
Myostatin Modulation in Spinal Muscular Atrophy: A Systematic Review of Preclinical and Clinical Evidence

 ,
,  , ,
, ,  and
and
Abstract
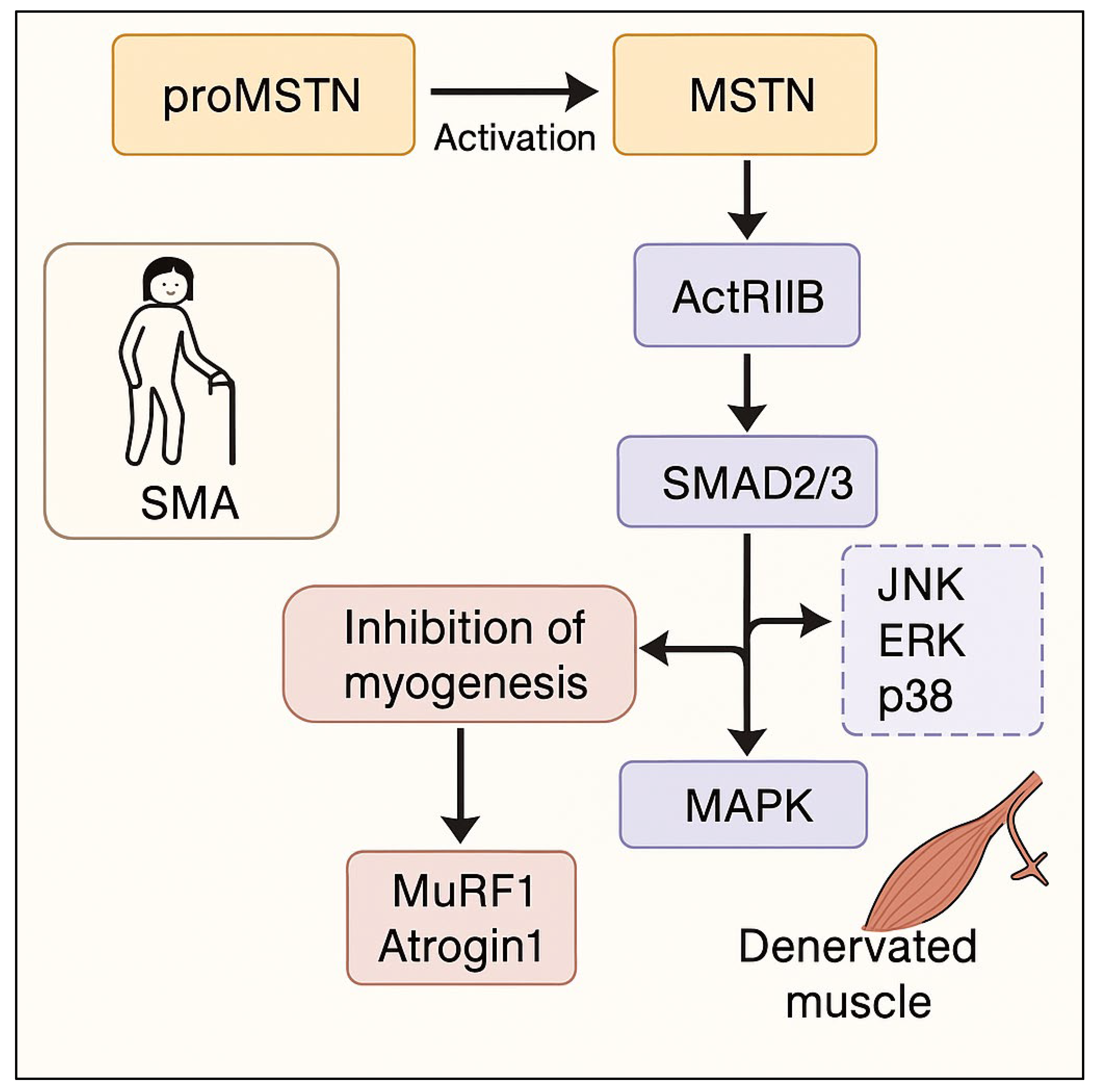
1. Introduction
2. Relevant Sections
2.1. Method
2.1.1. Search Strategy
2.1.2. Selection of the Studies
2.1.3. Data Extraction
2.1.4. Assessment of the Quality of Studies and Risk of Bias from the Review
2.2. Results
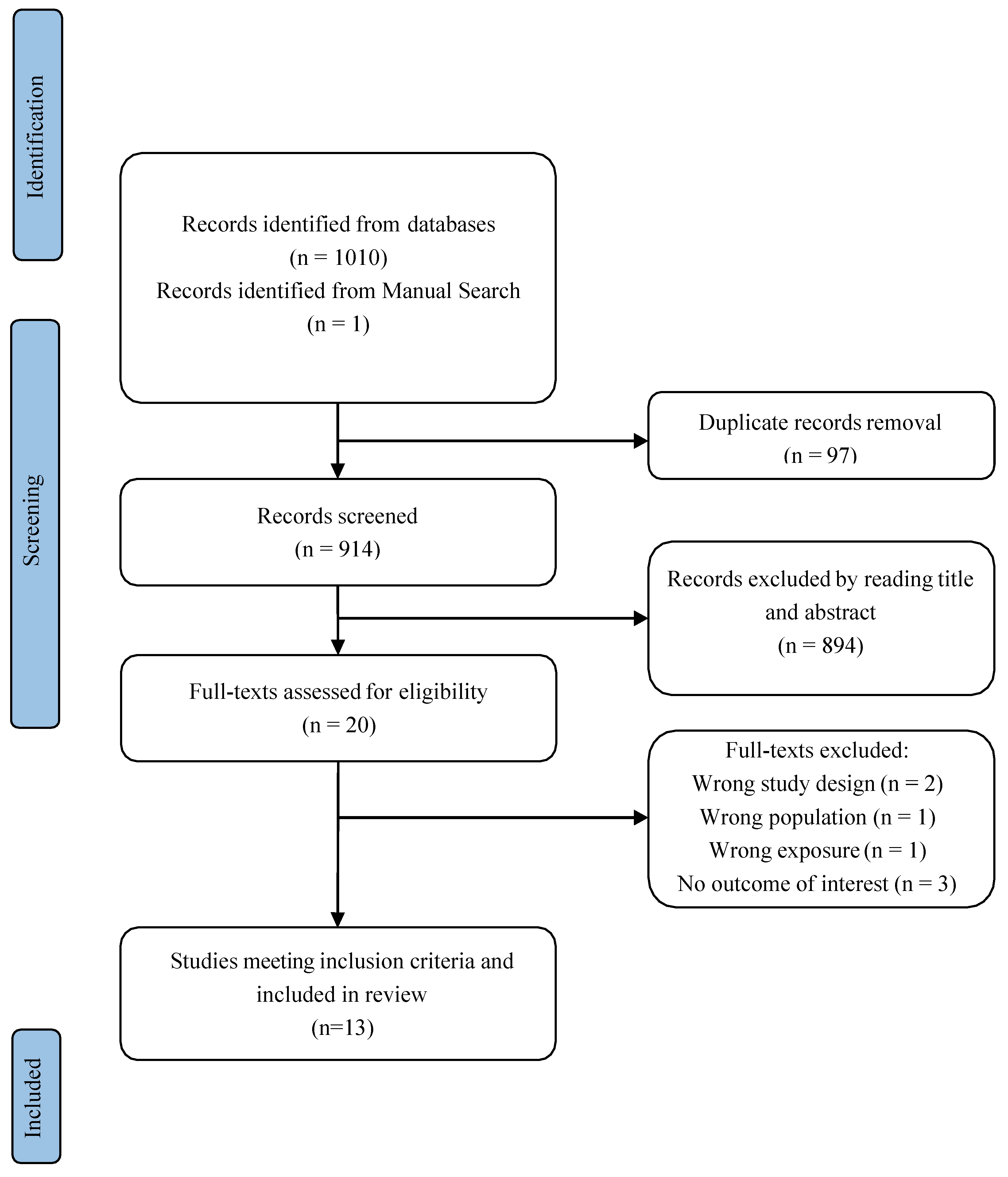
2.2.1. Study Selection
2.2.2. Preclinical and Clinical Evidence
3. Discussion
4. Conclusions
5. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Lee, S.-J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kambadur, R.; Matthews, K.G.; Somers, W.G.; Devlin, G.P.; Conaglen, J.V.; Fowke, P.J.; Bass, J.J. Myostatin, a transforming growth factor-β superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J. Cell. Physiol. 1999, 180, 1–9. [Google Scholar] [CrossRef]
- Kambadur, R.; Sharma, M.; Smith, T.P.; Bass, J.J. Mutations in myostatin (GDF8) in Double-Muscled Belgian Blue and Piedmontese Cattle. Genome Res. 1997, 7, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, A.; Bozzo, C.; Cacciani, N.; Caliaro, F.; Reggiani, C.; Mascarello, F.; Patruno, M. Effect of swimming on myostatin expression in white and red gastrocnemius muscle and in cardiac muscle of rats. Exp. Physiol. 2006, 91, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.S.; Quignon, P.; Bustamante, C.D.; Sutter, N.B.; Mellersh, C.S.; Parker, H.G.; A Ostrander, E.; Takahashi, J.S. A Mutation in the Myostatin Gene Increases Muscle Mass and Enhances Racing Performance in Heterozygote Dogs. PLoS Genet. 2007, 3, e79. [Google Scholar] [CrossRef] [PubMed]
- Clop, A.; Marcq, F.; Takeda, H.; Pirottin, D.; Tordoir, X.; Bibé, B.; Bouix, J.; Caiment, F.; Elsen, J.-M.; Eychenne, F.; et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006, 38, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.-J. Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Davies, M.V.; Koniaris, L.G.; Haynes, P.; Esquela, A.F.; Tomkinson, K.N.; McPherron, A.C.; Wolfman, N.M.; Lee, S.-J. Induction of Cachexia in Mice by Systemically Administered Myostatin. Science 2002, 296, 1486–1488. [Google Scholar] [CrossRef]
- Jiang, M.-S.; Liang, L.-F.; Wang, S.; Ratovitski, T.; Holmstrom, J.; Barker, C.; Stotish, R. Characterization and identification of the inhibitory domain of GDF-8 propeptide. Biochem. Biophys. Res. Commun. 2004, 315, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.R.; Lee, K. INVITED REVIEW: Inhibitors of myostatin as methods of enhancing muscle growth and development1. J. Anim. Sci. 2016, 94, 3125–3134. [Google Scholar] [CrossRef] [PubMed]
