
Refined Procedure to Purify and Sequence Circulating Cell-Free DNA in Prostate Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
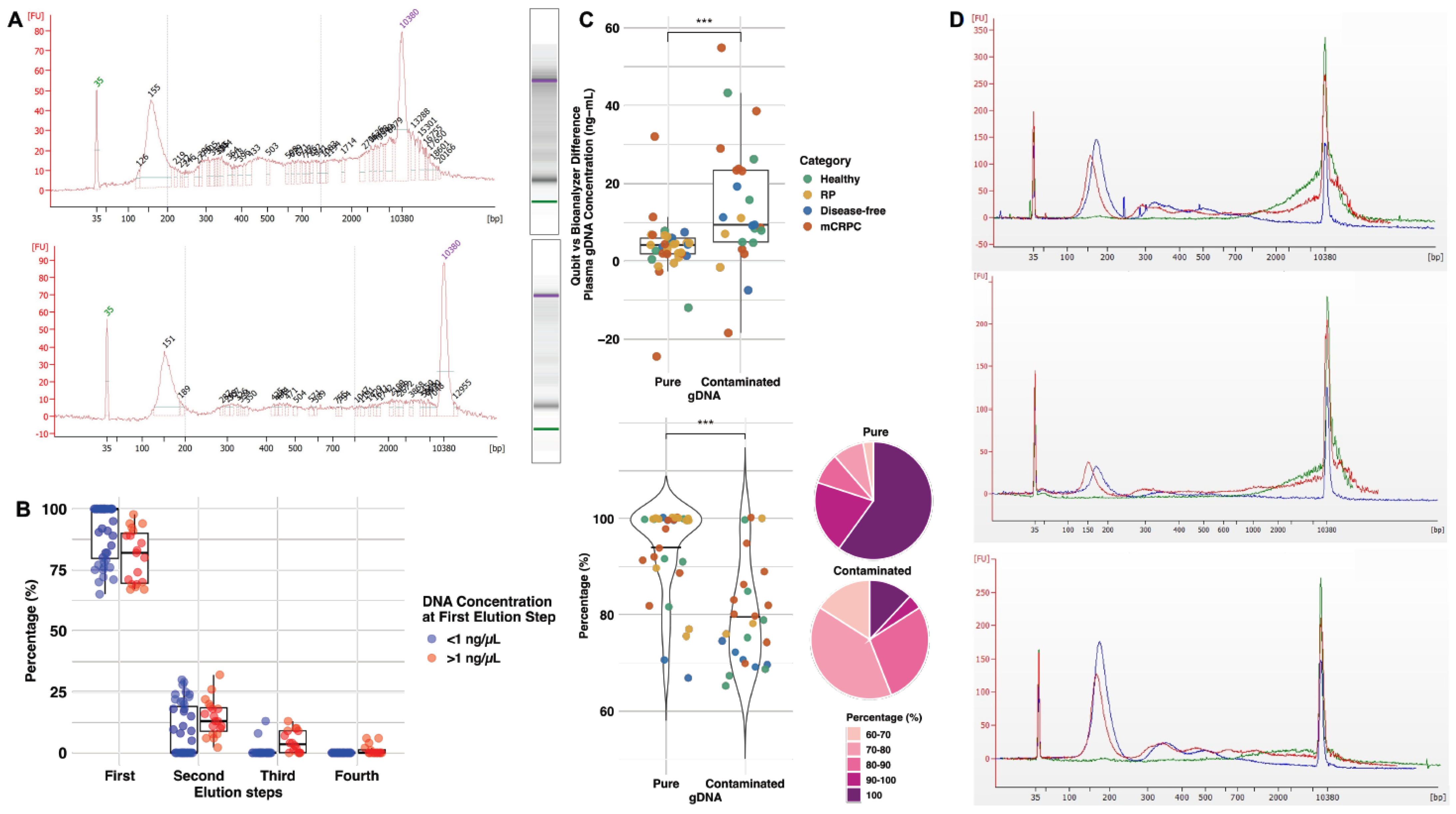
2.1. Optimization of cfDNA Purification Improves cfDNA Quality and Yield Using Less Plasma
2.2. The Recovery of High-Purity cfDNA Reaches over 90%
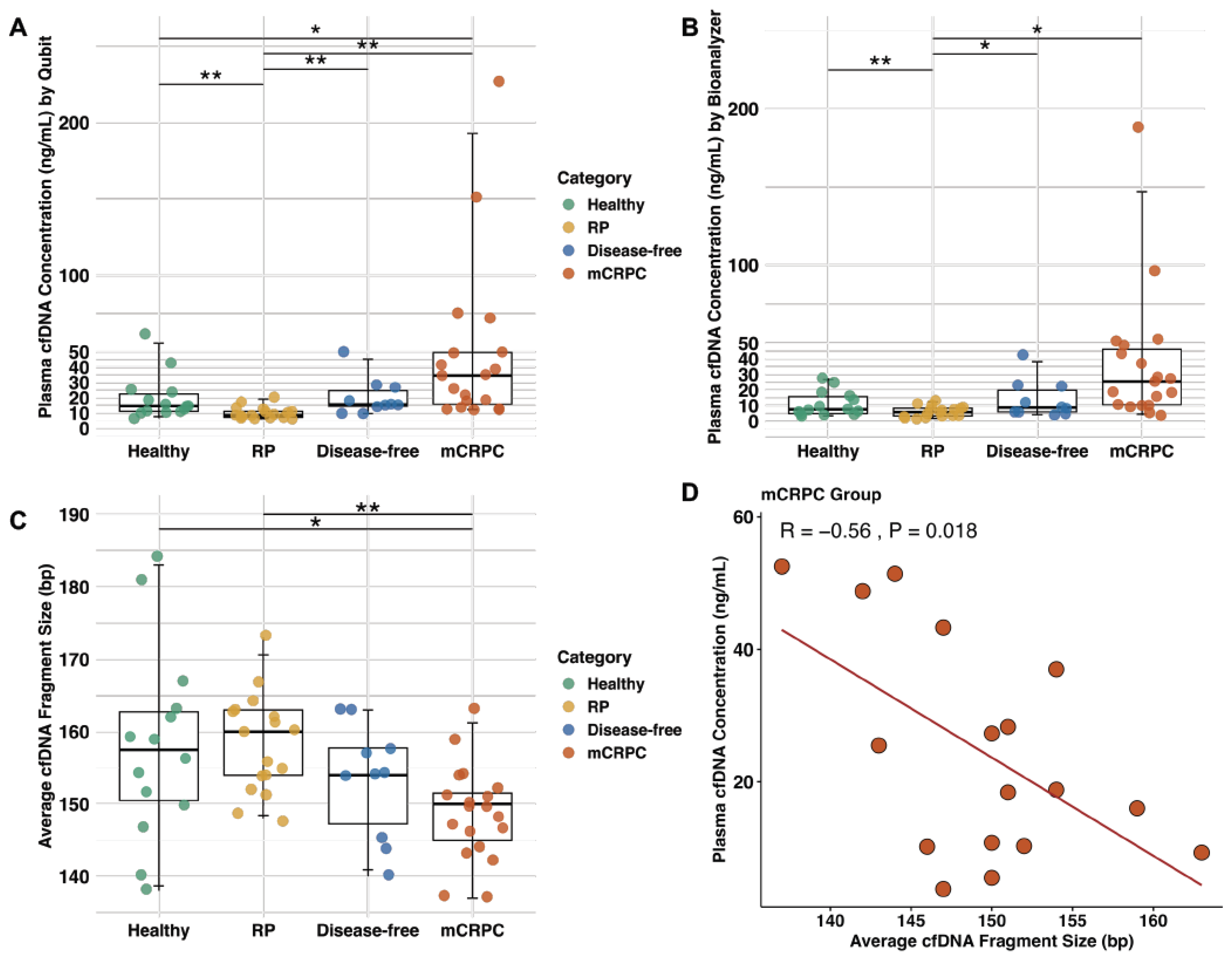
2.3. The Plasma of mCRPC Patients Contains Elevated cfDNA Levels Compared to Other Categories of Patients and Healthy Males
2.4. The cfDNA Fragments from mCRPC Patients Are Smaller than Other Patient Categories and Healthy Males
2.5. Exploring cfDNA Concentration of Patients with Their Clinical Features
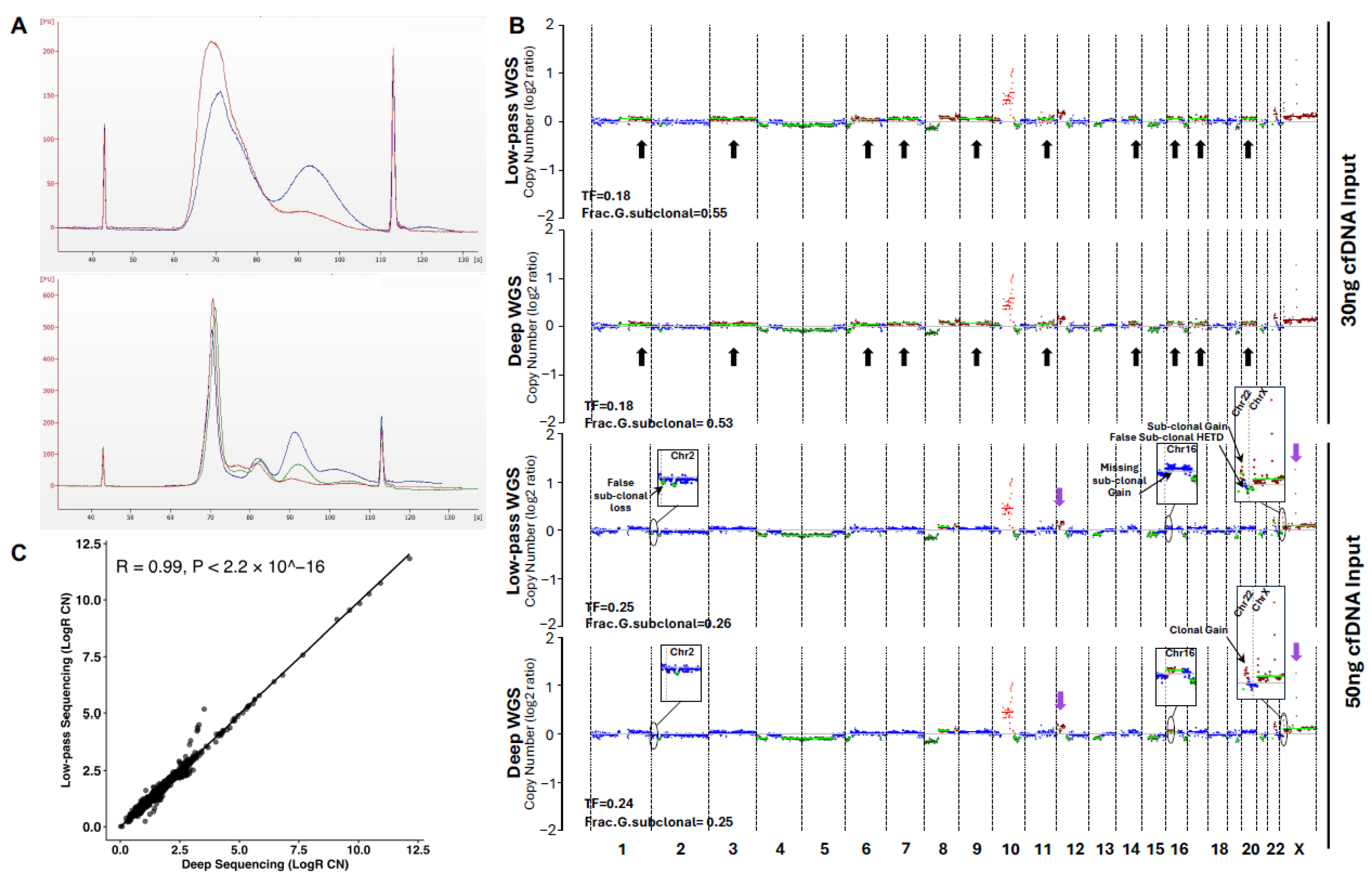
2.6. Further Protocol Refinements Lead to Highly Reliable Sequencing Data
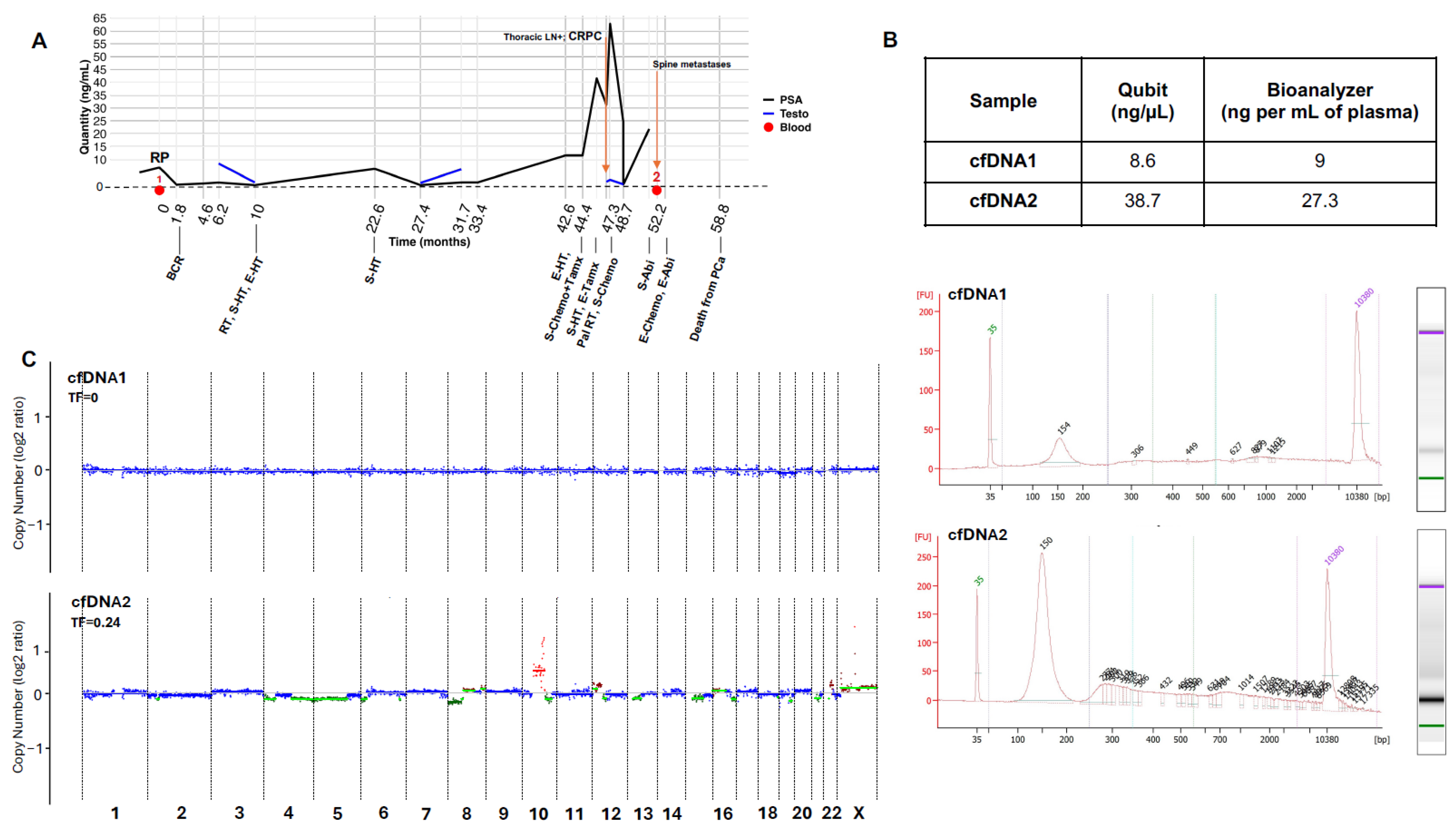
2.7. Clinical Application of the Overall Refined Protocol
3. Discussion
4. Material and Methods
4.1. Established Protocols on cfDNA in PCa
4.2. Ethical Review and Patient Consent
4.3. Blood Germline DNA Processing and Sequencing
4.4. Isolation and Quantification of cfDNA
4.5. Purification of cfDNA by Removal of Contaminating gDNA
4.6. cfDNA Library Preparation and Sequencing
4.7. Sequence Data Analysis
4.8. Analysis of CNVs
4.9. Analysis of Somatic Mutations
4.10. Downsampling
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berenguer, C.V.; Pereira, F.; Camara, J.S.; Pereira, J.A.M. Underlying Features of Prostate Cancer-Statistics, Risk Factors, and Emerging Methods for Its Diagnosis. Curr. Oncol. 2023, 30, 2300–2321. [Google Scholar] [CrossRef] [PubMed]
- Correas, J.M.; Halpern, E.J.; Barr, R.G.; Ghai, S.; Walz, J.; Bodard, S.; Dariane, C.; de la Rosette, J. Advanced ultrasound in the diagnosis of prostate cancer. World J. Urol. 2021, 39, 661–676. [Google Scholar] [CrossRef]
- Eldred-Evans, D.; Ahmed, H.U. Population-Based Prostate Cancer Screening With Magnetic Resonance Imaging or Ultrasonography-The IP1-PROSTAGRAM Study-Reply. JAMA Oncol. 2021, 7, 1575–1576. [Google Scholar] [CrossRef]
- Tikkinen, K.A.O.; Dahm, P.; Lytvyn, L.; Heen, A.F.; Vernooij, R.W.M.; Siemieniuk, R.A.C.; Wheeler, R.; Vaughan, B.; Fobuzi, A.C.; Blanker, M.H.; et al. Prostate cancer screening with prostate-specific antigen (PSA) test: A clinical practice guideline. BMJ 2018, 362, k3581. [Google Scholar] [CrossRef] [PubMed]
