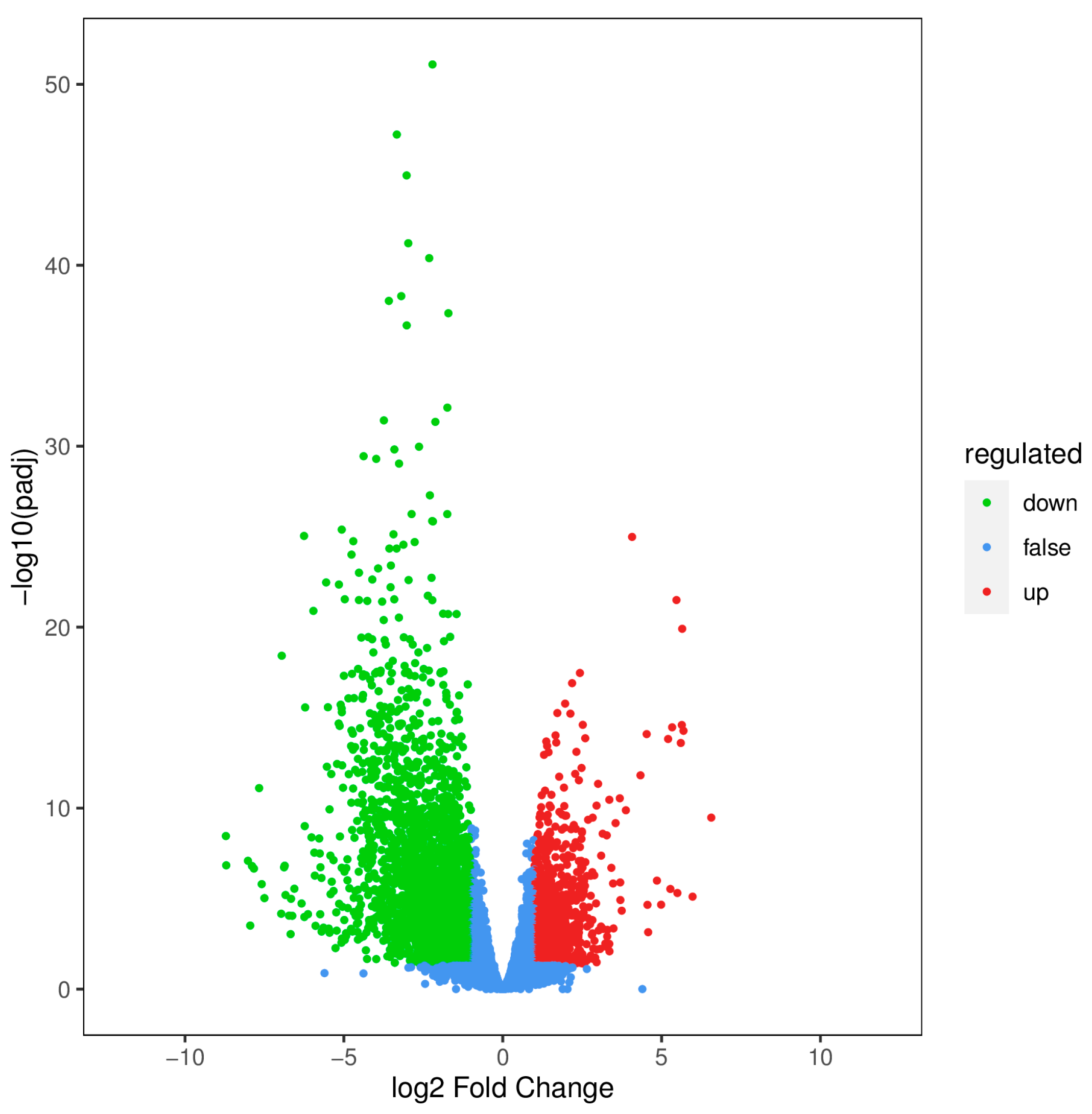
2.1.2. Screening and Analysis of DEGs
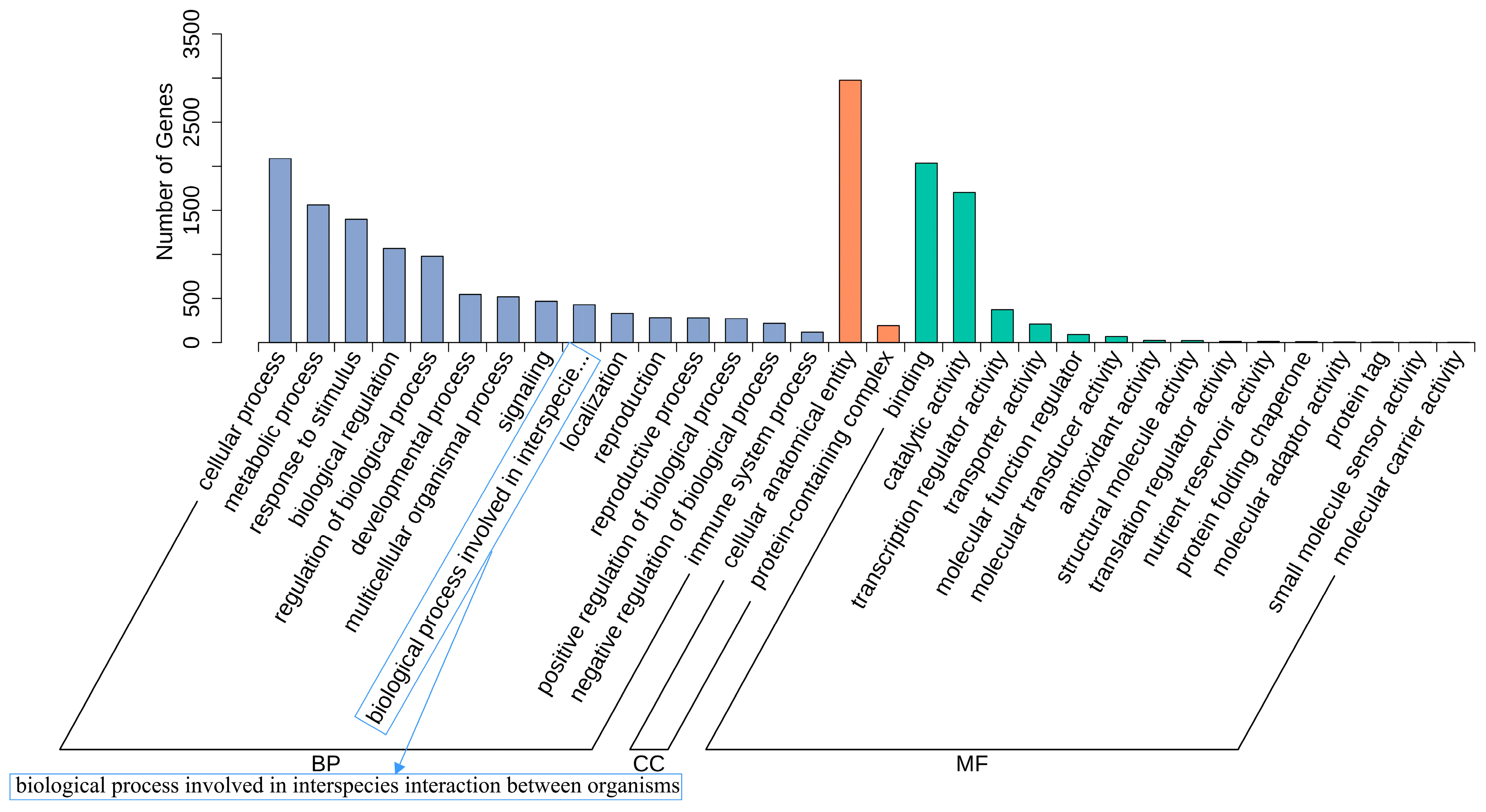
2.1.3. GO Enrichment Analysis of DEGs
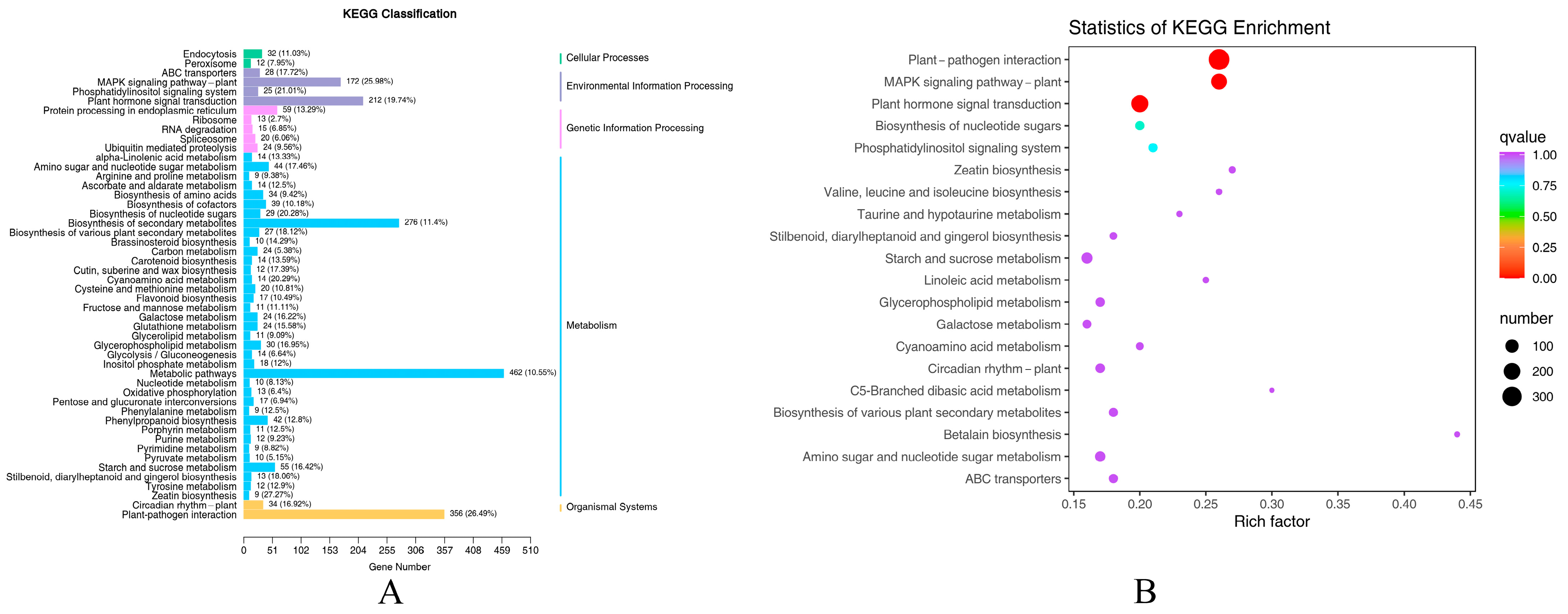
2.1.4. KEGG Enrichment Analysis of DEGs
2.1.5. Analysis of the Main KEGG Metabolic Pathways
2.1.6. Transcription Factor Analysis
2.2. Secondary Metabolome Analysis
2.2.1. Qualitative and Quantitative Analyses of Metabolites
2.2.2. PCA and OPLS-DA
2.2.3. Screening DSMs
2.2.4. Correlation Analysis of DSMs
2.2.5. KEGG Enrichment Analysis of DSMs
2.3. Integration Analyses of Transcriptome and Secondary Metabolome
2.3.1. KEGG Enrichment Analysis
2.3.2. Correlation Cluster Analysis
2.3.3. Typical Correlation Analysis
2.3.4. Summary Table for DEGs, DSMs, and Pathways
3. Discussion
3.1. Transcriptome Analysis of P. talassica × P. euphratica in Response to Leaf Damage
3.2. Transcription Factor Analysis of P. talassica × P. euphratica in Response to Leaf Damage
3.3. Secondary Metabolome Analysis of P. talassica × P. euphratica in Response to Leaf Damage
4. Materials and Methods
4.1. Site Description
4.2. Plant Material and Experimental Treatments
4.3. Transcriptome Analysis
4.4. Secondary Metabolome Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Abscisic Acid |
| CAT | Catalase |
| FDR | False Discovery Rate |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOG | Clusters of Orthologous Groups for Eukaryotic Complete Genomes |
| MRM | Multiple Reaction Monitoring |
| NR | Non-Redundant Protein Sequence Database |
| OPLS-DA | Orthogonal Partial Least Squares Discriminant Analysis |
| PAL | Phenylalanine ammonia-lyase |
| SOD | Superoxide Dismutase |
| VIP | Variable Importance in Projection |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Reads Mapped (%) | Clean Base (G) | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| CK-1 | 45,020,190 | 42,314,946 | 81.08 | 6.35 | 0.03 | 97.87 | 93.83 | 43.89 |
| CK-2 | 44,501,442 | 42,549,490 | 81.41 | 6.38 | 0.03 | 97.96 | 94.03 | 43.78 |
| CK-3 | 43,678,542 | 41,507,930 | 81.48 | 6.23 | 0.03 | 97.94 | 93.97 | 43.99 |
| CL75-1 | 47,133,128 | 46,083,746 | 82.46 | 6.91 | 0.03 | 98.08 | 94.34 | 43.97 |
| CL75-2 | 43,255,808 | 42,286,636 | 81.45 | 6.34 | 0.03 | 97.81 | 93.65 | 43.82 |
| CL75-3 | 53,348,448 | 50,859,102 | 81.84 | 7.63 | 0.03 | 97.77 | 93.55 | 43.97 |
| Soil Type | pH | Conductivity (μs·cm−1) | Salt Salinity (g·kg−1) | Organic Matter (g·kg−1) | Alkali-Hydrolyzed Nitrogen (mg·kg−1) | Available Potassium (mg·kg−1) | Available Phosphorus (mg·kg−1) |
