
Retinal Autophagy for Sustaining Retinal Integrity as a Proof of Concept for Age-Related Macular Degeneration

 ,
,  ,
, 
Abstract
1. General Introduction
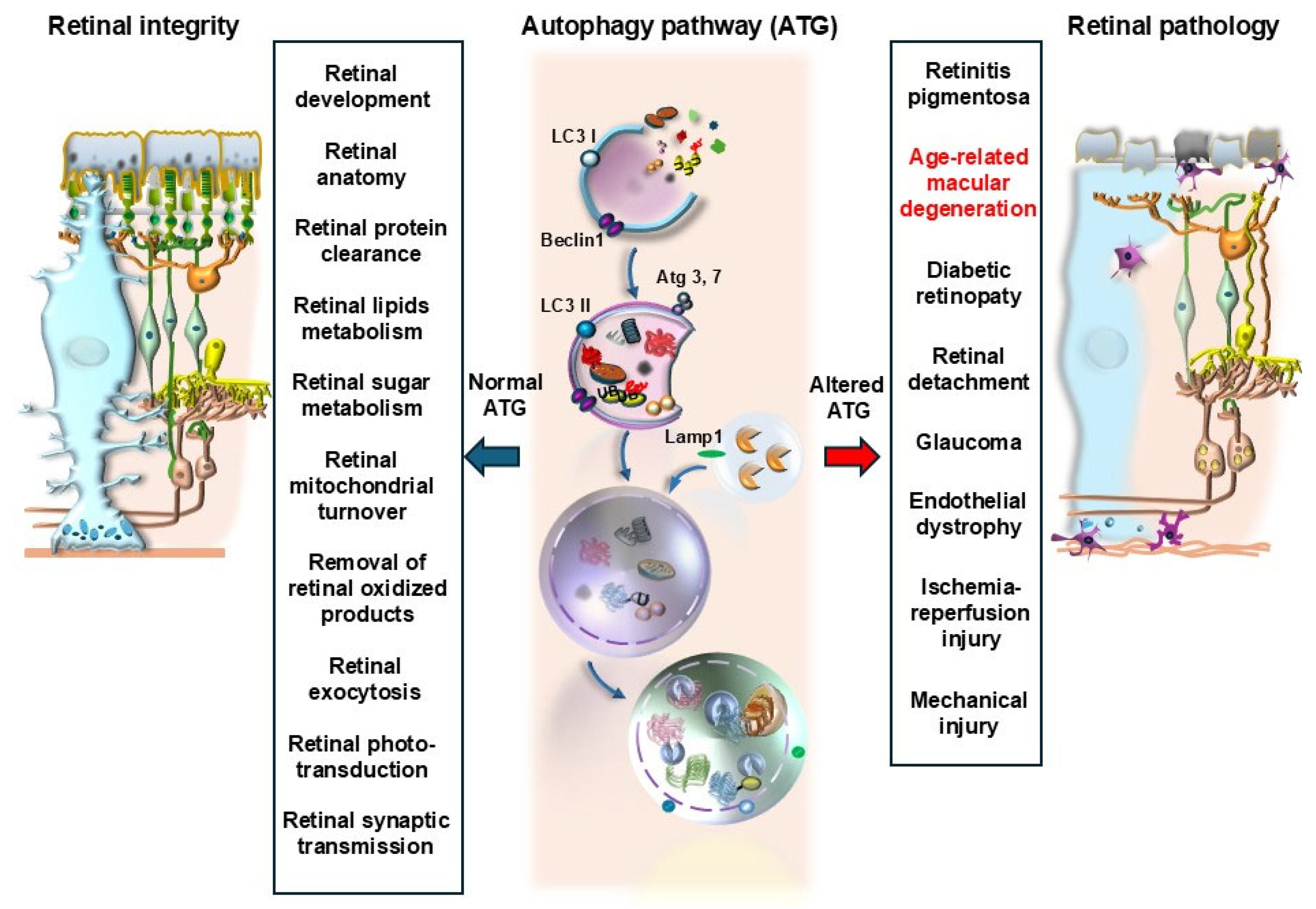
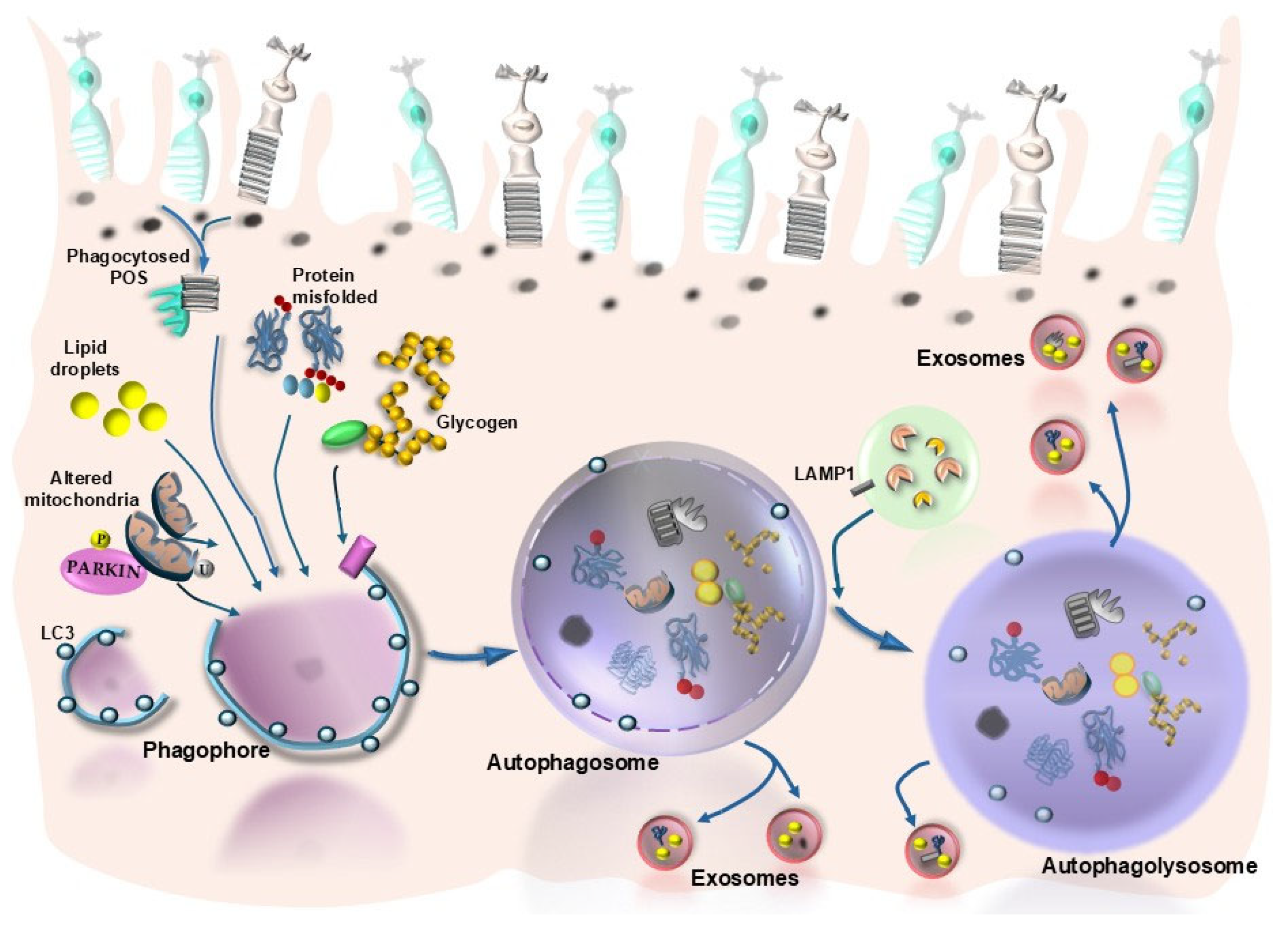
2. Introducing Autophagy in AMD
3. Protein Misfolding/Unfolding, Autophagy, and Retinal Degeneration
4. Lipids Autophagy (Lipophagy) and Retinal Degeneration
5. Glycation End-Products, Autophagy, and Retinal Degeneration
6. Mitochondria, Autophagy, and Retinal Degeneration
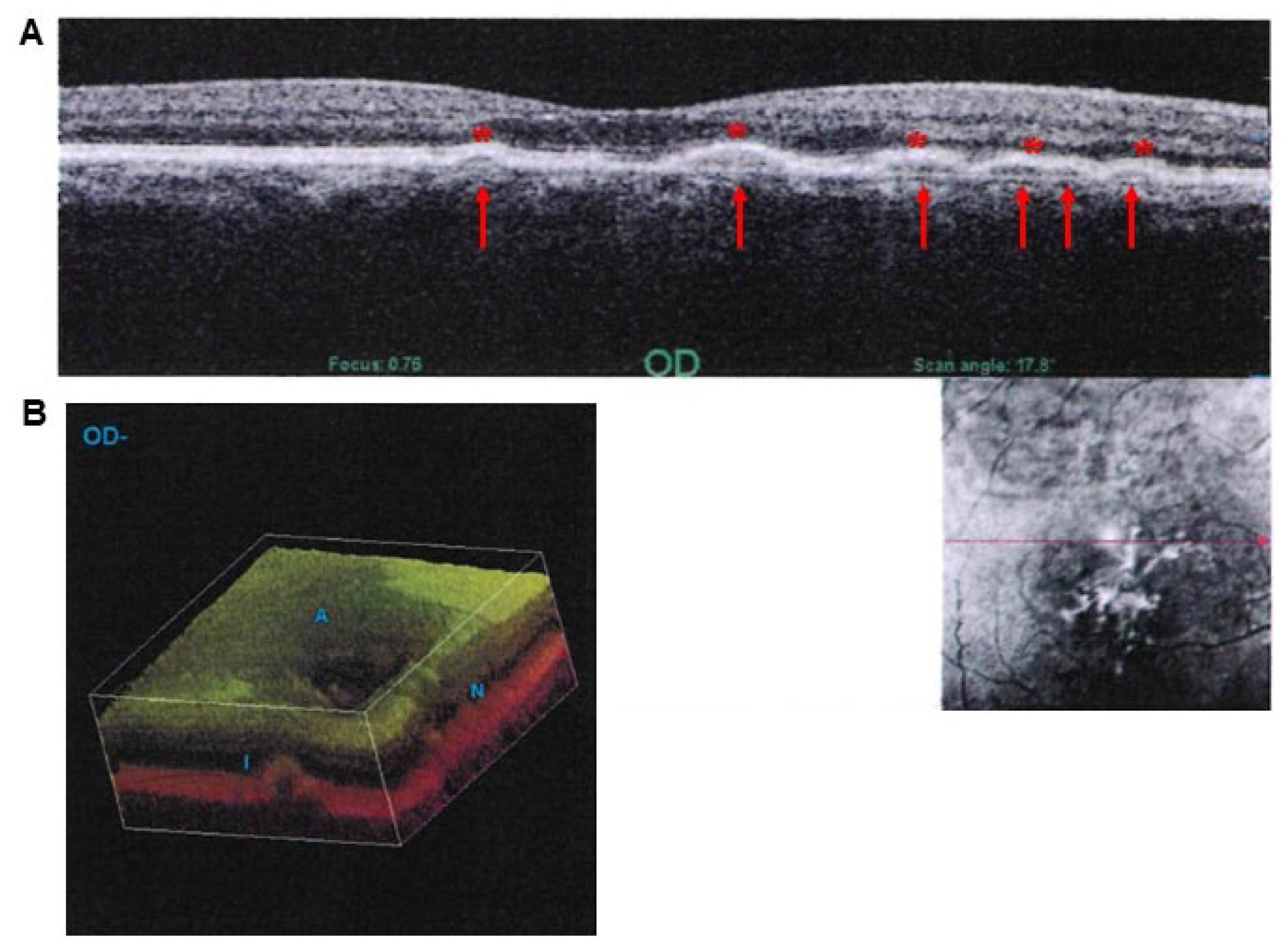
7. Intersections Between Ineffective Autophagy Clearance and Drusen Formation in AMD
8. Do Drusen Act as Innocent Bystanders or Effective Toxic Aggregates That Impair the Visual Process?
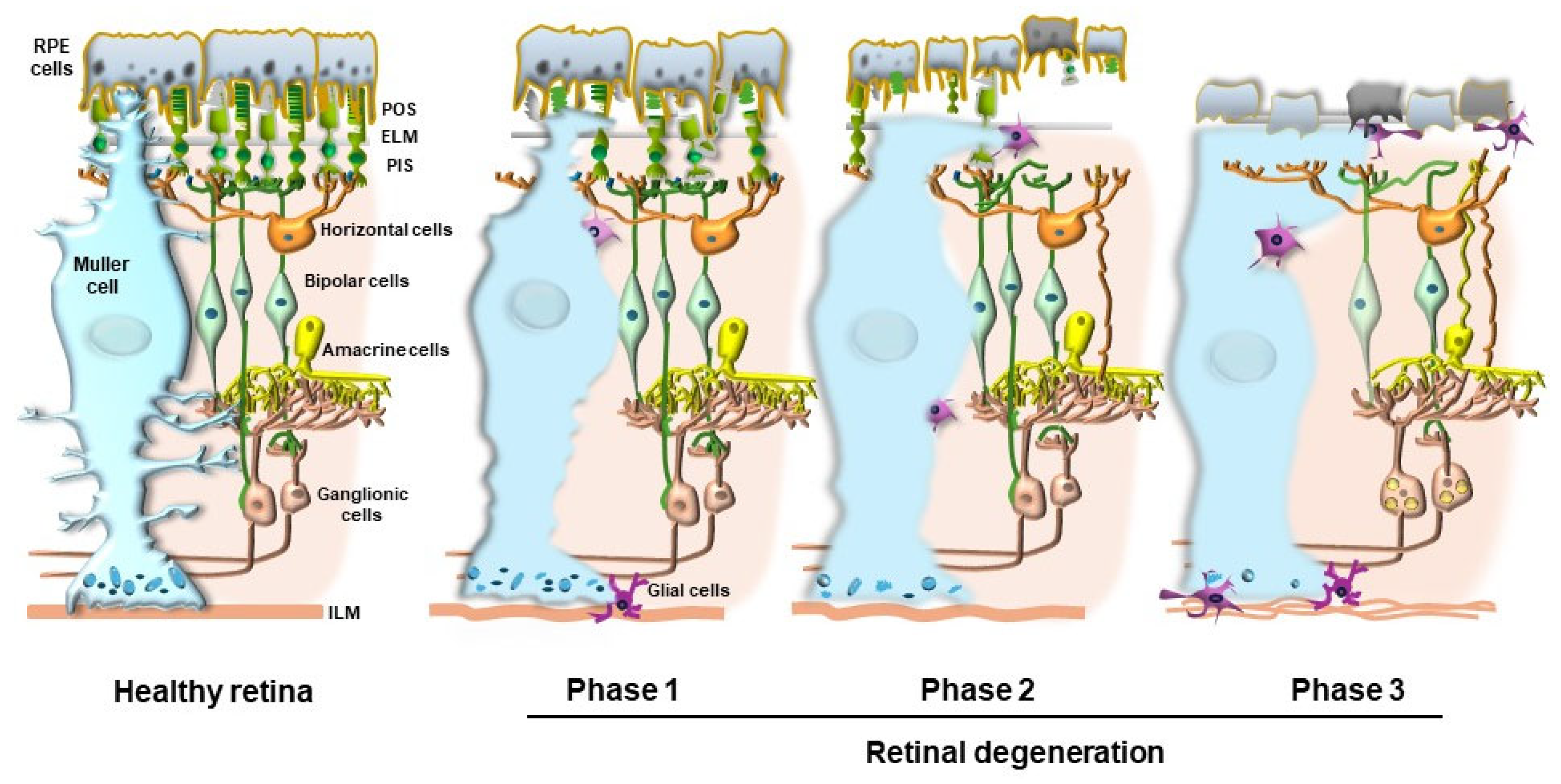
9. Commonalities Between Retinal and CNS Degeneration
10. Interaction Between Autophagy and Additional Mechanisms Involved in AMD
11. Implications for Therapeutic Strategies
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Markitantova, Y.; Simirskii, V. Retinal Pigment Epithelium Under Oxidative Stress: Chaperoning Autophagy and Beyond. Int. J. Mol. Sci. 2025, 26, 1193. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.; Schütt, F.; Holz, F.G.; Kopitz, J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004, 18, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, S. Effect of rapamycin on the fate of P23H opsin associated with retinitis pigmentosa (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 2006, 104, 517–529. [Google Scholar] [PubMed]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy 2007, 3, 433–441. [Google Scholar] [CrossRef]
- Mellén, M.A.; de la Rosa, E.J.; Boya, P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008, 15, 1279–1290. [Google Scholar] [CrossRef]
- Ryhänen, T.; Hyttinen, J.M.; Kopitz, J.; Rilla, K.; Kuusisto, E.; Mannermaa, E.; Viiri, J.; Holmberg, C.I.; Immonen, I.; Meri, S.; et al. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy mediated proteolysis in human retinal pigment epithelial cells. J. Cell. Mol. Med. 2009, 13, 3616–3631. [Google Scholar] [CrossRef]