- Thompson, I.M.; Ankerst, D.P.; Chi, C.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Feng, Z.; Parnes, H.L.; Coltman, C.A., Jr. Assessing prostate cancer risk: Results from the Prostate Cancer Prevention Trial. J. Natl. Cancer Inst. 2006, 98, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Tanaka, J.; Nakaya, Y.; Maeda, N.; Sakamoto, A.; Nakayama, A.; Satomura, H.; Sakai, M.; Konishi, K.; Yamamoto, Y.; et al. Whole-body MRI: Detecting bone metastases from prostate cancer. Jpn. J. Radiol. 2022, 40, 229–244. [Google Scholar] [CrossRef]
- Li, R.; Ravizzini, G.C.; Gorin, M.A.; Maurer, T.; Eiber, M.; Cooperberg, M.R.; Alemozzaffar, M.; Tollefson, M.K.; Delacroix, S.E.; Chapin, B.F. The use of PET/CT in prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 4–21. [Google Scholar] [CrossRef]
- Bjurlin, M.A.; Carter, H.B.; Schellhammer, P.; Cookson, M.S.; Gomella, L.G.; Troyer, D.; Wheeler, T.M.; Schlossberg, S.; Penson, D.F.; Taneja, S.S. Optimization of initial prostate biopsy in clinical practice: Sampling, labeling and specimen processing. J. Urol. 2013, 189, 2039–2046. [Google Scholar] [CrossRef]
- Trujillo, B.; Wu, A.; Wetterskog, D.; Attard, G. Blood-based liquid biopsies for prostate cancer: Clinical opportunities and challenges. Br. J. Cancer 2022, 127, 1394–1402. [Google Scholar] [CrossRef]
- Crocetto, F.; Russo, G.; Di Zazzo, E.; Pisapia, P.; Mirto, B.F.; Palmieri, A.; Pepe, F.; Bellevicine, C.; Russo, A.; La Civita, E.; et al. Liquid Biopsy in Prostate Cancer Management-Current Challenges and Future Perspectives. Cancers 2022, 14, 3272. [Google Scholar] [CrossRef]
- Kim, C.J.; Dong, L.; Amend, S.R.; Cho, Y.K.; Pienta, K.J. The role of liquid biopsies in prostate cancer management. Lab Chip 2021, 21, 3263–3288. [Google Scholar] [CrossRef] [PubMed]
- Garofoli, M.; Maiorano, B.A.; Bruno, G.; Giordano, G.; Falagario, U.G.; Necchi, A.; Carrieri, G.; Landriscina, M.; Conteduca, V. Circulating Tumor DNA: A New Research Frontier in Urological Oncology from Localized to Metastatic Disease. Eur. Urol. Oncol. 2024, 8, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Belic, J.; Graf, R.; Auer, M.; Lafer, I.; Fischereder, K.; Webersinke, G.; Pummer, K.; Augustin, H.; Pichler, M.; et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat. Commun. 2016, 7, 12008. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J. Natl. Cancer Inst. 2017, 109, djx118. [Google Scholar] [CrossRef]
