

Oxidative Stress in Neurodegenerative Disorders: A Key Driver in Impairing Skeletal Muscle Health


Abstract
1. Introduction
2. Molecular Mechanisms in Redox Homeostasis
2.1. Role of Pro-Oxidant Enzymes in Neurodegenerative Disorders
2.2. Role of Anti-Oxidant Enzymes in Neurodegenerative Disorders
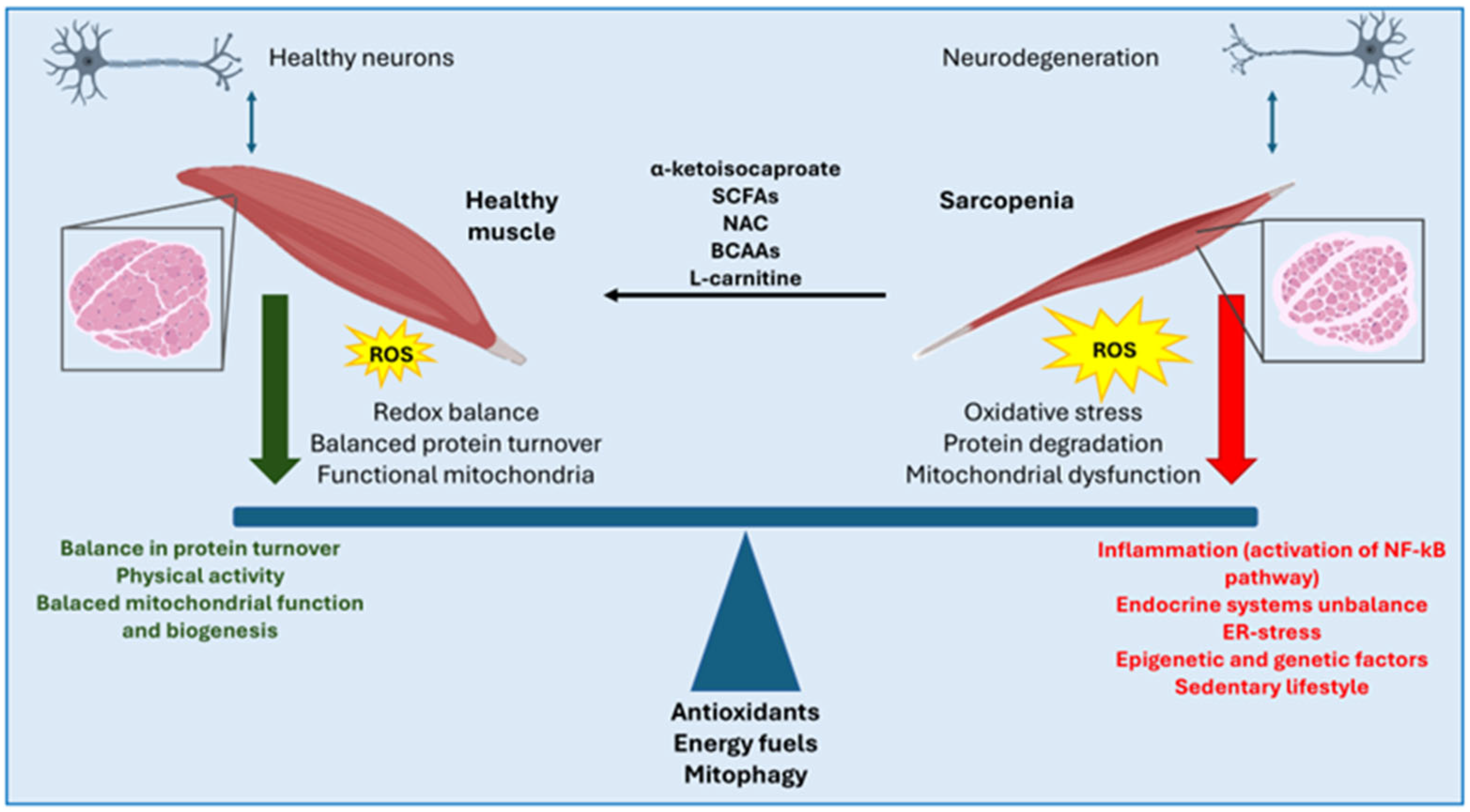
3. Metabolic Dysfunctions of Skeletal Muscle in Neurodegeneration
3.1. Role of Mitochondria in Skeletal Muscle Dysfunction Typical of Neurodegeneration
3.2. Role of Mitochondria Dynamics in Sarcopenia and Atrophy
3.3. Therapeutical Approaches to Sarcopenia/Atrophy
4. Oxidative Stress in Skeletal Muscle Dysfunction Associated with Neurodegenerative Diseases
4.1. Role of Oxidative Stress in ALS Skeletal Muscle
4.2. Role of Oxidative Stress in AD Skeletal Muscle
4.3. Role of Oxidative Stress in PD Skeletal Muscle
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Duranti, E.; Villa, C. Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 704. [Google Scholar] [CrossRef] [PubMed]
- Bastin, C.; Giacomelli, F.; Miévis, F.; Lemaire, C.; Guillaume, B.; Salmon, E. Anosognosia in Mild Cognitive Impairment: Lack of Awareness of Memory Difficulties Characterizes Prodromal Alzheimer’s Disease. Front. Psychiatry 2021, 12, 631518. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, C.P.; Pei, J.; Alugoju, P.; Anthikapalli, N.V.A.; Jayaraman, S.; Veeraraghavan, V.P.; Gopathy, S.; Roy, J.R.; Janaki, C.S.; Thalamati, D.; et al. New Strategies of Neurodegenerative Disease Treatment with Extracellular Vesicles (EVs) Derived from Mesenchymal Stem Cells (MSCs). Theranostics 2023, 13, 4138–4165. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Klenerman, D.; Rubinsztein, D.C. Developing Therapies for Neurodegenerative Disorders: Insights from Protein Aggregation and Cellular Stress Responses. Annu. Rev. Cell Dev. Biol. 2020, 36, 165–189. [Google Scholar] [CrossRef] [PubMed]
- Candelise, N.; Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C.; Manganelli, V.; Garofalo, T.; Sorice, M.; Misasi, R. Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications. Int. J. Mol. Sci. 2021, 22, 6016. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, C.-M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Hemerková, P.; Vališ, M. Role of Oxidative Stress in the Pathogenesis of Amyotrophic Lateral Sclerosis: Antioxidant Metalloenzymes and Therapeutic Strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative Stress in Alzheimer’s Disease: Current Knowledge of Signaling Pathways and Therapeutics. Mol. Biol. Rep. 2024, 51, 48. [Google Scholar] [CrossRef]
- Le Gal, K.; Schmidt, E.E.; Sayin, V.I. Cellular Redox Homeostasis. Antioxidants 2021, 10, 1377. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS Signalling: Adaptive Redox Switches through Oxidative/Nitrosative Protein Modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Houldsworth, A. Role of Oxidative Stress in Neurodegenerative Disorders: A Review of Reactive Oxygen Species and Prevention by Antioxidants. Brain Commun. 2024, 6, fcad356. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging Roles of Oxidative Stress in Brain Aging and Alzheimer’s Disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative Stress Factors in Parkinson’s Disease. Neural. Regen. Res. 2021, 16, 1383–1391. [Google Scholar] [CrossRef]