|---|---|---|---|---|---|---|---|
| loam | 8.03 | 609.43 | 2.87 | 21.34 | 17.08 | 143.07 | 22.05 |
References
- Su, M.X.; Zhang, M.; Liu, Y.; Han, Z.J. Abscisic acid, paclobutrazol, and salicylic acid alleviate salt stress in Populus talassica × Populus euphratica by modulating plant root architecture, photosynthesis, and the antioxidant defense system. Forests 2022, 13, 1864. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Y.; Su, M.X.; Han, Z.J.; Shi, J.Y. Evaluation of salt and drought tolerances of Populus talassica × Populus euphratica seedlings using leaf anatomical structures and physiological processes. Pak. J. Bot. 2023, 55, 1205–1214. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Jin, L.; Peng, R. Crosstalk between Ca2+ and other regulators assists plants in responding to abiotic stress. Plants 2022, 11, 1351. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.Z.; Ma, C.; Zhang, Y.; Zhang, Q.; Xu, G.; Yang, G. Decoyinine induced resistance in rice against small brown planthopper Laodelphax striatellus. Insects 2022, 13, 104. [Google Scholar] [CrossRef]
- An, Y.; Shen, Y.B.; Zhang, Z.X. Effects of mechanical damage and herbivore wounding on H2O2 metabolism and antioxidant enzyme activities in hybrid poplar leaves. J. For. Res. 2009, 20, 156–160. [Google Scholar] [CrossRef]
- Tang, F.; Zhao, W.L.; Gao, X.W. Communication between plants: Induced resistance in poplar seedlings following herbivore infestation, mechanical wounding, and volatile treatment of the neighbors. Entomol. Exp. Appl. 2013, 149, 110–117. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, G.; Sun, T.; Wang, L.; Xu, Q.; Liu, J.; Gao, B. Metabolites and plant hormones related to the resistance response to feeding stimulation and leaf clipping control in Chinese Pine (Pinus tabuliformis Carr.). Curr. Issues Mol. Biol. 2023, 45, 1086–1099. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, S.; He, W. Induction mechanism of ribosome inactivating protein CURCIN2 under mechanical wounding in Jatropha curcas L. Chin. J. Appl. Environ. Biol. 2023, 29, 52–56. (In Chinese) [Google Scholar]
- Colicchio, J.M.; Monnahan, P.J.; Kelly, J.K.; Hileman, L.C. Gene expression plasticity resulting from parental leaf damage in Mimulus guttatu. New Phytol. 2015, 205, 894–906. [Google Scholar] [CrossRef]
- Toyota, M.; Spencer, D.; Sawai-Toyota, S.; Wang, J.Q.; Zhang, T.; Koo, A.J.; Howe, G.A.; Gilroy, S. Glutamate triggers long-distance, calcium-based plant defense signaling. Science 2018, 361, 1112–1115. [Google Scholar] [CrossRef]