- De Laere, B.; van Dam, P.J.; Whitington, T.; Mayrhofer, M.; Diaz, E.H.; Van den Eynden, G.; Vandebroek, J.; Del-Favero, J.; Van Laere, S.; Dirix, L.; et al. Comprehensive Profiling of the Androgen Receptor in Liquid Biopsies from Castration-resistant Prostate Cancer Reveals Novel Intra-AR Structural Variation and Splice Variant Expression Patterns. Eur. Urol. 2017, 72, 192–200. [Google Scholar] [CrossRef]
- Herberts, C.; Annala, M.; Sipola, J.; Ng, S.W.S.; Chen, X.E.; Nurminen, A.; Korhonen, O.V.; Munzur, A.D.; Beja, K.; Schonlau, E.; et al. Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer. Nature 2022, 608, 199–208. [Google Scholar] [CrossRef]
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Tse, R.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Azad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S.; et al. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef]
- Vandekerkhove, G.; Struss, W.J.; Annala, M.; Kallio, H.M.L.; Khalaf, D.; Warner, E.W.; Herberts, C.; Ritch, E.; Beja, K.; Loktionova, Y.; et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur. Urol. 2019, 75, 667–675. [Google Scholar] [CrossRef]
- Herberts, C.; Murtha, A.J.; Fu, S.; Wang, G.; Schonlau, E.; Xue, H.; Lin, D.; Gleave, A.; Yip, S.; Angeles, A.; et al. Activating AKT1 and PIK3CA Mutations in Metastatic Castration-Resistant Prostate Cancer. Eur. Urol. 2020, 78, 834–844. [Google Scholar] [CrossRef]
- Del Re, M.; Conteduca, V.; Crucitta, S.; Gurioli, G.; Casadei, C.; Restante, G.; Schepisi, G.; Lolli, C.; Cucchiara, F.; Danesi, R.; et al. Androgen receptor gain in circulating free DNA and splicing variant 7 in exosomes predict clinical outcome in CRPC patients treated with abiraterone and enzalutamide. Prostate Cancer Prostatic Dis. 2021, 24, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Marquez, C.P.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J.; Simko, J.P.; et al. Cell-Free DNA Detection of Tumor Mutations in Heterogeneous, Localized Prostate Cancer Via Targeted, Multiregion Sequencing. JCO Precis. Oncol. 