- Cai, J.; Liao, F.; Mao, Y.; Liu, S.; Wu, X.; Tang, S.; Wang, S.; Shan, G.; Wu, S. Regulation of LAMTOR1 by oxidative stress in retinal pigment epithelium: Implications for age-related macular degeneration pathogenesis. Exp. Eye Res. 2024, 249, 110129. [Google Scholar] [CrossRef]
- Nashine, S.; Cohen, P.; Chwa, M.; Lu, S.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death Dis. 2017, 8, e2951. [Google Scholar] [CrossRef]
- Pinelli, R.; Bertelli, M.; Scaffidi, E.; Polzella, M.; Fulceri, F.; Biagioni, F.; Fornai, F. Nutraceuticals for dry age-related macular degeneration: A case report based on novel pathogenic and morphological insights. Arch. Ital. Biol. 2020, 158, 24–34. [Google Scholar] [CrossRef]
- Pinelli, R.; Biagioni, F.; Scaffidi, E.; Bumah, V.V.; Busceti, C.L.; Puglisi-Allegra, S.; Lazzeri, G.; Fornai, F. The potential effects of nutrients and light on autophagy-mediated visual function and clearance of retinal aggregates. Arch. Ital. Biol. 2022, 160, 115–135. [Google Scholar] [CrossRef]
- Pinelli, R.; Ferrucci, M.; Biagioni, F.; Berti, C.; Bumah, V.V.; Busceti, C.L.; Puglisi-Allegra, S.; Lazzeri, G.; Frati, A.; Fornai, F. Autophagy Activation Promoted by Pulses of Light and Phytochemicals Counteracting Oxidative Stress during Age-Related Macular Degeneration. Antioxidants 2023, 12, 1183. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, R.; Ferrucci, M.; Biagioni, F.; Bumah, V.; Scaffidi, E.; Puglisi-Allegra, S.; Fornai, F. Curcumin as a Perspective Protection for Retinal Pigment Epithelium during Autophagy Inhibition in the Course of Retinal Degeneration. Curr. Neuropharmacol. 2023, 21, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, R.; Ferrucci, M.; Berti, C.; Biagioni, F.; Scaffidi, E.; Bumah, V.V.; Busceti, C.L.; Lenzi, P.; Lazzeri, G.; Fornai, F. The Essential Role of Light-Induced Autophagy in the Inner Choroid/Outer Retinal Neurovascular Unit in Baseline Conditions and Degeneration. Int. J. Mol. Sci. 2023, 24, 8979. [Google Scholar] [CrossRef]