- Chen, Q.; Lu, X.; Guo, X.; Liu, J.; Liu, Y.; Guo, Q.; Tang, Z. The specific responses to mechanical wound in leaves and roots of Catharanthus roseus seedlings by metabolomics. J. Plant Interact. 2018, 13, 450–460. [Google Scholar] [CrossRef]
- Bender, K.W.; Snedden, W.A. Calmodulin-related proteins step out from the shadow of their namesake. Plant Physiol. 2013, 163, 486–495. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, Z.; Xu, F.; Ahmad, W.; Lu, G.; Su, Y.; Xu, L. Genome-wide identification of LRR-RLK family in Saccharum and expression analysis in response to biotic and abiotic stress. Curr. Issues Mol. Biol. 2021, 43, 1632–1651. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Y.; Yin, H.; Gao, M.; Zhang, Q.; Chen, Y. Genome-wide identification and characterization of the LRR-RLK gene family in two Vernicia species. Int. J. Genom. 2015, 2015, 823427. [Google Scholar]
- MAPK Group; Ichimura, K.; Shinozaki, K.; Tena, G.; Sheen, J.; Henry, Y.; Champion, A.; Kreis, M.; Zhang, S.; Hirt, H.; et al. Mitogen-activated protein kinase cascades in plants: A new nomenclature. Trends Plant Sci. 2002, 7, 301–308. [Google Scholar]
- Zhang, M.; Su, J.; Zhang, Y.; Xu, J.; Zhang, S. Conveying endogenous and exogenous signals: MAPK cascades in plant growth and defense. Curr. Opin. Plant Biol. 2018, 45, 1–10. [Google Scholar] [CrossRef]
- Gomez-Osuna, A.; Calatrava, V.; Galvan, A.; Fernandez, E.; Llamas, A. Identification of the MAPK Cascade and its Relationship with Nitrogen Metabolism in the Green Alga Chlamydomonas reinhardtii. Int. J. Mol. Sci. 2020, 21, 3417. [Google Scholar] [CrossRef]
- Edward, E.F.; Debora, G.; Iván, F.A. The squeeze cell hypothesis for the activation of jasmonate synthesis in response to wounding. New Phytol. 2014, 204, 282–288. [Google Scholar]
- Sözen, C.; Schenk, S.T.; Boudsocq, M.; Chardin, C.; Almeida-Trapp, M.; Krapp, A.; Hirt, H.; Mithöfer, A.; Colcombet, J. Wounding and insect feeding trigger two independent MAPK pathways with distinct regulation and kinetics. Plant Cell. 2020, 32, 1988–2003. [Google Scholar] [CrossRef]
- von Dahl, C.C.; Baldwin, I.T. Deciphering the role of ethylene in plant-herbivore interactions. J. Plant Growth Regul. 2007, 26, 201–209. [Google Scholar] [CrossRef]