2021, 5, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, N.M.; Maurice-Dror, C.; Herberts, C.; Tu, W.; Fan, W.; Murtha, A.J.; Kollmannsberger, C.; Kwan, E.M.; Parekh, K.; Schonlau, E.; et al. Prediction of plasma ctDNA fraction and prognostic implications of liquid biopsy in advanced prostate cancer. Nat. Commun. 2024, 15, 1828. [Google Scholar] [CrossRef]
- Xia, S.; Kohli, M.; Du, M.; Dittmar, R.L.; Lee, A.; Nandy, D.; Yuan, T.; Guo, Y.; Wang, Y.; Tschannen, M.R.; et al. Plasma genetic and genomic abnormalities predict treatment response and clinical outcome in advanced prostate cancer. Oncotarget 2015, 6, 16411–16421. [Google Scholar] [CrossRef] [PubMed]
- Eagle, S.H.C.; Robertson, J.; Bastedo, D.P.; Liu, K.; Nash, J.H.E. Evaluation of five commercial DNA extraction kits using Salmonella as a model for implementation of rapid Nanopore sequencing in routine diagnostic laboratories. Access Microbiol. 2023, 5, 000468.v3. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef]
- Chapman, L.; Ledet, E.M.; Barata, P.C.; Cotogno, P.; Manogue, C.; Moses, M.; Christensen, B.R.; Steinwald, P.; Ranasinghe, L.; Layton, J.L.; et al. TP53 Gain-of-Function Mutations in Circulating Tumor DNA in Men With Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 148–154. [Google Scholar] [CrossRef]
- Ye, S.; Wang, H.; He, K.; Peng, M.; Wang, Y.; Li, Y.; Jiang, S.; Li, J.; Yi, L.; Cui, R. Clinical Characterization of Mismatch Repair Gene-Deficient Metastatic Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 533282. [Google Scholar] [CrossRef]
- Fan, L.; Fei, X.; Zhu, Y.; Pan, J.; Sha, J.; Chi, C.; Gong, Y.; Du, X.; Zhou, L.; Dong, B.; et al. Comparative Analysis of Genomic Alterations across Castration Sensitive and Castration Resistant Prostate Cancer via Circulating Tumor DNA Sequencing. J. Urol. 2021, 205, 461–469. [Google Scholar] [CrossRef]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Marquez, C.P.; Chu, C.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J.; et al. Cell-free DNA concentration and fragment size as a biomarker for prostate cancer. Sci. Rep. 2021, 11, 5040. [Google Scholar] [CrossRef]