- Burtscher, J.; Millet, G.P.; Place, N.; Kayser, B.; Zanou, N. The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection. Int. J. Mol. Sci. 2021, 22, 6479. [Google Scholar] [CrossRef]
- Duranti, E.; Villa, C. From Brain to Muscle: The Role of Muscle Tissue in Neurodegenerative Disorders. Biology 2024, 13, 719. [Google Scholar] [CrossRef]
- Porcelli, S.; Bellistri, G.; Ramaglia, M.; Rasica, L.; Veronesi, F.; Marzorati, M. Evaluation of Skeletal Muscle Oxidative Metabolism in Alzheimer’s Disease: 2360 Board #107 May 29, 11: 00 AM–12: 30 PM. Med. Sci. Sports Exerc. 2015, 47, 633. [Google Scholar] [CrossRef]
- Mukhamedyarov, M.A.; Grishin, S.N.; Yusupova, E.R.; Zefirov, A.L.; Palotás, A. Alzheimer’s Beta-Amyloid-Induced Depolarization of Skeletal Muscle Fibers: Implications for Motor Dysfunctions in Dementia. Cell Physiol. Biochem. 2009, 23, 109–114. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, Y.; Zhao, C.; Pang, S.; Lu, J.; Chan, P. α-Synuclein Aggregation Causes Muscle Atrophy through Neuromuscular Junction Degeneration. J. Cachexia Sarcopenia Muscle 2023, 14, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, H.K.; Pereira, M.; Rajavelu, I.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Rajasekaran, J.J. Oxidative Stress: Fundamentals and Advances in Quantification Techniques. Front. Chem. 2024, 12, 1470458. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, A.; Viviano, M.; Feoli, A.; Milite, C.; Sarno, G.; Castellano, S.; Sbardella, G. NADPH Oxidases: From Molecular Mechanisms to Current Inhibitors. J. Med. Chem. 2023, 66, 11632–11655. [Google Scholar] [CrossRef]
- Massari, M.; Nicoll, C.R.; Marchese, S.; Mattevi, A.; Mascotti, M.L. Evolutionary and Structural Analyses of the NADPH Oxidase Family in Eukaryotes Reveal an Initial Calcium Dependency. Redox Biol. 2022, 56, 102436. [Google Scholar] [CrossRef]
- Pecchillo Cimmino, T.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef]
- Begum, R.; Thota, S.; Abdulkadir, A.; Kaur, G.; Bagam, P.; Batra, S. NADPH Oxidase Family Proteins: Signaling Dynamics to Disease Management. Cell Mol. Immunol. 2022, 19, 660–686. [Google Scholar] [CrossRef]
- Dustin, C.M.; Shiva, S.S.; Vazquez, A.; Saeed, A.; Pascoal, T.; Cifuentes-Pagano, E.; Pagano, P.J. NOX2 in Alzheimer’s and Parkinson’s Disease. Redox Biol. 2024, 78, 103433. [Google Scholar] [CrossRef]
- Ndrepepa, G. Myeloperoxidase—A Bridge Linking Inflammation and Oxidative Stress with Cardiovascular Disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef]
- Kargapolova, Y.; Geißen, S.; Zheng, R.; Baldus, S.; Winkels, H.; Adam, M. The Enzymatic and Non-Enzymatic Function of Myeloperoxidase (MPO) in Inflammatory Communication. Antioxidants 2021, 10, 562. [Google Scholar] [CrossRef]