- Cotton, T.R.; Fischer, G.; Wang, X.; McCoy, J.C.; Czepnik, M.; Thompson, T.B.; Hyvönen, M. Structure of the human myostatin precursor and determinants of growth factor latency. EMBO J. 2018, 37, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Rebbapragada, A.; Benchabane, H.; Wrana, J.L.; Celeste, A.J.; Attisano, L. Myostatin Signals through a Transforming Growth Factor β-Like Signaling Pathway to Block Adipogenesis. Mol. Cell. Biol. 2003, 23, 7230–7242. [Google Scholar] [CrossRef]
- Wolfman, N.M.; McPherron, A.C.; Pappano, W.N.; Davies, M.V.; Song, K.; Tomkinson, K.N.; Wright, J.F.; Zhao, L.; Sebald, S.M.; Greenspan, D.S.; et al. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc. Natl. Acad. Sci. USA 2003, 100, 15842–15846. [Google Scholar] [CrossRef]
- McFarlane, C.; Hui, G.Z.; Amanda, W.Z.W.; Lau, H.Y.; Lokireddy, S.; XiaoJia, G.; Mouly, V.; Butler-Browne, G.; Gluckman, P.D.; Sharma, M.; et al. Human myostatin negatively regulates human myoblast growth and differentiation. Am. J. Physiol. Physiol. 2011, 301, C195–C203. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Vernus, B.; Chelh, I.; Cassar-Malek, I.; Gabillard, J.C.; Sassi, A.H.; Seiliez, I.; Picard, B.; Bonnieu, A. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell. Mol. Life Sci. 2014, 71, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, Y.; Diedrich, D.A.; Li, J.; Luo, W.; Zhao, X.; Guo, Y.; Luo, Y.; Zhang, T.; Wang, X.; et al. A horizontal and perpendicular interlaminar approach for intrathecal nusinersen injection in patients with spinal muscular atrophy and scoliosis: An observational study. Orphanet J. Rare Dis. 2024, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Qaisar, R.; Bose, B.; Sudheer, S.P. The elusive role of myostatin signaling for muscle regeneration and maintenance of muscle and bone homeostasis. Osteoporos. Sarcopenia 2023, 9, 1–7. [Google Scholar] [CrossRef]
- Philip, B.; Lu, Z.; Gao, Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell. Signal. 2004, 17, 365–375. [Google Scholar] [CrossRef]
- Wetzlich, B.; Nyakundi, B.B.; Yang, J. Therapeutic applications and challenges in myostatin inhibition for enhanced skeletal muscle mass and functions. Mol. Cell. Biochem. 2024, 480, 1535–1553. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef]
- Crawford, T.O.; Darras, B.T.; Day, J.W.; Young, S.D.; Duong, T.; Nelson, L.L.; Barrett, D.; Song, G.; Bilic, S.; Cote, S.; et al. Safety and Efficacy of Apitegromab in Patients with Spinal Muscular Atrophy Types 2 and 3. Neurology 2024, 102, e209151. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Day, J.W.; De Vivo, D.C.; Krueger, J.M.; Mercuri, E.; Nascimento, A.; Pasternak, A.; Mazzone, E.S.; Duong, T.; Song, G.; et al. Long-term efficacy, safety, and patient-reported outcomes of apitegromab in patients with spinal muscular atrophy: Results from the 36-month TOPAZ study. Front. Neurol. 2024, 15, 1419791. [Google Scholar] [CrossRef]