- Kohli, M.; Tan, W.; Zheng, T.; Wang, A.; Montesinos, C.; Wong, C.; Du, P.; Jia, S.; Yadav, S.; Horvath, L.G.; et al. Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine 2020, 54, 102728. [Google Scholar] [CrossRef] [PubMed]
- Fettke, H.; Kwan, E.M.; Docanto, M.M.; Bukczynska, P.; Ng, N.; Graham, L.K.; Mahon, K.; Hauser, C.; Tan, W.; Wang, X.H.; et al. Combined Cell-free DNA and RNA Profiling of the Androgen Receptor: Clinical Utility of a Novel Multianalyte Liquid Biopsy Assay for Metastatic Prostate Cancer. Eur. Urol. 2020, 78, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Reimers, M.A.; Yip, S.M.; Zhang, L.; Cieslik, M.; Dhawan, M.; Montgomery, B.; Wyatt, A.W.; Chi, K.N.; Small, E.J.; Chinnaiyan, A.M.; et al. Clinical Outcomes in Cyclin-dependent Kinase 12 Mutant Advanced Prostate Cancer. Eur. Urol. 2020, 77, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.; Aggarwal, S.; Bhardwaj, S.; Ramakrishna, G.; Pandey, C.K. Serial perioperative cell-free DNA levels in donors and recipients undergoing living donor liver transplantation. Acta Anaesthesiol. Scand. 2017, 61, 1084–1094. [Google Scholar] [CrossRef]
- Du, M.; Tian, Y.; Tan, W.; Wang, L.; Wang, L.; Kilari, D.; Huang, C.C.; Wang, L.; Kohli, M. Plasma cell-free DNA-based predictors of response to abiraterone acetate/prednisone and prognostic factors in metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 705–713. [Google Scholar] [CrossRef]
- Liu, H.; Gao, Y.; Vafaei, S.; Gu, X.; Zhong, X. The Prognostic Value of Plasma Cell-Free DNA Concentration in the Prostate Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 599602. [Google Scholar] [CrossRef]
- Mouliere, F.; Robert, B.; Arnau Peyrotte, E.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef]
- Knutson, T.P.; Luo, B.; Kobilka, A.; Lyman, J.; Guo, S.; Munro, S.A.; Li, Y.; Heer, R.; Gaughan, L.; Morris, M.J.; et al. AR alterations inform circulating tumor DNA detection in metastatic castration resistant prostate cancer patients. Nat. Commun. 2024, 15, 10648. [Google Scholar] [CrossRef]
- Sonpavde, G.; Agarwal, N.; Pond, G.R.; Nagy, R.J.; Nussenzveig, R.H.; Hahn, A.W.; Sartor, O.; Gourdin, T.S.; Nandagopal, L.; Ledet, E.M.; et al. Circulating tumor DNA alterations in patients with metastatic castration-resistant prostate cancer. Cancer 2019, 125, 1459–1469. [Google Scholar] [CrossRef]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef]