- Rivera Antonio, A.M.; Padilla Martínez, I.I.; Torres-Ramos, M.A.; Rosales-Hernández, M.C. Myeloperoxidase as a Therapeutic Target for Oxidative Damage in Alzheimer’s Disease. J. Enzym. Inhib. Med. Chem. 2025, 40, 2456282. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.; Jamil, R.T. Biochemistry, Xanthine Oxidase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine Oxidoreductase: One Enzyme for Multiple Physiological Tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Liu, Y.; Zhang, G.; Yang, Z.; Xu, W.; Chen, Q. The Applications and Mechanisms of Superoxide Dismutase in Medicine, Food, and Cosmetics. Antioxidants 2023, 12, 1675. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Anand, N.; Varma, S.R.; Ramamurthy, S.; Vichitra, C.; Sharma, A.; Mahalakshmi, A.M.; Essa, M.M. Superoxide Dismutase and Neurological Disorders. IBRO Neurosci. Rep. 2024, 16, 373–394. [Google Scholar] [CrossRef]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef]
- Fetherolf, M.M.; Boyd, S.D.; Winkler, D.D.; Winge, D.R. Oxygen-Dependent Activation of Cu,Zn-Superoxide Dismutase-1. Metallomics 2017, 9, 1047–1059. [Google Scholar] [CrossRef]
- Rasheed, Z. Therapeutic Potentials of Catalase: Mechanisms, Applications, and Future Perspectives. Int. J. Health Sci. 2024, 18, 1–6. [Google Scholar]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Anwar, S.; Alrumaihi, F.; Sarwar, T.; Babiker, A.Y.; Khan, A.A.; Prabhu, S.V.; Rahmani, A.H. Exploring Therapeutic Potential of Catalase: Strategies in Disease Prevention and Management. Biomolecules 2024, 14, 697. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1G93A Mice Predates Disease Onset and Is a Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Geng, J.; Zeng, X.; Han, R.; Huh, Y.E.; Peng, J. Exploring Causal Effects of Sarcopenia on Risk and Progression of Parkinson Disease by Mendelian Randomization. NPJ Park. Dis. 2024, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, S.M.; Orchard, K.J.A. Sarcopenia as a Risk Factor for Alzheimer’s Disease: Genetic and Epigenetic Perspectives. Genes 2024, 15, 561. [Google Scholar] [CrossRef] [PubMed]
- Garneau, L.; Aguer, C. Role of Myokines in the Development of Skeletal Muscle Insulin Resistance and Related Metabolic Defects in Type 2 Diabetes. Diabetes Metab. 2019, 45, 505–516. [Google Scholar] [CrossRef]
- Chung, H.K.; Ryu, D.; Kim, K.S.; Chang, J.Y.; Kim, Y.K.; Yi, H.-S.; Kang, S.G.; Choi, M.J.; Lee, S.E.; Jung, S.-B.; et al. Growth Differentiation Factor 15 Is a Myomitokine Governing Systemic Energy Homeostasis. J. Cell Biol. 2017, 216, 149–165. [Google Scholar] [CrossRef]
- Jo, D.; Yoon, G.; Kim, O.Y.; Song, J. A New Paradigm in Sarcopenia: Cognitive Impairment Caused by Imbalanced Myokine Secretion and Vascular Dysfunction. Biomed. Pharmacother. 2022, 147, 112636. [Google Scholar] [CrossRef]
- Kwon, Y.N.; Yoon, S.S. Sarcopenia: Neurological Point of View. J. Bone Metab. 2017, 24, 83–89. [Google Scholar] [CrossRef]