- Muramatsu, H.; Kuramochi, T.; Katada, H.; Ueyama, A.; Ruike, Y.; Ohmine, K.; Shida-Kawazoe, M.; Miyano-Nishizawa, R.; Shimizu, Y.; Okuda, M.; et al. Novel myostatin-specific antibody enhances muscle strength in muscle disease models. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Lair, L.L.; Connolly, A.M.; Byrne, B.J.; Chen, K.S.; Coric, V.; Qureshi, I.; Durham, S.; Campbell, D.J.; Maclaine, G.; et al. Taldefgrobep Alfa and the Phase 3 RESILIENT Trial in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2024, 25, 10273. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. Declaración PRISMA 2020: Una guía actualizada para la publicación de revisiones sistemáticas. Rev. Espanola De Cardiol. 2021, 74, 790–799. [Google Scholar] [CrossRef]
- Barrett, D.; Bilic, S.; Chyung, Y.; Cote, S.M.; Iarrobino, R.; Kacena, K.; Kalra, A.; Long, K.; Nomikos, G.; Place, A.; et al. A Randomized Phase 1 Safety, Pharmacokinetic and Pharmacodynamic Study of the Novel Myostatin Inhibitor Apitegromab (SRK-015): A Potential Treatment for Spinal Muscular Atrophy. Adv. Ther. 2021, 38, 3203–3222. [Google Scholar] [CrossRef] [PubMed]
- de Albuquerque, A.L.A.; Chadanowicz, J.K.; Giudicelli, G.C.; Staub, A.L.P.; Weber, A.C.; Silva, J.M.D.S.; Becker, M.M.; Kowalski, T.W.; Siebert, M.; Saute, J.A.M. Serum myostatin as a candidate disease severity and progression biomarker of spinal muscular atrophy. Brain Commun. 2024, 6, fcae062. [Google Scholar] [CrossRef]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F.; et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef]
- Liu, M.; Hammers, D.W.; Barton, E.R.; Sweeney, H.L.; Singh, R.N. Activin Receptor Type IIB Inhibition Improves Muscle Phenotype and Function in a Mouse Model of Spinal Muscular Atrophy. PLoS ONE 2016, 11, e0166803. [Google Scholar] [CrossRef]
- Long, K.K.; O’sHea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef]
- Mackels, L.; Mariot, V.; Buscemi, L.; Servais, L.; Dumonceaux, J. Impact of Disease Severity and Disease-Modifying Therapies on Myostatin Levels in SMA Patients. Int. J. Mol. Sci. 2024, 25, 8763. [Google Scholar] [CrossRef] [PubMed]
- Piemonte, F.; Petrillo, S.; Capasso, A.; Coratti, G.; D'AMico, A.; Catteruccia, M.; Pera, M.C.; Palermo, C.; Pane, M.; Abiusi, E.; et al. Myostatin Levels in SMA Following Disease-Modifying Treatments: A Multi-Center Study. Ann. Clin. Transl. Neurol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Rindt, H.; Buckley, D.M.; Vale, S.M.; Krogman, M.; Rose, F.F.; Garcia, M.L.; Lorson, C.L. Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy. Neuromuscul. Disord. 2012, 22, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Rose, F.F.; Mattis, V.B.; Rindt, H.; Lorson, C.L. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2009, 18, 997–1005. [Google Scholar] [CrossRef]