- von Loon, L.C.; Geraats, B.P.J.; Linthorst, H.J.M. Ethylene as a modulator of disease resistance in plants. Trends Plant Sci. 2006, 11, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Parkin, I.; Lydiate, D.; Hannoufa, A. An auxin-responsive SCARECROW-like transcriptional activator interacts with histone deacetylase. Plant Mol. Biol. 2004, 55, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lei, W.; Zhou, Z.; Li, Y.; Zhang, D.; Lin, H. Transcription factor GLK1 promotes anthocyanin biosynthesis via an MBW complex-dependent pathway in Arabidopsis thaliana. J. Integr. Plant Biol. 2023, 65, 1521–1535. [Google Scholar] [CrossRef]
- Liu, F.; Li, X.; Wang, M.; Wen, J.; Yi, B.; Shen, J.; Ma, C.; Fu, T.; Tu, J. Interactions of WRKY15 and WRKY33 transcription factors and their roles in the resistance of oilseed rape to Sclerotinia infection. Plant Biotechnol. J. 2018, 16, 911–925. [Google Scholar] [CrossRef]
- Hu, Y.; Dong, Q.; Yu, D. Arabidopsis WRKY46 coordinates with WRKY70 and WRKY53 in basal resistance against pathogen Pseudomonas syringa. Plant Sci. 2012, 185–186, 288–297. [Google Scholar] [CrossRef]
- Hara, K.; Yagi, M.; Kusano, T.; Sano, H. Rapid systemic accumulation of transcripts encoding a tobacco WRKY transcription factor upon wounding. Mol. Gen. Genet. 2000, 263, 30–37. [Google Scholar] [CrossRef]
- Chen, M.; She, Z.Y.; Aslam, M.; Liu, T.; Wang, Z.; Qi, J.; Niu, X. Genomic insights of the WRKY genes in kenaf (Hibiscus cannabinus L.) reveal that HcWRKY44 improves the plant’s tolerance to the salinity stress. Front. Plant Sci. 2022, 13, 984233. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Li, M.; Jia, H.; Wei, F.; Xia, Z.; Zhang, X.; Chang, J.; Wang, Z. Overexpression of the WRKY transcription factor gene NtWRKY65 enhances salt tolerance in tobacco (Nicotiana tabacum). BMC Plant Biol. 2024, 24, 326. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Chen, Z.; Fang, Z.; Meng, J.; Jun, T.; Zhao, D. PoWRKY69-PoVQ11 module positively regulates drought tolerance by accumulating fructose in Paeonia ostii. Plant J. 2024, 119, 1782–1799. [Google Scholar] [CrossRef]
- Ding, Z.J.; Yan, J.Y.; Li, C.X.; Li, G.X.; Wu, Y.R.; Zheng, S.J. Transcription factor WRKY46 modulates the development of Arabidopsis lateral roots in osmotic/salt stress conditions via regulation of ABA signaling and auxin homeostasis. Plant J. 2015, 84, 56–69. [Google Scholar] [CrossRef]
- Zhao, F.; Zeng, H. The related transcriptional factors in the transcriptome of mechanically-damaged Zoysia japonica. Biotechnol. Bull. 2019, 35, 7–12. (In Chinese) [Google Scholar]