- Ciccarone, F.; Castelli, S.; Lazzarino, G.; Scaricamazza, S.; Mangione, R.; Bernardini, S.; Apolloni, S.; D’Ambrosi, N.; Ferri, A.; Ciriolo, M.R. Lipid Catabolism and Mitochondrial Uncoupling Are Stimulated in Brown Adipose Tissue of Amyotrophic Lateral Sclerosis Mouse Models. Genes Dis. 2022, 10, 321–324. [Google Scholar] [CrossRef]
- Dong, H.; Tsai, S.-Y. Mitochondrial Properties in Skeletal Muscle Fiber. Cells 2023, 12, 2183. [Google Scholar] [CrossRef]
- Castelli, S.; Desideri, E.; Laureti, L.; Felice, F.; De Cristofaro, A.; Scaricamazza, S.; Lazzarino, G.; Ciriolo, M.R.; Ciccarone, F. N-Acetylaspartate Promotes Glycolytic-to-Oxidative Fiber-Type Switch and Resistance to Atrophic Stimuli in Myotubes. Cell Death Dis. 2024, 15, 686. [Google Scholar] [CrossRef]
- Felice, F.; De Falco, P.; Milani, M.; Castelli, S.; Ragnini-Wilson, A.; Lazzarino, G.; D’Ambrosi, N.; Ciccarone, F.; Ciriolo, M.R. N-Acetylaspartate Mitigates pro-Inflammatory Responses in Microglial Cells by Intersecting Lipid Metabolism and Acetylation Processes. Cell Commun. Signal. 2024, 22, 564. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Massimino, M.L.; Biancotto, G.; Angeletti, R.; Reggiani, C.; Sorgato, M.C.; Bertoli, A.; Stella, R. Absolute Quantification of Myosin Heavy Chain Isoforms by Selected Reaction Monitoring Can Underscore Skeletal Muscle Changes in a Mouse Model of Amyotrophic Lateral Sclerosis. Anal. Bioanal. Chem. 2017, 409, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Fiber-Type Shifting in Sarcopenia of Old Age: Proteomic Profiling of the Contractile Apparatus of Skeletal Muscles. Int. J. Mol. Sci. 2023, 24, 2415. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic Profiling of Fast-To-Slow Muscle Transitions during Aging. Front. Physiol. 2011, 2, 105. [Google Scholar] [CrossRef]
- Vinokurov, A.Y.; Bazhenov, P.A.; Pogonyalova, M.Y.; Seryogina, E.S.; Vetrova, E.A.; Andreeva, L.; Abramov, A.Y.; Angelova, P.R. Enhancement of Energy Metabolism in Skeletal Myocytes Protects Against Age-Related Sarcopenia. J. Cell Mol. Med. 2025, 29, e70588. [Google Scholar] [CrossRef]
- Zhang, F.-M.; Wu, H.-F.; Wang, K.-F.; Yu, D.-Y.; Zhang, X.-Z.; Ren, Q.; Chen, W.-Z.; Lin, F.; Yu, Z.; Zhuang, C.-L. Transcriptome Profiling of Fast/Glycolytic and Slow/Oxidative Muscle Fibers in Aging and Obesity. Cell Death Dis. 2024, 15, 459. [Google Scholar] [CrossRef]
- Ji, L.L.; Kang, C. Role of PGC-1α in Sarcopenia: Etiology and Potential Intervention—A Mini-Review. Gerontology 2015, 61, 139–148. [Google Scholar] [CrossRef]
- Wiedmer, P.; Jung, T.; Castro, J.P.; Pomatto, L.C.D.; Sun, P.Y.; Davies, K.J.A.; Grune, T. Sarcopenia—Molecular Mechanisms and Open Questions. Ageing Res. Rev. 2021, 65, 101200. [Google Scholar] [CrossRef]
- Li, Y.-P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-Alpha Acts via P38 MAPK to Stimulate Expression of the Ubiquitin Ligase Atrogin1/MAFbx in Skeletal Muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef]
- Changchien, C.-Y.; Lin, Y.-H.; Cheng, Y.-C.; Chang, H.-H.; Peng, Y.-S.; Chen, Y. Indoxyl Sulfate Induces Myotube Atrophy by ROS-ERK and JNK-MAFbx Cascades. Chem. Biol. Interact. 2019, 304, 43–51. [Google Scholar] [CrossRef]