- Ding, X.; Cao, S.; Wang, Q.; Du, B.; Lu, K.; Qi, S.; Cheng, Y.; Tuo, Q.Z.; Liang, W.; Lei, P. DNALI1 Promotes Neurodegeneration after Traumatic Brain Injury via Inhibition of Autophagosome-Lysosome Fusion. Adv. Sci. 2024, 11, e2306399. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2023, 19, 388–400. [Google Scholar] [CrossRef]
- Pinelli, R.; Biagioni, F.; Limanaqi, F.; Bertelli, M.; Scaffidi, E.; Polzella, M.; Busceti, C.L.; Fornai, F. A Re-Appraisal of Pathogenic Mechanisms Bridging Wet and Dry Age-Related Macular Degeneration Leads to Reconsider a Role for Phytochemicals. Int. J. Mol. Sci. 2020, 21, 5563. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Koskela, A.; Blasiak, J.; Kaarniranta, K. Autophagy in drusen biogenesis secondary to age-related macular degeneration. Acta Ophthalmol. 2024, 102, 759–772. [Google Scholar] [CrossRef]
- Wang, X.L.; Gao, Y.X.; Yuan, Q.Z.; Zhang, M. NLRP3 and autophagy in retinal ganglion cell inflammation in age-related macular degeneration: Potential therapeutic implications. Int. J. Ophthalmol. 2024, 17, 1531–1544. [Google Scholar] [CrossRef]
- Prieto-Garcia, C.; Matkovic, V.; Mosler, T.; Li, C.; Liang, J.; Oo, J.A.; Haidle, F.; Mačinković, I.; Cabrera-Orefice, A.; Berkane, R.; et al. Pathogenic proteotoxicity of cryptic splicing is alleviated by ubiquitination and ER-phagy. Science 2024, 386, 768–776. [Google Scholar] [CrossRef]
- Azam, M.; Jastrzebska, B. Mechanisms of Rhodopsin-Related Inherited Retinal Degeneration and Pharmacological Treatment Strategies. Cells 2025, 14, 49. [Google Scholar] [CrossRef]
- Awad, A.M.; Seetharaman, A.T.M.; Hossain, M.S.; Elshaer, S.L.; Abdelaziz, R.R.; Nader, M.A.; Gangaraju, R. Cysteine Leukotriene Receptor Antagonist-Montelukast Effects on Diabetic Retinal Microvascular Endothelial Cells Curtail Autophagy. Investig. Ophthalmol. Vis. Sci. 2024, 65, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Feng, J.; Pan, Y.; Ouyang, L.; He, T.; Xing, Y. Mettl3-Mediated N6-Methyladenosine Modification Mitigates Ganglion Cell Loss and Retinal Dysfunction in Retinal Ischemia-Reperfusion Injury by Inhibiting FoxO1-Mediated Autophagy. Investig. Ophthalmol. Vis. Sci. 2025, 66, 58. [Google Scholar] [CrossRef]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin. Eye Res. 2020, 74, 100771. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Müller Cell Metabolic Signatures: Evolutionary Conservation and Disruption in Disease. Trends Endocrinol. Metab. 2020, 31, 320–329. [Google Scholar] [CrossRef]
- Pfeiffer, R.L.; Anderson, J.R.; Dahal, J.; Garcia, J.C.; Yang, J.H.; Sigulinsky, C.L.; Rapp, K.; Emrich, D.P.; Watt, C.B.; Johnstun, H.A.; et al. A pathoconnectome of early neurodegeneration: Network changes in retinal degeneration. Exp. Eye Res. 2020, 199, 108196. [Google Scholar] [CrossRef]
- Chowdhury, O.; Bammidi, S.; Gautam, P.; Babu, V.S.; Liu, H.; Shang, P.; Xin, Y.; Mahally, E.; Nemani, M.; Koontz, V.; et al. Activated mTOR Signaling in the RPE Drives EMT, Autophagy, and Metabolic Disruption, Resulting in AMD-Like Pathology in Mice. Aging Cell 2025, 17, e70018. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Notomi, S.; Wu, G.; Fukuda, Y.; Maehara, Y.; Fukushima, M.; Murakami, Y.; Takahashi, M.; Izumi, Y.; Sonoda, K.H. Altered fatty acid distribution in lysosome-associated membraneprotein-2 deficient mice. Biochem. Biophys. Rep. 2024, 40, 101822. [Google Scholar] [CrossRef]
- Shatz, N.; Chohan, Y.; Klionsky, D.J. ATG14 and STX18: Gatekeepers of lipid droplet degradation and the implications for disease modulation. Autophagy 2024, 20, 1697–1699. [Google Scholar] [CrossRef]
- Deng, G.; Moran, E.P.; Cheng, R.; Matlock, G.; Zhou, K.; Moran, D.; Chen, D.; Yu, Q.; Ma, J.X. Therapeutic Effects of a Novel Agonist of Peroxisome Proliferator-Activated Receptor Alpha for the Treatment of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5030–5042. [Google Scholar] [CrossRef]
- Zhu, L.; Guo, L.; Xu, J.; Xiang, Q.; Tan, Y.; Tian, F.; Du, X.; Zhang, S.; Wen, T.; Liu, L. Postprandial Triglyceride-Rich Lipoproteins-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Inflammation in White Adipocytes. J. Nutr. 2024, 154, 1619–1630. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Romagnoli, A.; Refolo, G.; Vescovo, T.; Ciccosanti, F.; Zuchegna, C.; Lozzi, F.; Occhigrossi, L.; Piacentini, M.; Fimia, G.M. Role of AMBRA1 in mitophagy regulation: Emerging evidence in aging-related diseases. Autophagy 2024, 20, 2602–2615. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Maugeri, G.; Magrì, B.; Bucolo, C.; D’Agata, V. Targeting the PINK1/Parkin pathway: A new perspective in the prevention and therapy of diabetic retinopathy. Exp. Eye Res. 2024, 247, 110024. [Google Scholar] [CrossRef]
- Zhang, J.; Li, W.; Liu, Z.; Chen, Y.; Wei, X.; Peng, L.; Xu, M.; Ji, Y. Defective post-transcriptional modification of tRNA disrupts mitochondrial homeostasis in Leber’s hereditary optic neuropathy. J. Biol. Chem. 2024, 300, 107728. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.Y.; Valapala, M. Role of TFEB in Diseases Associated with Lysosomal Dysfunction. Adv. Exp. Med. Biol. 2023, 1415, 319–325. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, J.; Liang, Y.; Huang, J.; Fu, Y.; Chen, N.; Lu, B.; Zhao, C. Clearance of lipid droplets by chimeric autophagy-tethering compound ameliorates the age- related macular degeneration phenotype in mice lacking APOE. Autophagy 2023, 19, 2668–2681. [Google Scholar] [CrossRef]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed]
- Rosato, A.S.; Tang, R.; Grimm, C. Two-pore and TRPML cation channels: Regulators of phagocytosis, autophagy and lysosomal exocytosis. Pharmacol. Ther. 2021, 220, 107713. [Google Scholar] [CrossRef]
- Shahhossein-Dastjerdi, S.; Koina, M.E.; Fatseas, G.; Arfuso, F.; Chan-Ling, T. Autophagy and Exocytosis of Lipofuscin Into the Basolateral Extracellular Space of Human Retinal Pigment Epithelium From Fetal Development to Adolescence. Investig. Ophthalmol. Vis. Sci. 2024, 65, 32. [Google Scholar] [CrossRef]
- Sundaramurthi, H.; Roche, S.L.; Grice, G.L.; Moran, A.; Dillion, E.T.; Campiani, G.; Nathan, J.A.; Kennedy, B.N. Selective Histone Deacetylase 6 Inhibitors Restore Cone Photoreceptor Vision or Outer Segment Morphology in Zebrafish and Mouse Models of Retinal Blindness. Front. Cell Dev. Biol. 2020, 8, 689. [Google Scholar] [CrossRef]