- Alabarce, F.S.; Dillenburg, L.R. A possible ontogenetic trade-off between defense and tolerance in response to simulated herbivory in seedlings and saplings of Araucaria angustifolia. Theor. Exp. Plant Physiol. 2014, 26, 147–156. [Google Scholar] [CrossRef]
- Li, T.M.; Juan, W.L.; Shu, Y.H.; Zhou, Y.M.; Tian, L.; Zhang, X.G.; Liu, F.L.; Liu, P. TMT based proteomic profiling of Sophora alopecuroides leaves reveal flavonoid biosynthesis processes in response to salt stress. J. Proteom. 2021, 253, 104457. [Google Scholar]
- Murata, K.; Kitano, T.; Yoshimoto, R.; Takata, R.; Ube, N.; Ueno, K.; Ueno, M.; Yabuta, Y.; Teraishi, M.; Holland, C.K.; et al. Natural variation in the expression and catalytic activity of a naringenin 7-O-methyltransferase influences antifungal defenses in diverse rice cultivars. Plant J. 2020, 101, 1103–1117. [Google Scholar] [CrossRef]
- Yu, W.; Liu, H.; Luo, J.; Zhang, S. Partial root-zone simulated drought induces greater flavonoid accumulation than full root-zone simulated water deficiency in the leaves of Ginkgo biloba. Environ. Exp. Bot. 2022, 201, 104998. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.; Antonescu, C.; Chang, T.-C.; Mendell, J.; Salzberg, S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.-Y.; Dillies, M.-A. SARTools: A DESeq2- and EdgeR-based r pipeline for comprehensive differential analysis of RNA-seq data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]

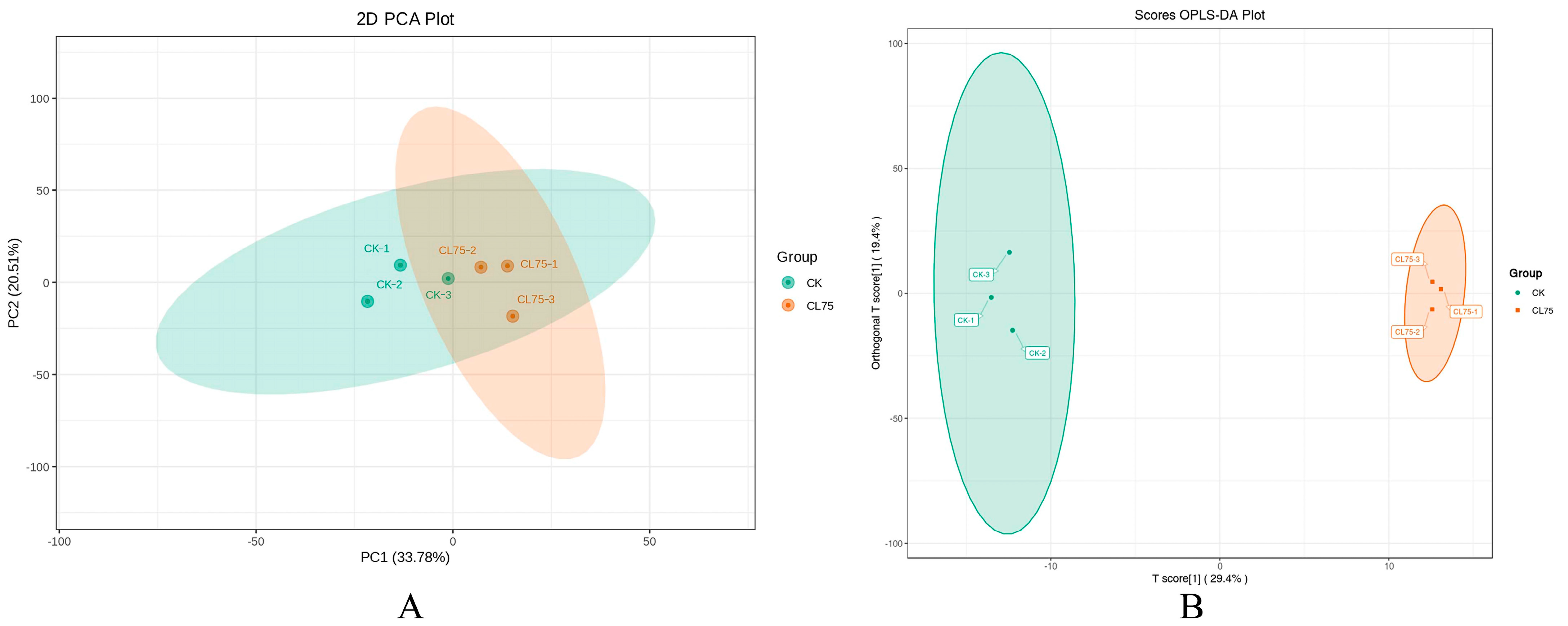
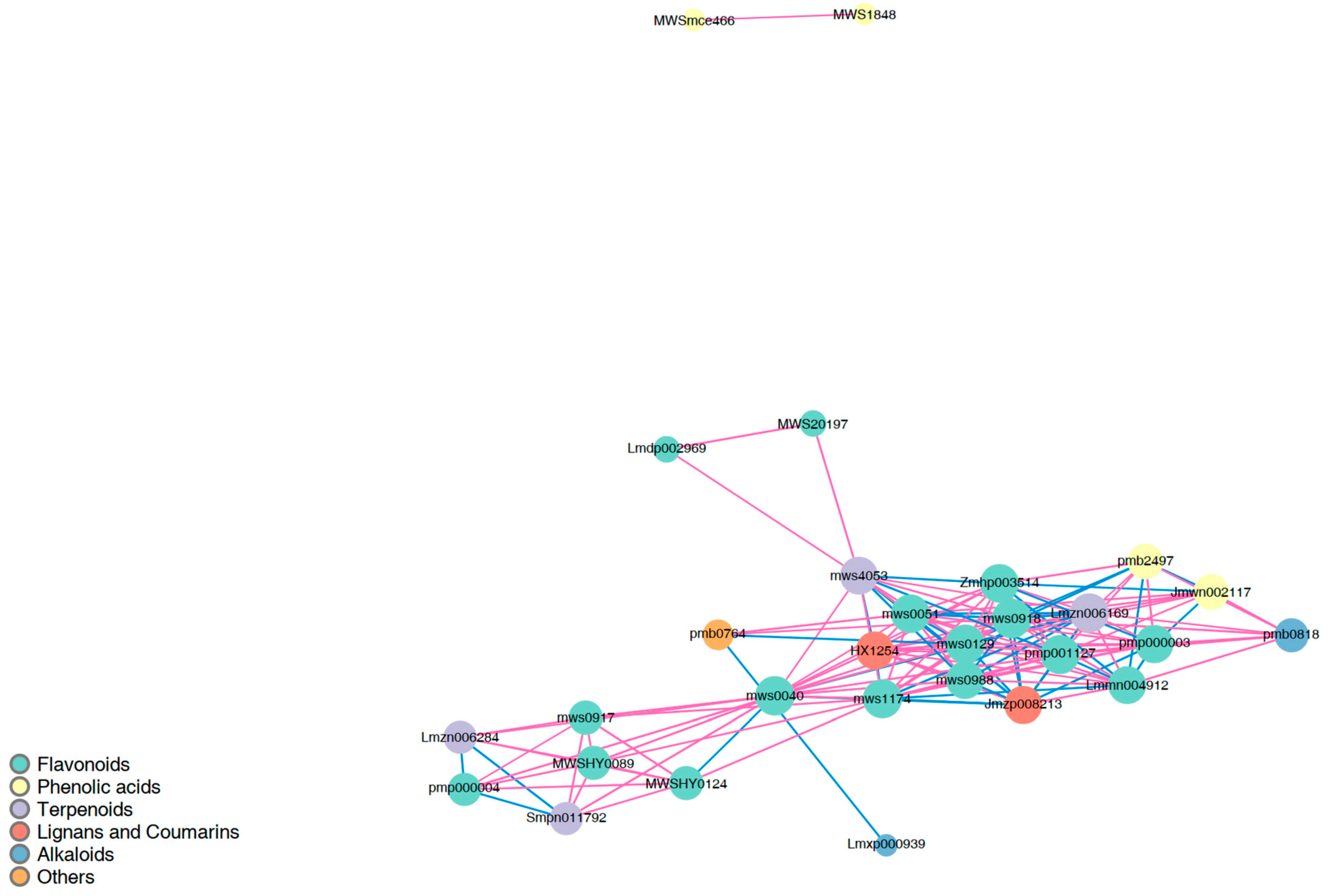


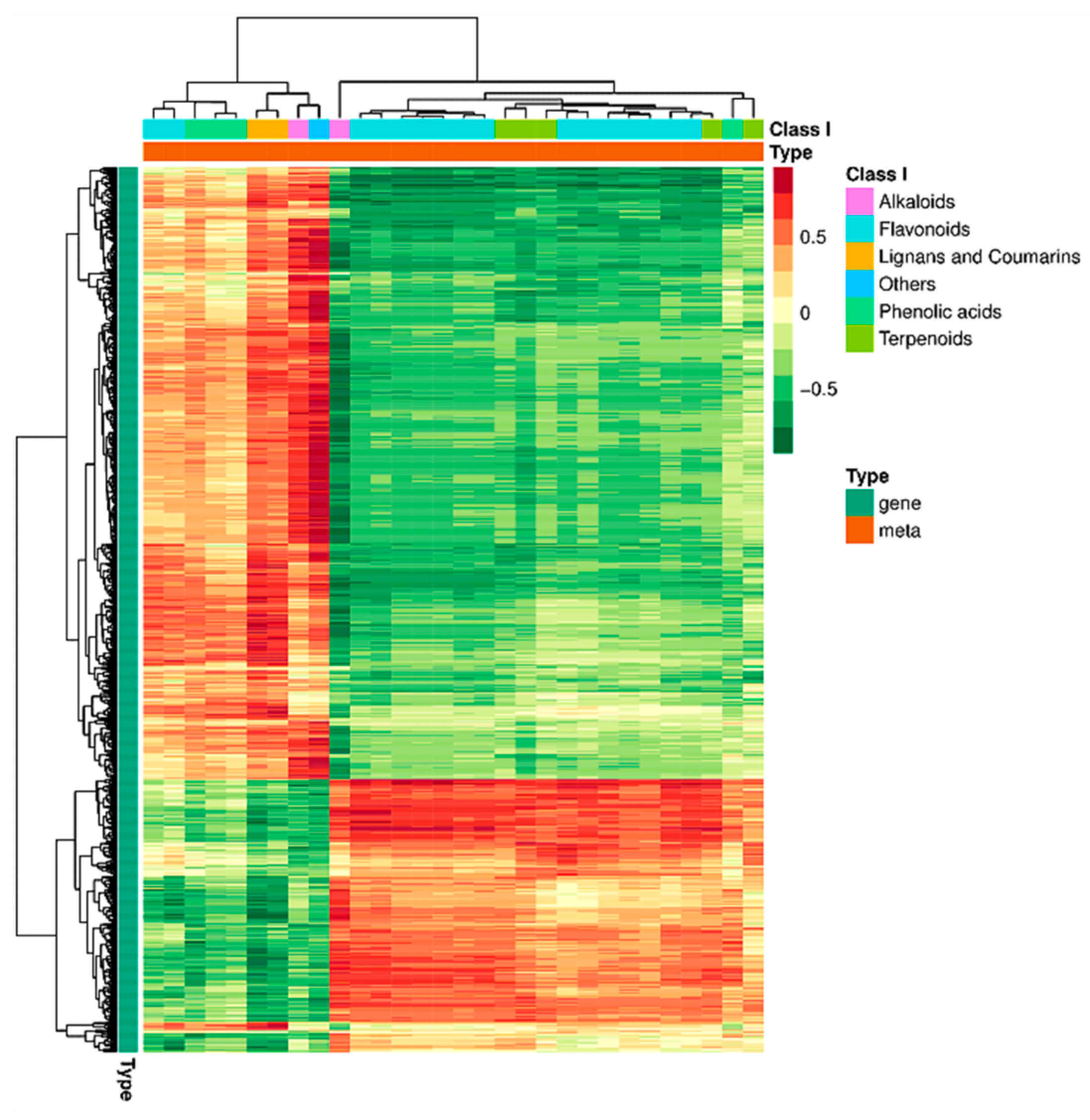
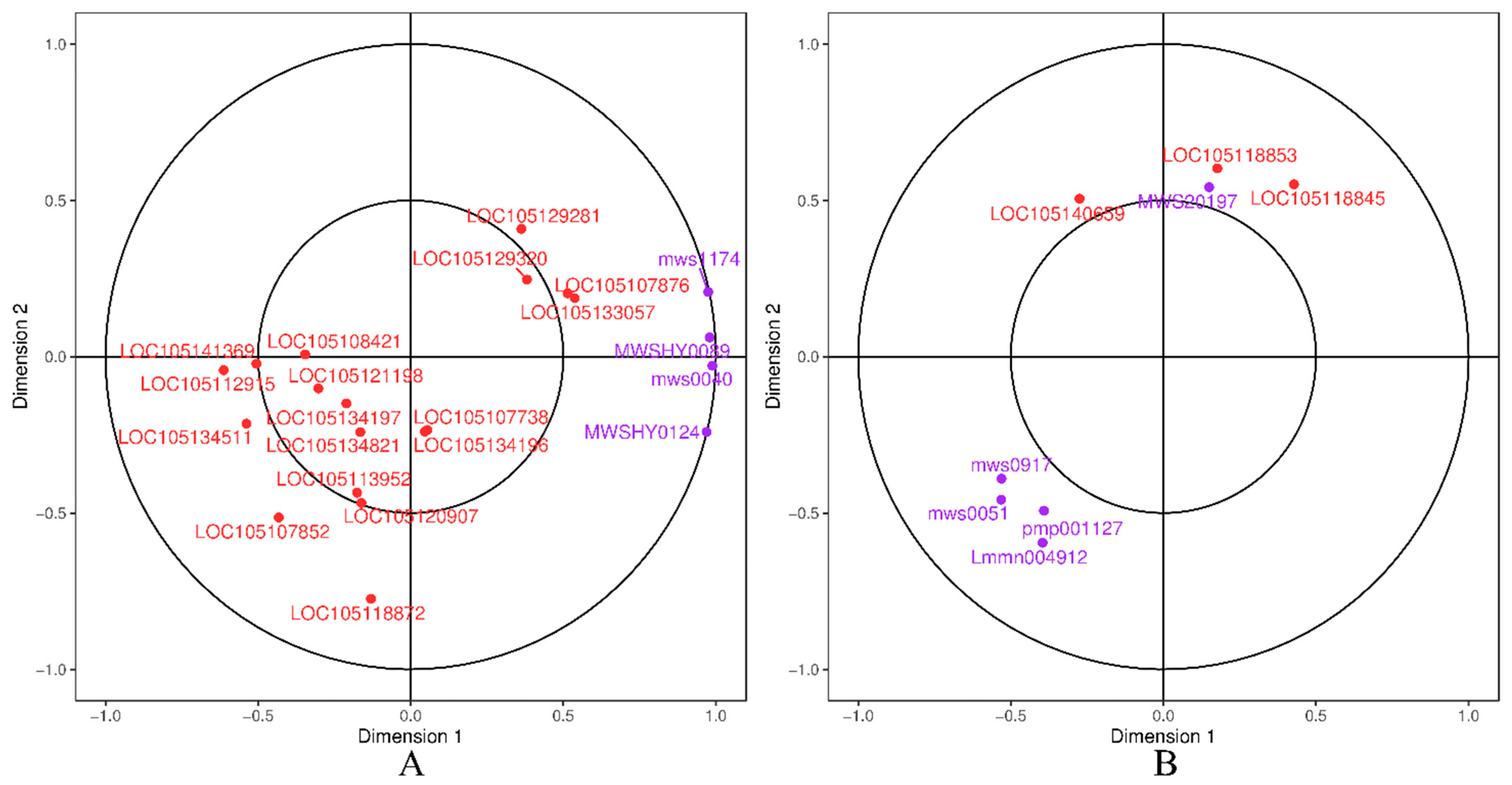
| Pathway | Gene ID | log2 Fold Change | Nr Functional Annotation |
|---|---|---|---|
| plant–pathogen interaction | LOC105111058 | 2.39 | PREDICTED: STS14 protein-like [P. euphratica] |
| LOC105127365 | 2.35 | PREDICTED: ethylene-responsive transcription factor CRF1 [P. euphratica] | |
| LOC105138044 | 2.11 | PREDICTED: probable LRR receptor-like serine/threonine-protein kinase At1g67720 [P. euphratica] | |
| LOC105132085 | 1.68 | PREDICTED: elongation factor Tu, mitochondrial [P. euphratica] | |
| LOC105138760 | 1.58 | PREDICTED: protein STRUBBELIG-RECEPTOR FAMILY 3 isoform X1 [P. euphratica] | |
| LOC105124176 | −5.13 | PREDICTED: probable LRR receptor-like serine/threonine-protein kinase At3g47570 [P. euphratica] | |
| LOC105124276 | −5.16 | PREDICTED: probable WRKY transcription factor 41 [P. euphratica] | |
| LOC105112185 | −5.43 | PREDICTED: receptor-like cytosolic serine/threonine-protein kinase RBK2 isoform X1 [P. euphratica] | |
| LOC105110565 | −6.22 | PREDICTED: calcium-binding protein CML38 [P. euphratica] | |