- Dolan, E.; Artioli, G.G.; Pereira, R.M.R.; Gualano, B. Muscular Atrophy and Sarcopenia in the Elderly: Is There a Role for Creatine Supplementation? Biomolecules 2019, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.S.; Shelar, S.B.; Jones, D.P.; Hoidal, J.R. Reductive Stress Impairs Myogenic Differentiation. Redox. Biol. 2020, 34, 101492. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Bilgel, M.; Walker, K.A.; Moghekar, A.R.; Fishbein, K.W.; Spencer, R.G.; Resnick, S.M.; Ferrucci, L. Skeletal Muscle Mitochondrial Function Predicts Cognitive Impairment and Is Associated with Biomarkers of Alzheimer’s Disease and Neurodegeneration. Alzheimers Dement. 2023, 19, 4436–4445. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Lo Buglio, A.; Vendemiale, G. Mitochondrial Impairment in Sarcopenia. Biology 2021, 10, 31. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 Deficiency Links Age-related Sarcopenia and Impaired Autophagy to Activation of an Adaptive Mitophagy Pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Dulac, M.; Leduc-Gaudet, J.-P.; Cefis, M.; Ayoub, M.-B.; Reynaud, O.; Shams, A.; Moamer, A.; Nery Ferreira, M.F.; Hussain, S.N.; Gouspillou, G. Regulation of Muscle and Mitochondrial Health by the Mitochondrial Fission Protein Drp1 in Aged Mice. J. Physiol. 2021, 599, 4045–4063. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.-P.; Hussain, S.N.A.; Barreiro, E.; Gouspillou, G. Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging. Int. J. Mol. Sci. 2021, 22, 8179. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.-P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin Overexpression Protects from Ageing-Related Loss of Muscle Mass and Strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Lim, P.; Woo, S.W.; Han, J.; Lee, Y.L.; Shim, J.H.; Kim, H.S. Effects of Alpha-Ketoisocaproate in Oxidative Stress-Induced C2C12 Myotubes via Inhibition of P38 MAPK and ERK1/2. Biochem. Biophys. Rep. 2025, 41, 101955. [Google Scholar] [CrossRef]
- Sestili, P.; Martinelli, C.; Colombo, E.; Barbieri, E.; Potenza, L.; Sartini, S.; Fimognari, C. Creatine as an Antioxidant. Amino. Acids 2011, 40, 1385–1396. [Google Scholar] [CrossRef]
- Gui, M.; Lv, L.; Hu, S.; Qin, L.; Wang, C. Sarcopenia in Parkinson’s Disease: From Pathogenesis to Interventions. Metabolism 2025, 169, 156272. [Google Scholar] [CrossRef] [PubMed]
- Cacciatore, S.; Calvani, R.; Esposito, I.; Massaro, C.; Gava, G.; Picca, A.; Tosato, M.; Marzetti, E.; Landi, F. Emerging Targets and Treatments for Sarcopenia: A Narrative Review. Nutrients 2024, 16, 3271. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Wang, Q.; Wang, Y.; Lou, S. The Function of Previously Unappreciated Exerkines Secreted by Muscle in Regulation of Neurodegenerative Diseases. Front. Mol. Neurosci. 2023, 16, 1305208. [Google Scholar] [CrossRef] [PubMed]