- Sun, K.; Chen, J.; Fan, Y.; Cai, J.; Jiang, X.; Liu, W.; Zhu, X. Lack of retinal degeneration in a Dram2 knockout mouse model. Vision Res. 2025, 226, 108509. [Google Scholar] [CrossRef]
- Nag, T.C. Accumulation of autophagosomes in aging human photoreceptor cell synapses. Exp. Eye Res. 2025, 251, 110240. [Google Scholar] [CrossRef] [PubMed]
- Tyszka, A.; Szypulski, K.; Pyza, E.; Damulewicz, M. Autophagy in the retina affects photoreceptor synaptic plasticity and behavior. J. Insect. Physiol. 2025, 161, 104741. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Helotera, H.; Ionov, M.; Derwich, M.; Kaarniranta, K. Potential of autophagy in subretinal fibrosis in neovascular age-related macular degeneration. Cell Mol. Biol. Lett. 2025, 30, 54. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.L.; Thompson, D.A.; et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 2015, 11, 939–953. [Google Scholar] [CrossRef]
- Giorgianni, F.; Beranova-Giorgianni, S. Oxidized low-density lipoprotein causes ribosome reduction and inhibition of protein synthesis in retinal pigment epithelial cells. Biochem. Biophys. Rep. 2022, 32, 101345. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hose, S.; Sinha, D. AKT2-mediated lysosomal dysfunction promotes secretory autophagy in retinal pigment epithelium (RPE) cells. Autophagy 2024, 20, 2841–2842. [Google Scholar] [CrossRef]
- Dalvi, S.; Roll, M.; Chatterjee, A.; Kumar, L.K.; Bhogavalli, A.; Foley, N.; Arduino, C.; Spencer, W.; Reuben-Thomas, C.; Ortolan, D.; et al. Human iPSC-based disease modeling studies identify a common mechanistic defect and potential therapies for AMD and related macular dystrophies. Dev. Cell 2024, 59, 3290–3305.e9. [Google Scholar] [CrossRef]
- Vujosevic, S.; Alovisi, C.; Chakravarthy, U. Epidemiology of geographic atrophy and its precursor features of intermediate age-related macular degeneration. Acta Ophthalmol. 2023, 101, 839–856. [Google Scholar] [CrossRef]