| LOC105135772 | −7.59 | PREDICTED: 3-ketoacyl-CoA synthase 1-like [P. euphratica] | |
| plant hormone signal transduction | LOC105141486 | 4.56 | PREDICTED: hypothetical protein POPTR_009G126000v3 [P. trichocarpa] |
| LOC105121906 | 3.88 | PREDICTED: abscisic acid receptor PYL4-like [P. euphratica] | |
| LOC105136244 | 2.47 | PREDICTED: probable carboxylesterase 8 [P. euphratica] | |
| LOC105139037 | 2.43 | PREDICTED: transcription activator GLK1-like isoform X1 [P. euphratica] | |
| LOC105119527 | 2.43 | PREDICTED: scarecrow-like protein 4 [P. euphratica] | |
| LOC105121293 | −4.68 | PREDICTED: probable xyloglucan endotransglucosylase/hydrolase protein 23 [P. euphratica] | |
| LOC105112185 | −5.43 | PREDICTED: receptor-like cytosolic serine/threonine-protein kinase RBK2 isoform X1 [P. euphratica] | |
| LOC105129715 | −5.70 | PREDICTED: leucine-rich repeat receptor protein kinase EXS-like [P. euphratica] | |
| LOC105121294 | −5.74 | PREDICTED: probable xyloglucan endotransglucosylase/hydrolase protein 23 [P. euphratica] | |
| LOC105141318 | −5.93 | PREDICTED: probably inactive leucine-rich repeat receptor-like protein kinase IMK2 [P. euphratica] | |
| MAPK signaling pathway—plant | LOC105140947 | 5.65 | PREDICTED: endochitinase WIN8 isoform X1 [P. euphratica] |
| LOC105121906 | 3.88 | PREDICTED: abscisic acid receptor PYL4-like [P. euphratica] | |
| LOC105125000 | 3.36 | PREDICTED: transcription factor bHLH94-like [P. euphratica] | |
| LOC105140224 | 3.00 | PREDICTED: acidic endochitinase WIN6 [P. euphratica] | |
| LOC105140204 | 2.66 | PREDICTED: acidic endochitinase WIN6-like [P. euphratica] | |
| LOC105131648 | −4.71 | PREDICTED: receptor-like protein kinase isoform X1 [P. euphratica] | |
| LOC105131478 | −5.07 | PREDICTED: probable WRKY transcription factor 33 [P. euphratica] | |
| LOC105124176 | −5.13 | PREDICTED: probable LRR receptor-like serine/threonine-protein kinase At3g47570 [P. euphratica] | |
| LOC105124276 | −5.16 | PREDICTED: probable WRKY transcription factor 41 [P. euphratica] | |
| LOC105112185 | −5.43 | PREDICTED: receptor-like cytosolic serine/threonine-protein kinase RBK2 isoform X1 [P. euphratica] |
| Transcription Factor | Number of DEGs | Up | Down | Transcription Factor | Number of DEGs | Up | Down |
|---|---|---|---|---|---|---|---|
| Alfin-like | 1 | 1 | 0 | IWS1 | 1 | 0 | 1 |
| AP2/ERF-AP2 | 2 | 2 | 0 | Jumonji | 4 | 0 | 4 |
| AP2/ERF-ERF | 62 | 4 | 58 | LOB | 7 | 3 | 4 |
| AP2/ERF-RAV | 2 | 0 | 2 | MADS-M-type | 1 | 0 | 1 |