- Kubat, G.B.; Picone, P. Skeletal Muscle Dysfunction in Amyotrophic Lateral Sclerosis: A Mitochondrial Perspective and Therapeutic Approaches. Neurol. Sci. 2024, 45, 4121–4131. [Google Scholar] [CrossRef]
- Barone, R.; Bramato, G.; Gnoni, V.; Giugno, A.; Urso, D.; Zecca, C.; Nigro, S.; Filardi, M.; Logroscino, G. Sarcopenia in Subjects with Alzheimer’s Disease: Prevalence and Comparison of Agreement between EGWSOP1, EGWSOP2, and FNIH Criteria. BMC Geriatr. 2024, 24, 278. [Google Scholar] [CrossRef]
- Cai, Y.; Feng, F.; Wei, Q.; Jiang, Z.; Ou, R.; Shang, H. Sarcopenia in Patients with Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 598035. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef]
- Léger, B.; Vergani, L.; Sorarù, G.; Hespel, P.; Derave, W.; Gobelet, C.; D’Ascenzio, C.; Angelini, C.; Russell, A.P. Human Skeletal Muscle Atrophy in Amyotrophic Lateral Sclerosis Reveals a Reduction in Akt and an Increase in Atrogin-1. FASEB J. 2006, 20, 583–585. [Google Scholar] [CrossRef]
- Chen, W.; Guo, L.; Li, M.; Wei, C.; Li, S.; Xu, R. The Pathogenesis of Amyotrophic Lateral Sclerosis: Mitochondrial Dysfunction, Protein Misfolding and Epigenetics. Brain Res. 2022, 1786, 147904. [Google Scholar] [CrossRef]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Cicardi, M.E.; Giorgetti, E.; Onesto, E.; Messi, E.; Poletti, A. ALS-Related Misfolded Protein Management in Motor Neurons and Muscle Cells. Neurochem. Int. 2014, 79, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, C.; Yi, J.; Wu, S.; Luo, G.; Xu, X.; Lin, P.-H.; Sun, J.; Zhou, J. Suppressed Autophagy Flux in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model during Disease Progression. Physiol. Rep. 2015, 3, e12271. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Girgenrath, M. The Role of Apoptosis in Neuromuscular Diseases and Prospects for Anti-Apoptosis Therapy. Trends Mol. Med. 2006, 12, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Scaricamazza, S.; Bracaglia, A.; D’Ercole, C.; Parisi, C.; D’Angelo, P.; Proietti, D.; Cappelletti, C.; Macone, A.; Lozanoska-Ochser, B.; et al. Polyamine Metabolism Dysregulation Contributes to Muscle Fiber Vulnerability in ALS. Cell Rep. 2025, 44, 115123. [Google Scholar] [CrossRef]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Moore, V.D.G.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of Early Pathogenesis in the SOD1G93A Mouse Model of ALS: Part II, Results and Discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef]
- Genin, E.C.; Abou-Ali, M.; Paquis-Flucklinger, V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes 2023, 14, 1981. [Google Scholar] [CrossRef]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-Related Mitochondrial Dysfunction in Skeletal Muscle of an ALS Mouse Model during the Disease Progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef]
- Motataianu, A.; Serban, G.; Barcutean, L.; Balasa, R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors. Int. J. Mol. Sci. 2022, 23, 9339. [Google Scholar] [CrossRef]