- Welsh, B.T.; Cote, S.M.; Meshulam, D.; Jackson, J.; Pal, A.; Lansita, J.; Kalra, A. Preclinical Safety Assessment and Toxicokinetics of Apitegromab, an Antibody Targeting Proforms of Myostatin for the Treatment of Muscle-Atrophying Disease. Int. J. Toxicol. 2021, 40, 322–336. [Google Scholar] [CrossRef]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachex- Sarcopenia Muscle 2020, 11, 768–782. [Google Scholar] [CrossRef]
- Vandenbroucke, J.P.; von Elm, E.; Altman, D.G.; Gøtzsche, P.C.; Mulrow, C.D.; Pocock, S.J.; Poole, C.; Schlesselman, J.J.; Egger, M.; STROBE Initiative. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): Explanation and elaboration. Ann. Intern. Med. 2007, 147, 8. [Google Scholar] [CrossRef]
- Whiting, P.; Savović, J.; Higgins, J.P.T.; Caldwell, D.M.; Reeves, B.C.; Shea, B.; Davies, P.; Kleijnen, J.; Churchill, R.; ROBIS group. ROBIS: A new tool to assess risk of bias in systematic reviews was developed. J. Clin. Epidemiol. 2016, 69, 225–234. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Author(s), Year | Country | Study Design | Population Type | SMA Type |
|---|---|---|---|---|
| Barrett et al., 2021 [26] | USA | Phase 1, randomized, double-blind, placebo-controlled, sequential cohort, two-part, single ascending dose (SAD) and multiple ascending dose (MAD) study. | Healthy adult subjects. | No SMA patients were involved in this study. It aimed to gather safety data to support future trials in SMA patients. |
| Crawford et al., 2024 [21] | USA | Phase 2 TOPAZ clinical trial. | Human cases. | Types 2 and 3 SMA. |
| de Albuquerque et al., 2024 [27] | Brazil | Clinical study (cross-sectional case-control followed by 12-month cohort) alongside a bioinformatic study. | Clinical study involved 27 SMA patients and 27 healthy controls. Bioinformatic study involved data from mice, and two studies with humans. | SMA cases were 5q SMA, types 1, 2, and 3. |
| Feng et al., 2016 [28] | USA | Experimental study. | A7 mice were treated suboptimally with SMN-C3, then subjected to follistatin treatment, in an effort to rescue phenotypes. | This pharmacologically induced model displays features reminiscent of human SMA but presents a unique scenario, enabling a broader examination of what happens. The features of this model indicate a less severe phenotype that shows the pathological responses of human 2 and 3 types. |
| Liu et al., 2016 [29] | USA | Experimental animal study. | Male C/C SMA model mice. | Type 3 SMA. |
| Long et al., 2019 [30] | USA | Experimental study. | Used A7 (SMNA7) mouse models. | NA |
| Mackels et al., 2024 [31] | Belgium and the United Kingdom | Retrospective study. | SMA patients. | Thirteen patients with SMA 1, six patients with SMA 2, and six patients with SMA 3. |
| Piemonte et al., 2025 [32] | Italy (Multi-center study) | Prospective, multi-center study incorporating cross-sectional and longitudinal components. | Both symptomatic SMA patients receiving DMTs and presymptomatic patients identified through neonatal screening. | Types 2, 2, 3, and presymptomatic. |