- Dang, H.X.; Chauhan, P.S.; Ellis, H.; Feng, W.; Harris, P.K.; Smith, G.; Qiao, M.; Dienstbach, K.; Beck, R.; Atkocius, A.; et al. Cell-free DNA alterations in the AR enhancer and locus predict resistance to AR-directed therapy in patients with metastatic prostate cancer. JCO Precis. Oncol. 2020, 4, 680–713. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Madison, R.W.; Chung, J.H.; Gjoerup, O.V.; Severson, E.A.; Dennis, L.; Fendler, B.J.; Morley, S.; Zhong, L.; Graf, R.P.; et al. Genomic Analysis of Circulating Tumor DNA in 3,334 Patients with Advanced Prostate Cancer Identifies Targetable BRCA Alterations and AR Resistance Mechanisms. Clin. Cancer Res. 2021, 27, 3094–3105. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Mizuno, K.; Yamasaki, T.; Miyazaki, Y.; Makino, Y.; Okasho, K.; Li, X.; Utsunomiya, N.; Goto, T.; Kobayashi, T.; et al. Clinical utility of androgen receptor gene aberrations in circulating cell-free DNA as a biomarker for treatment of castration-resistant prostate cancer. Sci. Rep. 2019, 9, 4030. [Google Scholar] [CrossRef] [PubMed]
- Romanel, A.; Gasi Tandefelt, D.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015, 7, 312re10. [Google Scholar] [CrossRef]
- Kohli, M.; Li, J.; Du, M.; Hillman, D.W.; Dehm, S.M.; Tan, W.; Carlson, R.; Campion, M.B.; Wang, L.; Wang, L.; et al. Prognostic association of plasma cell-free DNA-based androgen receptor amplification and circulating tumor cells in pre-chemotherapy metastatic castration-resistant prostate cancer patients. Prostate Cancer Prostatic Dis. 2018, 21, 411–418. [Google Scholar] [CrossRef]
- Kang, M.; Cho, E.; Jang, J.; Lee, J.; Jeon, Y.; Jeong, B.C.; Seo, S.I.; Jeon, S.S.; Lee, H.M.; Choi, H.Y.; et al. Genomic analysis of Korean patients with advanced prostate cancer by use of a comprehensive next-generation sequencing panel and low-coverage, whole-genome sequencing. Investig. Clin. Urol. 2019, 60, 227–234. [Google Scholar] [CrossRef]