- Agrón, E.; Domalpally, A.; Chen, Q.; Lu, Z.; Chew, E.Y.; Keenan, T.D.L.; AREDS and AREDS2 Research Groups. An Updated Simplified Severity Scale for Age-Related Macular Degeneration Incorporating Reticular Pseudodrusen: Age-Related Eye Disease Study Report Number 42. Ophthalmology 2024, 131, 1164–1174. [Google Scholar] [CrossRef]
- Anderson, D.M.G.; Kotnala, A.; Migas, L.G.; Patterson, N.H.; Tideman, L.; Cao, D.; Adhikari, B.; Messinger, J.D.; Ach, T.; Tortorella, S.; et al. Lysolipids are prominent in subretinal drusenoid deposits, a high-risk phenotype in age-related macular degeneration. Front. Ophthalmol. 2023, 3, 1258734. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Loygorri, J.I.; Boya, P. Recycling the recyclers: Lysophagy emerges as anew pharmacological target for retinal degeneration. Autophagy 2024, 20, 2589–2590. [Google Scholar] [CrossRef] [PubMed]
- Bammidi, S.; Ghosh, S.; Chowdhury, O.; Babu, V.S.; Dutta, P.; Hose, S.; Sinha, D. MLST8 overexpression in RPE cells disrupts autophagy through novel mechanisms affecting AMD pathogenesis. Autophagy 2025, 9, 1–3. [Google Scholar] [CrossRef]
- Wei, J.; Chen, X.; Xiong, Y.; Gao, Y. Advances in Ubiquitination and Proteostasis in Retinal Degeneration. Front. Biosci. Landmark Ed. 2024, 29, 260. [Google Scholar] [CrossRef]
- Feng, L.; Li, X.; Shen, W.; Gao, C. Stress granules as transient reservoirs for autophagy proteins: A key mechanism for plant recovery from heat stress. Autophagy 2025, 19, 1–3. [Google Scholar] [CrossRef]
- Mautone, L.; Cordella, F.; Soloperto, A.; Ghirga, S.; Di Gennaro, G.; Gigante, Y.; Di Angelantonio, S. Understanding retinal tau pathology through functional 2D and 3D iPSC-derived in vitro retinal models. Acta Neuropathol. Commun. 2025, 13, 19. [Google Scholar] [CrossRef]
- Fathinajafabadi, A.; Pérez-Jiménez, E.; Riera, M.; Knecht, E.; Gonzàlez-Duarte, R. CERKL, a retinal disease gene, encodes an mRNA-binding protein that localizes in compact and untranslated mRNPs associated with microtubules. PLoS ONE 2014, 9, e87898. [Google Scholar] [CrossRef]
- Hu, X.; Lu, Z.; Yu, S.; Reilly, J.; Liu, F.; Jia, D.; Qin, Y.; Han, S.; Liu, X.; Qu, Z.; et al. CERKL regulates autophagy via the NAD-dependent deacetylase SIRT1. Autophagy 2019, 15, 453–465. [Google Scholar] [CrossRef] [PubMed]