| Rindt et al., 2012 [33] | USA | Experimental model construction and analysis. | Smn−/−; Mstn+/+ Smn−/−; Mstn−/− (or Smn-KO; Mstn-WT) and (Smn- KO; Mstn-KO). | A7 SMA model by genetically invaliding MSTN in conjunction. |
| Rose Jr et al., 2009 [34] | USA | Experimental study. | hSMN2+/+; hSMN∆7+/+; Smn−/− mice. | NA |
| Servais et al., 2024 [24] | Belgium, UK, USA | This manuscript reviews the role of myostatin in muscle, explores the preclinical and clinical development of taldefgrobep and introduces the phase 3 RESILIENT trial of taldefgrobep in SMA. | Patients with SMA. | NA |
| Welsh et al., 2021 [35] | USA | Preclinical toxicology and toxicokinetic (TK) studies. | Cynomolgus monkeys (adult); Sprague Dawley rats (adult and juvenile). | NA |
| Zhou et al., 2020 [36] | London UK | Experimental animal study, with longitudinal assessment of treated data. | 47 mouse models which were constructed from genetic variants to mimic some traits in human SMN. | Severe SMA. |
| Author(s), Year | SMA Type | Treatment Route and Dosage | Key Molecular Findings | Functional Outcomes | Histological Outcomes |
|---|---|---|---|---|---|
| Barrett et al., 2021 [26] | NA | Intravenous (IV) infusion Part A (SAD): Single ascending doses of apitegromab: 1, 3, 10, 20, 30 mg/kg or placebo. Part B (MAD): Multiple ascending doses of apitegromab: 10, 20, 30 mg/kg or placebo every 2 weeks for a total of three doses. | Dose-dependent and sustained increases in serum latent myostatin were seen. | Apitegromab was safe and well-tolerated. No clinically meaningful changes in baseline vital signs, electrocardiograms, or clinical laboratory parameters and no anti-drug antibody formation. | NA |
| Crawford et al., 2024 [21] | Types 2 and 3 SMA. | Apitegrob was injected by intravenous treatment every 4 weeks for 12 months. | The elevated target measure also demonstrated quick reached in a dose | Improved effect. | NA |
| de Albuquerque et al., 2024 [27] | SMA cases were 5q SMA, types 1, 2, and 3. | Data from treated and un-treated individuals analyzed, although treatment with Nusinersen occurred. | Bioinformatic Study: Skeletal muscle gene expression of Mstn decreased and of Fst increased. Clinical Studies: Serum myostatin levels show promise as a novel biomarker for evaluating the severity and progression of spinal muscular atrophy. | A correlation of negative association with clinical severity (CHOP INTEND HFMSE and RULM) was found. | NA |