- Ritch, E.; Fu, S.Y.F.; Herberts, C.; Wang, G.; Warner, E.W.; Schonlau, E.; Taavitsainen, S.; Murtha, A.J.; Vandekerkhove, G.; Beja, K.; et al. Identification of Hypermutation and Defective Mismatch Repair in ctDNA from Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Barnicle, A.; Sibilla, C.; Lai, Z.; Corcoran, C.; Barrett, J.C.; Adelman, C.A.; Qiu, P.; Easter, A.; Dearden, S.; et al. Detection of BRCA1, BRCA2, and ATM Alterations in Matched Tumor Tissue and Circulating Tumor DNA in Patients with Prostate Cancer Screened in PROfound. Clin. Cancer Res. 2023, 29, 81–91. [Google Scholar] [CrossRef]
- Chung, S.; Tamura, K.; Furihata, M.; Uemura, M.; Daigo, Y.; Nasu, Y.; Miki, T.; Shuin, T.; Fujioka, T.; Nakamura, Y.; et al. Overexpression of the potential kinase serine/ threonine/tyrosine kinase 1 (STYK 1) in castration-resistant prostate cancer. Cancer Sci. 2009, 100, 2109–2114. [Google Scholar] [CrossRef]
- Wang, Z.; Qu, L.; Deng, B.; Sun, X.; Wu, S.; Liao, J.; Fan, J.; Peng, Z. STYK1 promotes epithelial-mesenchymal transition and tumor metastasis in human hepatocellular carcinoma through MEK/ERK and PI3K/AKT signaling. Sci. Rep. 2016, 6, 33205. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, D.; Li, W.; Di, S.; Zhang, Z.; Zhang, J.; Xu, L.; Guo, K.; Zhu, Y.; Han, J.; et al. STYK1 promotes tumor growth and metastasis by reducing SPINT2/HAI-2 expression in non-small cell lung cancer. Cell Death Dis. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Valenca, I.; Ferreira, A.R.; Correia, M.; Kuhl, S.; van Roermund, C.; Waterham, H.R.; Maximo, V.; Islinger, M.; Ribeiro, D. Prostate Cancer Proliferation Is Affected by the Subcellular Localization of MCT2 and Accompanied by Significant Peroxisomal Alterations. Cancers 2020, 12, 3152. [Google Scholar] [CrossRef]
- Costa, E.T.; Barnabe, G.F.; Li, M.; Dias, A.A.; Machado, T.R.; Asprino, P.F.; Cavalher, F.P.; Ferreira, E.N.; Del Mar Inda, M.; Nagai, M.H.; et al. Intratumoral heterogeneity of ADAM23 promotes tumor growth and metastasis through LGI4 and nitric oxide signals. Oncogene 2015, 34, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Solorzano, S.R.; Imaz-Rosshandler, I.; Camacho-Arroyo, I.; Garcia-Tobilla, P.; Morales-Montor, G.; Salazar, P.; Arena-Ortiz, M.L.; Rodriguez-Dorantes, M. GABA promotes gastrin-releasing peptide secretion in NE/NE-like cells: Contribution to prostate cancer progression. Sci. Rep. 2018, 8, 10272. [Google Scholar] [CrossRef]