| AUX/IAA | 6 | 3 | 3 | MADS-MIKC | 1 | 0 | 1 |
| B3 | 8 | 0 | 8 | MBF1 | 1 | 0 | 1 |
| B3-ARF | 3 | 2 | 1 | mTERF | 5 | 5 | 0 |
| bHLH | 26 | 7 | 19 | MYB | 33 | 12 | 21 |
| bZIP | 9 | 5 | 4 | MYB-related | 13 | 9 | 4 |
| C2C2-CO-like | 8 | 6 | 2 | NAC | 38 | 3 | 35 |
| C2C2-Dof | 8 | 8 | 0 | NF-YA | 3 | 2 | 1 |
| C2C2-GATA | 7 | 1 | 6 | NF-YB | 2 | 2 | 0 |
| C2H2 | 17 | 6 | 11 | NF-YC | 2 | 0 | 2 |
| C3H | 12 | 3 | 9 | Others | 14 | 4 | 10 |
| CAMTA | 4 | 0 | 4 | Pseudo ARR-B | 3 | 3 | 0 |
| CSD | 1 | 1 | 0 | SBP | 5 | 2 | 3 |
| DBB | 2 | 1 | 1 | SET | 4 | 4 | 0 |
| EIL | 1 | 0 | 1 | SNF2 | 3 | 0 | 3 |
| FAR1 | 1 | 1 | 0 | SWI/SNF-BAF60b | 1 | 0 | 1 |
| GARP-ARR-B | 2 | 1 | 1 | TAZ | 1 | 1 | 0 |
| GARP-G2-like | 8 | 6 | 2 | TCP | 3 | 2 | 1 |
| GNAT | 4 | 1 | 3 | Tify | 7 | 1 | 6 |
| GRAS | 22 | 9 | 13 | TRAF | 3 | 0 | 3 |
| GRF | 1 | 1 | 0 | Trihelix | 4 | 1 | 3 |
| HB-BELL | 1 | 0 | 1 | TUB | 1 | 0 | 1 |
| HB-HD-ZIP | 5 | 3 | 2 | WRKY | 45 | 5 | 40 |
| HB-WOX | 1 | 0 | 1 | zf-HD | 4 | 3 | 1 |
| HSF | 7 | 2 | 5 | / |
| Index | Class I | Compounds | Formula | Type |
|---|---|---|---|---|
| MWSHY0089 | Flavonoids | 5,4′-Dihydroxy-7-methoxyflavanone (Sakuranetin) | C16H14O5 | up |
| MWSHY0124 | Flavonoids | Pinocembrin (Dihydrochrysin) | C15H12O4 | up |
| Zmhp003514 | Flavonoids | 6,7,8-Tetrahydroxy-5-methoxyflavone | C16H12O6 | up |
| mws0988 | Flavonoids | Rhamnetin; 3,5,3′,4′-Tetrahydroxy-7-Methoxyflavone | C16H12O7 | up |
| mws0918 | Flavonoids | Prunetin (5,4′-Dihydroxy-7-methoxyisoflavone) | C16H12O5 | up |
| mws1174 | Flavonoids | 3-O-Acetylpinobanksin | C17H14O6 | up |
| Lmmn004912 | Flavonoids | 3-O-Methylquercetin | C16H12O7 | up |
| mws0129 | Flavonoids | Genkwanin (Apigenin 7-methyl ether) | C16H12O5 | up |
| mws0040 | Flavonoids | Chrysin | C15H10O4 | up |
| mws0051 | Flavonoids | Acacetin | C16H12O5 | up |
| pmp001127 | Flavonoids | Chrysoeriol; 5,7,4′-Trihydroxy-3′-Methoxyflavone | C16H12O6 | up |
| pmp000003 | Flavonoids | Nepetin (5,7,3′,4′-Tetrahydroxy-6-methoxyflavone) | C16H12O7 | up |
| mws0917 | Flavonoids | 3,7-Di-O-methylquercetin | C17H14O7 | up |
| pmp000004 | Flavonoids | 4′,5,7-Trihydroxy-3′,6-dimethoxyflavone (Jaceosidin) | C17H14O7 | up |
| MWS20197 | Flavonoids | Quercetin-3-O-rhamnoside (Quercitrin) | C21H20O11 | down |
| Lmdp002969 | Flavonoids | Myricetin-3-O-galactoside | C21H20O13 | down |
| mws4053 | Terpenoids | 3-Hydroxyurs-12-en-28-oic acid (Ursolic acid) | C30H48O3 | up |
| Smpn011792 | Terpenoids | 2,3-Dihydroxy-12-ursen-28-oic acid | C30H48O4 | up |