- Loeffler, J.-P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.-L. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular Mechanisms of Neurodegeneration in Alzheimer’s Disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s Disease: Pathogenesis, Diagnostics, and Therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, E.; Resende, E.D.P.F.; Gomes, K.B. Physiopathological Mechanisms Underlying Alzheimer’s Disease: A Narrative Review. Dement. Neuropsychol. 2024, 18, e2024VR01. [Google Scholar] [CrossRef] [PubMed]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef] [PubMed]
- Sehar, U.; Rawat, P.; Reddy, A.P.; Kopel, J.; Reddy, P.H. Amyloid Beta in Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12924. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The Clinical Symptoms of Parkinson’s Disease. J. Neurochem. 2016, 139 (Suppl. S1), 318–324. [Google Scholar] [CrossRef]
- Murphy, K.T.; Lynch, G.S. Impaired Skeletal Muscle Health in Parkinsonian Syndromes: Clinical Implications, Mechanisms and Potential Treatments. J. Cachexia Sarcopenia Muscle 2023, 14, 1987–2002. [Google Scholar] [CrossRef]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease Gene SNCA: Evolutionary and Structural Insights with Pathological Implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef]
- Mizuno, Y. More than 20 Years of the Discovery of Park2. Neurosci. Res. 2020, 159, 3–8. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef]
- Skou, L.D.; Johansen, S.K.; Okarmus, J.; Meyer, M. Pathogenesis of DJ-1/PARK7-Mediated Parkinson’s Disease. Cells 2024, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving Neurodegeneration: Common Mechanisms and Strategies for New Treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Lim, K.-L.; Tan, J.M.M. Role of the Ubiquitin Proteasome System in Parkinson’s Disease. BMC Biochem. 2007, 8 (Suppl. S1), S13. [Google Scholar] [CrossRef]
- Lavin, K.M.; Sealfon, S.C.; McDonald, M.-L.N.; Roberts, B.M.; Wilk, K.; Nair, V.D.; Ge, Y.; Lakshman Kumar, P.; Windham, S.T.; Bamman, M.M. Skeletal Muscle Transcriptional Networks Linked to Type I Myofiber Grouping in Parkinson’s Disease. J. Appl. Physiol. 2020, 128, 229–240. [Google Scholar] [CrossRef]
- Kelly, N.A.; Hammond, K.G.; Bickel, C.S.; Windham, S.T.; Tuggle, S.C.; Bamman, M.M. Effects of Aging and Parkinson’s Disease on Motor Unit Remodeling: Influence of Resistance Exercise Training. J. Appl. Physiol. 2018, 124, 888–898. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Seo, M.H.; Yeo, S. Triadin Decrease Impairs the Expression of E-C Coupling Related Proteins in Muscles of MPTP-Induced Parkinson’s Disease Mice. Front. Neurosci. 2021, 15, 649688. [Google Scholar] [CrossRef]
- Henrich, M.T.; Oertel, W.H.; Surmeier, D.J.; Geibl, F.F. Mitochondrial Dysfunction in Parkinson’s Disease—A Key Disease Hallmark with Therapeutic Potential. Mol. Neurodegener. 2023, 18, 83. [Google Scholar] [CrossRef]