- Chen, J.Y.; Chang, C.F.; Huang, S.P.; Huang, C.Y.; Yu, C.C.; Lin, V.C.; Geng, J.H.; Li, C.Y.; Lu, T.L.; Bao, B.Y. Integrated analysis identifies GABRB3 as a biomarker in prostate cancer. BMC Med. Genom. 2024, 17, 41. [Google Scholar] [CrossRef]
- Bourgey, M.; Dali, R.; Eveleigh, R.; Chen, K.C.; Letourneau, L.; Fillon, J.; Michaud, M.; Caron, M.; Sandoval, J.; Lefebvre, F.; et al. GenPipes: An open-source framework for distributed and scalable genomic analyses. Gigascience 2019, 8, giz037. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Bergmann, E.A.; Chen, B.J.; Arora, K.; Vacic, V.; Zody, M.C. Conpair: Concordance and contamination estimator for matched tumor-normal pairs. Bioinformatics 2016, 32, 3196–3198. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2022. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahimirad, S.; Derderian, S.; Hamel, L.; Scarlata, E.; McKercher, G.; Brimo, F.; Rajan, R.; Rompre-Brodeur, A.; Kassouf, W.; Sanchez-Salas, R.; et al. Refined Procedure to Purify and Sequence Circulating Cell-Free DNA in Prostate Cancer. Int. J. Mol. Sci. 2025, 26, 5839. https://doi.org/10.3390/ijms26125839
Rahimirad S, Derderian S, Hamel L, Scarlata E, McKercher G, Brimo F, Rajan R, Rompre-Brodeur A, Kassouf W, Sanchez-Salas R, et al. Refined Procedure to Purify and Sequence Circulating Cell-Free DNA in Prostate Cancer. International Journal of Molecular Sciences. 2025; 26(12):5839. https://doi.org/10.3390/ijms26125839
Chicago/Turabian StyleRahimirad, Samira, Seta Derderian, Lucie Hamel, Eleonora Scarlata, Ginette McKercher, Fadi Brimo, Raghu Rajan, Alexis Rompre-Brodeur, Wassim Kassouf, Rafael Sanchez-Salas, and et al. 2025. "Refined Procedure to Purify and Sequence Circulating Cell-Free DNA in Prostate Cancer" International Journal of Molecular Sciences 26, no. 12: 5839. https://doi.org/10.3390/ijms26125839
APA StyleRahimirad, S., Derderian, S., Hamel, L., Scarlata, E., McKercher, G., Brimo, F., Rajan, R., Rompre-Brodeur, A., Kassouf, W., Sanchez-Salas, R., Aprikian, A., & Chevalier, S. (2025). Refined Procedure to Purify and Sequence Circulating Cell-Free DNA in Prostate Cancer. International Journal of Molecular Sciences, 26(12), 5839. https://doi.org/10.3390/ijms26125839