| Lmzn006169 | Terpenoids | 3,19-Dihydroxyurs-12-en-28-oic acid (Pomolic acid) | C30H48O4 | up |
| Wbmn009702 | Terpenoids | 2,3,23-Trihydroxyurs-12-en-28-oic acid | C30H48O5 | up |
| Lmzn006284 | Terpenoids | 2-Hydroxyursolic acid | C30H48O4 | up |
| pmb2497 | Phenolic acids | 4-Hydroxy-3-methoxymandelate | C9H10O5 | up |
| MWS1848 | Phenolic acids | Phenyl acetate | C8H8O2 | down |
| Jmwn002117 | Phenolic acids | 2-(3,4-dihydroxyphenyl)ethanediol 1-O-β-D-glucopyranoside | C14H20O9 | down |
| MWSmce466 | Phenolic acids | 4-Hydroxyacetophenone | C8H8O2 | down |
| pmb0818 | Alkaloids | Methoxyindoleacetic acid | C11H11NO3 | up |
| Lmxp000939 | Alkaloids | Zarzissine | C5H5N5 | down |
| HX1254 | Lignans and Coumarins | trans-1,2-dihydrodehydroguaiaretic acid | C20H22O4 | down |
| Jmzp008213 | Lignans and Coumarins | Dehydrodiisoeugenol | C20H22O4 | down |
| pmb0764 | Others | 4-Methyl-5-thiazoleethanol | C6H9NOS | down |
| Leaf Damage | |
|---|---|
| transcription factors | AP2/ERF, WRKY, MYB, NAC |
| DEGs | LOC105111058, LOC105125000, LOC105125713, LOC105132625, LOC105138044, LOC105129033, LOC105129145, LOC105135969, LOC105110708, LOC105142478 |
| DSMs | flavonoids, terpenoids, phenolic acids |
| metabolic pathways | flavone and flavonol biosynthesis, thiamine metabolism, flavonoid biosynthesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, M.; Han, Z.; Liu, Y.; Liu, M.; Guo, L.; Wu, J.; Wu, X. Transcriptome and Metabolome Analyses of Short-Term Responses of Populus talassica × Populus euphratica to Leaf Damage. Int. J. Mol. Sci. 2025, 26, 5869. https://doi.org/10.3390/ijms26125869
Su M, Han Z, Liu Y, Liu M, Guo L, Wu J, Wu X. Transcriptome and Metabolome Analyses of Short-Term Responses of Populus talassica × Populus euphratica to Leaf Damage. International Journal of Molecular Sciences. 2025; 26(12):5869. https://doi.org/10.3390/ijms26125869
Chicago/Turabian StyleSu, Mengxu, Zhanjiang Han, Ying Liu, Meilin Liu, Lu Guo, Jiaju Wu, and Xiaofeng Wu. 2025. "Transcriptome and Metabolome Analyses of Short-Term Responses of Populus talassica × Populus euphratica to Leaf Damage" International Journal of Molecular Sciences 26, no. 12: 5869. https://doi.org/10.3390/ijms26125869
APA StyleSu, M., Han, Z., Liu, Y., Liu, M., Guo, L., Wu, J., & Wu, X. (2025). Transcriptome and Metabolome Analyses of Short-Term Responses of Populus talassica × Populus euphratica to Leaf Damage. International Journal of Molecular Sciences, 26(12), 5869. https://doi.org/10.3390/ijms26125869