| Feng et al., 2016 [28] | This pharmacologically induced model displays features reminiscent of human SMA but presents a unique scenario, enabling a broader examination of what happens. The features of this model indicate a less severe phenotype that shows the pathological responses of human 2 and 3 types. | SMN-C3: Used to initially generate symptomatic adult A7 mice—a suboptimal dose was used to allow survival to adulthood. Regimens reported a greater rescue effect with the higher concentrations during the SMN treatment. After initial findings SMN was stabilized and Recombinant AAV1-follistatin (FS344) then was administered with a dose with PND4 at 5 × 1011 Viral particles. | After SMN modification, levels of functional full-length SMN2 were elevated and the treatment was sustained. | Our findings illustrate that this combination improves mean lifespan of transgenic SMA through some sort of rescue mechanism. | Improved or restored synapse and NMJ function as well as an increase in myofiber cross-sectional area. |
| Liu et al., 2016 [29] | Type 3 SMA. | The 47 mice “were treated intraperitoneally with ~1012 genome copies” either via extra-cellular domain protein or w/protease. | Elevated levels of SMAD3; increased CDKN1A. | A7 increased “all limb muscle that is the SMAC or 55”. | NA |
| Long et al., 2019 [30] | NA | SMN2 modifier SMN-C1 added at PND1 w/IP. Dosage depends on whether it was low or high. Once the mice reached the high-dose concentration, an intermuscular injection of murSRK-015P was given. This concentration and route of action was repeated for 4 weeks. | After SRK-015 Treatment vGLUT1 synapses levels as baseline non-SMA Control, levels of “myostatin are directly linked to activity”. | When administered to a low-high SMN model, “that the Low-High treatment results in an increase of Synaptic inputs” and increased VHC function as “evidenced by positive trophic factors”. | This report suggests that multiple approaches towards function could be used or targeted to enhance SMN with specific functions in an independent and effective manner. |
| Mackels et al., 2024 [31] | Thirteen patients with SMA 1, six patients with SMA 2, and six patients with SMA 3. | Intrathecal injection. Doses depended on individual, as research focuses on how levels are altered after specific doses. Data were collected prior to treatment and after 2, 6, 10, 18, and 30 months of treatment. | This found an inverse relation (or that there was decreased volume in key chemicals, as well as their expression). | Lower myostatin levels (meaning more SMN effectiveness) with 32-item Motor Function Measure (MFM32) (n = 12, rho = 0.83, 95% CI [0.31; 0.99], p < 0.001). | NA |
| Piemonte et al., 2025 [32] | Types 1, 2, 3, and presymptomatic. | Not specified, but mentioned available DMTs (Nusinersen, Risdiplam, Onasemnogene abeparvovec). | Baseline myostatin levels significantly lower in SMA patients compared to controls. Significant difference in myostatin levels according to functional status in the whole cohort and also across all SMA types. Changes in myostatin in pre-symptomatic before and after treatment. | Significant difference between HFMSE and myostatin levels. Significant difference between CHOP INTEND and myostatin levels. | NA |