- Mendes, D.; Peixoto, F.; Oliveira, M.M.; Andrade, P.B.; Videira, R.A. Mitochondrial Dysfunction in Skeletal Muscle of Rotenone-Induced Rat Model of Parkinson’s Disease: SC-Nanophytosomes as Therapeutic Approach. Int. J. Mol. Sci. 2023, 24, 16787. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Y.; Zhou, T.-T.; Zhang, Y.; Chen, N.-H.; Yuan, Y.-H. Distribution of α-Synuclein Aggregation in the Peripheral Tissues. Neurochem. Res. 2022, 47, 3627–3634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, J.; Shen, F.-F.; Yuan, Y.-S.; Li, X.; Ji, P.; Zhu, L.; Sun, L.; Ding, J.; Niu, Q.; et al. Activated Schwann Cells and Increased Inflammatory Cytokines IL-1β, IL-6, and TNF-α in Patients’ Sural Nerve Are Lack of Tight Relationship with Specific Sensory Disturbances in Parkinson’s Disease. CNS Neurosci. Ther. 2020, 26, 518–526. [Google Scholar] [CrossRef]
- Dzamko, N. Cytokine Activity in Parkinson’s Disease. Neuronal Signal. 2023, 7, NS20220063. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]

 = increase
= increase  = decrease.
= increase = decrease.
= decrease.
= increase = decrease.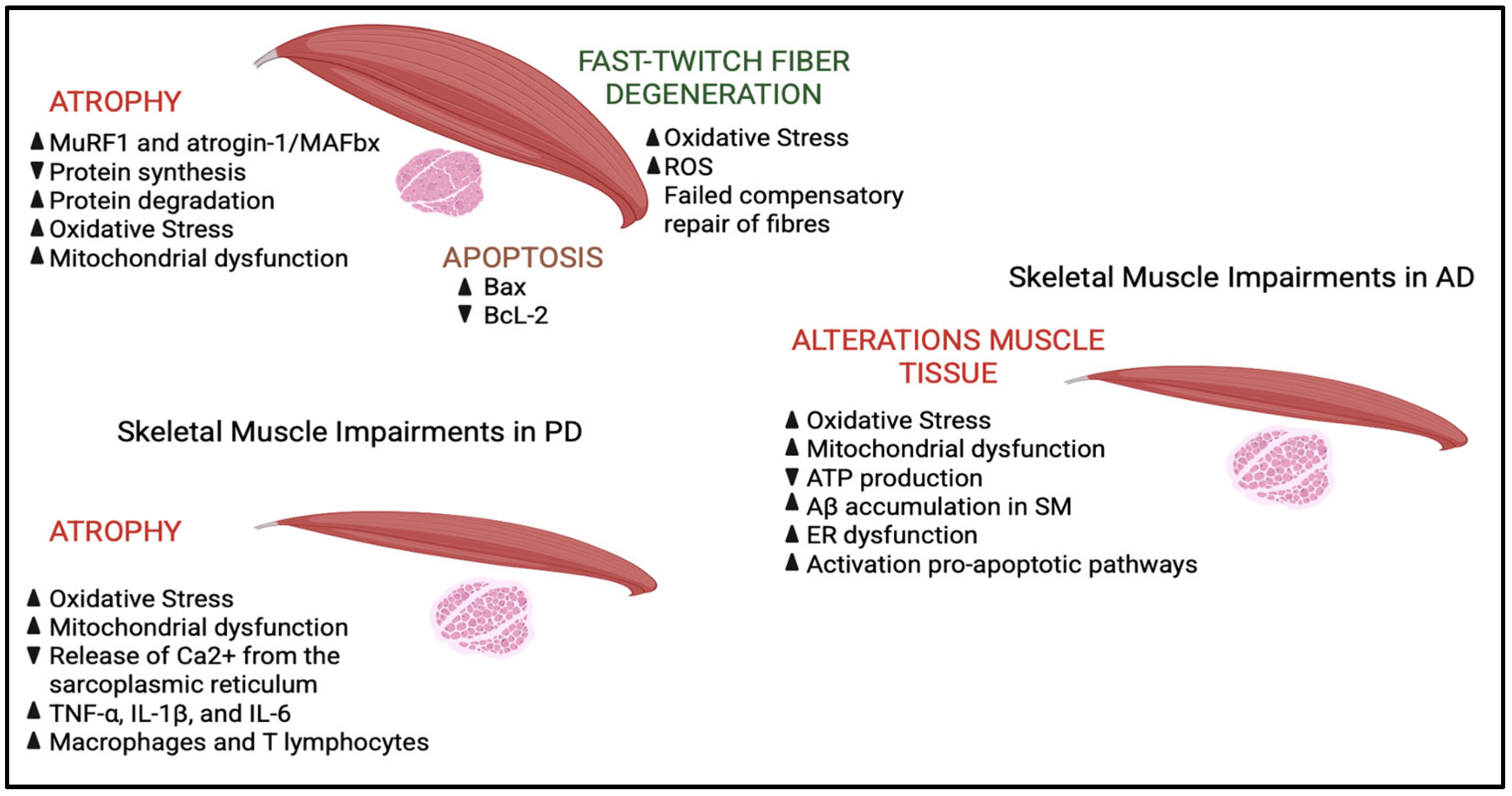

{kind=link}
{kind=link}
{kind=link}
| Enzyme | Classification | EC Number | Main Substrate(s) | Product(s) | Cellular Role |
|---|---|---|---|---|---|
| NADPH oxydase (NOX) | pro-oxidant | EC 1.6.3.1 | NADPH + O2 | Superoxide (O2•−) + NADP+ + H+ | Host defense, signaling, and Cell differentiation |
| Myeloperoxidase (MPO) | pro-oxidant | EC 1.11.2.2 | H2O2 + Cl− (or Br−) | Hypochlorous (or hypobromous acid) acid (HOCl, HOBr) + H2O | Antimicrobial activity |
| Xanthine oxidase (XOR) | pro-oxidant | EC 1.17.3.2 | Hypoxanthine/Xanthine + O2 | Uric acid + Superoxide (O2•−·) + Hydrogen peroxide H2O2 | Catabolism of purine nucleotides |
| Superoxide dismutases (SODs) | anti-oxidant | EC 1.15.1.1 | Superoxide (O2•−) | Hydrogen peroxide H2O2 + O2 | ROS detoxification, Antioxidant defense, Redox signaling modulation, Cell survival and homeostasis, Inflammation regulation |
| Catalase (CAT) | anti-oxidant | EC 1.11.1.6 | Hydrogen peroxide (H2O2) | Water (H2O) + O2 | Antioxidant defense, Redox homeostasis, Redox signaling modulation, Cell survival, Inflammation control |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castelli, S.; Carinci, E.; Baldelli, S. Oxidative Stress in Neurodegenerative Disorders: A Key Driver in Impairing Skeletal Muscle Health. Int. J. Mol. Sci. 2025, 26, 5782. https://doi.org/10.3390/ijms26125782
Castelli S, Carinci E, Baldelli S. Oxidative Stress in Neurodegenerative Disorders: A Key Driver in Impairing Skeletal Muscle Health. International Journal of Molecular Sciences. 2025; 26(12):5782. https://doi.org/10.3390/ijms26125782
Chicago/Turabian StyleCastelli, Serena, Emily Carinci, and Sara Baldelli. 2025. "Oxidative Stress in Neurodegenerative Disorders: A Key Driver in Impairing Skeletal Muscle Health" International Journal of Molecular Sciences 26, no. 12: 5782. https://doi.org/10.3390/ijms26125782
APA StyleCastelli, S., Carinci, E., & Baldelli, S. (2025). Oxidative Stress in Neurodegenerative Disorders: A Key Driver in Impairing Skeletal Muscle Health. International Journal of Molecular Sciences, 26(12), 5782. https://doi.org/10.3390/ijms26125782