| Rindt et al., 2012 [33] | A7 SMA model by genetically invaliding MSTN in conjunction. | NA | NA | NA | These can be used to identify areas where muscle damage could be detected, and as stated earlier there was little change noted with the process. |
| Rose Jr et al., 2009 [34] | NA | Recombinant human follistatin administered via intraperitoneal injection. | SMN protein levels in spinal cord and muscle were unchanged in follistatin-treated SMA mice compared with vehicle-treated controls. This indicates an SMN-independent mechanism of action. Plastin-3 levels in spinal cord were similar in follistatin-treated, PBS-treated, and wild-type mice. | Treated animals had improved motor function compared to controls by testing righting reflex, and an increase in mean lifespan by ~30% which equated to 4.6 days. | Ventral horn cell (VHC) number: increased number of VHCs as a result of administration; VHC sections were slightly greater that those of untreated. |
| Servais et al., 2024 [24] | NA | Taldefgrobep | This study demonstrates little effects of the compound itself, and highlights the complex nature of patients, SMN variants, and function for all treated participants | NA | NA |
| Welsh et al., 2021 [35] | NA | IV bolus, once weekly. Cynomolgus monkeys: 0, 10, 30, 100 mg/kg; SD rats (adult and juvenile): 0, 30, 100, 300 mg/kg | Confirmed target engagement by measuring serum latent myostatin levels; baseline levels of latent myostatin ranged from approximately 20 to 100 ng/mL. Apitegromab exposure led to higher levels of latent myostatin across multiple timepoints in a dose-dependent manner. | Apitegromab-related increase in muscle weights, ranging to a mean of 3–32% over controls. | Minimal to slight hypertrophy of muscle fibers (limited to right/left biceps brachii), consistent with increased muscle weights. |
| Zhou et al., 2020 [36] | Severe SMA. | PM025 which acts as an SMN and AAV-MPRO, with subcutaneous injections where both were set between 40 g/g and around a baseline expression. | We can look at full-length SMN transcript, what effects those had in expression and at what rate it occurred. Results from data where “Mstn mRNA level was 80% in skeletal”. | After tests concerning motor skill and function, these areas showed “two-fold increase in time for righting-reflex-skill” also improved with “38% growth of muscle mass”. | After several dissections and “fiber areas” for muscles and neuronal structure results came in with: “a 50% increase” to the neuronal structure’s function, |
| Outcome | Preclinical Studies | Clinical Studies |
|---|---|---|
| % Increases in muscle mass | Up to +38% muscle mass increase in mouse models (Zhou et al., 2020) [36]; +3–32% in primate and rodent models (Welsh et al., 2021) [35]. | Muscle mass increase not directly quantified; implied via improved strength and motor scores (TOPAZ study). |
| Motor function improvement | Improved righting reflex and lifespan (+4.6 days) in SMA mice (Rose et al., 2009) [34]; enhanced synaptic inputs (Long et al., 2019) [30]. | HFMSE improvement of +3.6 points at 12 months, sustained at 36 months (Crawford et al., 2024 [21]; TOPAZ trial). |
| NMJ Architecture/Function | Enhanced NMJ integrity and vGLUT1 synapse density (Long et al., 2019) [30]; increased cross-sectional area of myofibers (Feng et al., 2016 [28]). | NMJ structure not assessed directly; clinical improvements observed, but structural endpoints not reported. |
| Histological/Structural outcomes | Muscle fiber hypertrophy and preserved motor neurons (Rose et al., 2009 [34]; Feng et al., 2016 [28]). | Structural outcomes not evaluated; no histological data available. |
| Serum myostatin modulation | Reduction in Mstn mRNA and increase in Fst in muscle (de Albuquerque et al., 2024 [27]). | Dose-dependent increase in serum latent myostatin after Apitegromab (Barrett et al., 2021 [26]; Mackels et al., 2024 [31]). |
| Duration and persistence of effect | Short-term survival benefit and structural rescue in animal models (Rose et al., 2009 [34]; Zhou et al., 2020 [36]). | Motor gains sustained over 36 months in SMA types 2 and 3 (Crawford et al., 2024 [21]). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnazzo, M.; Pisanò, G.; Baldini, V.; Giacomelli, G.; Scullin, S.; Piccolo, B.; Turco, E.C.; Esposito, S.; Pera, M.C. Myostatin Modulation in Spinal Muscular Atrophy: A Systematic Review of Preclinical and Clinical Evidence. Int. J. Mol. Sci. 2025, 26, 5858. https://doi.org/10.3390/ijms26125858
Gnazzo M, Pisanò G, Baldini V, Giacomelli G, Scullin S, Piccolo B, Turco EC, Esposito S, Pera MC. Myostatin Modulation in Spinal Muscular Atrophy: A Systematic Review of Preclinical and Clinical Evidence. International Journal of Molecular Sciences. 2025; 26(12):5858. https://doi.org/10.3390/ijms26125858
Chicago/Turabian StyleGnazzo, Martina, Giulia Pisanò, Valentina Baldini, Giovanna Giacomelli, Silvia Scullin, Benedetta Piccolo, Emanuela Claudia Turco, Susanna Esposito, and Maria Carmela Pera. 2025. "Myostatin Modulation in Spinal Muscular Atrophy: A Systematic Review of Preclinical and Clinical Evidence" International Journal of Molecular Sciences 26, no. 12: 5858. https://doi.org/10.3390/ijms26125858
APA StyleGnazzo, M., Pisanò, G., Baldini, V., Giacomelli, G., Scullin, S., Piccolo, B., Turco, E. C., Esposito, S., & Pera, M. C. (2025). Myostatin Modulation in Spinal Muscular Atrophy: A Systematic Review of Preclinical and Clinical Evidence. International Journal of Molecular Sciences, 26(12), 5858. https://doi.org/10.3390/ijms26125858