- García-Arroyo, R.; Gavaldà-Navarro, A.; Villarroya, F.; Marfany, G.; Mirra, S. Overexpression of CERKL Protects Retinal Pigment Epithelium Mitochondria from Oxidative Stress Effects. Antioxidants 2021, 10, 2018. [Google Scholar] [CrossRef]
- Biswas, P.; Woodard, D.R.; Hollingsworth, T.J.; Khan, N.W.; Lazaro, D.R.; Berry, A.M.; Dagar, M.; Pan, Y.; Garland, D.; Shaw, P.X.; et al. Ablation of Htra1 leads to sub-RPE deposits and photoreceptor abnormalities. JCI Insight. 2025, 10, e178827. [Google Scholar] [CrossRef]
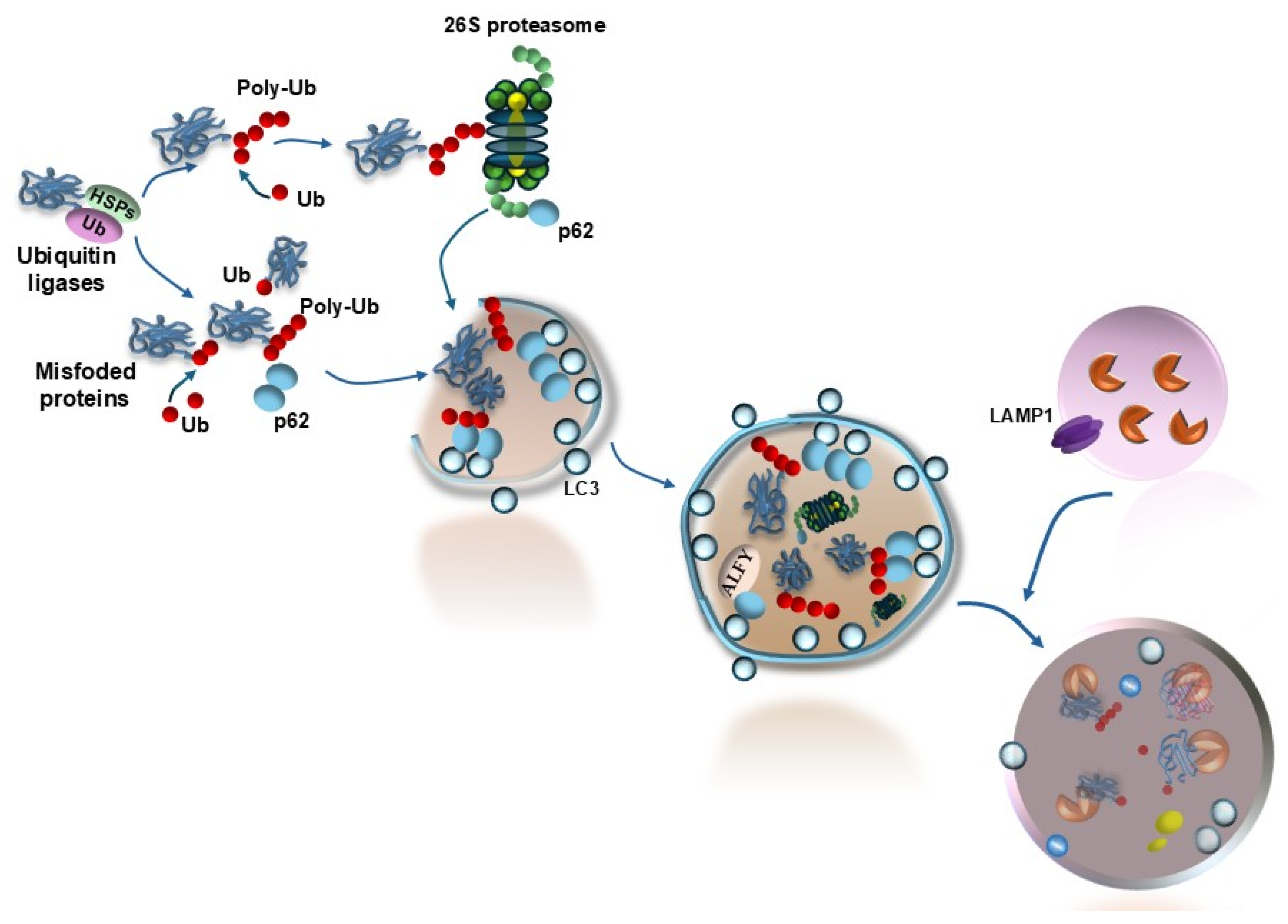
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Ciechanover, A.; Livneh, I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget 2016, 7, 83833–83834. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, S.; Biagioni, F.; Ferese, R.; Busceti, C.L.; Frati, A.; Novelli, G.; Ruggieri, S.; Fornai, F. Vacuolar Protein Sorting Genes in Parkinson’s Disease: A Re- appraisal of Mutations Detection Rate and Neurobiology of Disease. Front. Neurosci. 2016, 10, 532. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Salvetti, A.; Puglisi-Allegra, S.; Lenzi, P.; Fornai, F. Morphology, clearing efficacy, and mTOR dependency of the organelle autophagoproteasome. Eur. J. Histochem. 2021, 65, 3220. [Google Scholar] [CrossRef]
- Leger, F.; Fernagut, P.O.; Canron, M.H.; Léoni, S.; Vital, C.; Tison, F.; Bezard, E.; Vital, A. Protein aggregation in the aging retina. J. Neuropathol. Exp. Neurol. 2011, 70, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, R.; Biagioni, F.; Bertelli, M.; Busceti, C.L.; Scaffidi, E.; Ryskalin, L.; Fornai, F. Retinal Degeneration Following Chronic Administration of the Parkinsonism-Inducing Neurotoxin MPTP. Arch. Ital. Biol. 2021, 159, 64–81. [Google Scholar] [CrossRef]
- Anderson, D.H.; Talaga, K.C.; Rivest, A.J.; Barron, E.; Hageman, G.S.; Johnson, L.V. Characterization of beta amyloid assemblies in drusen: The deposits associated with aging and age-related macular degeneration. Exp. Eye Res. 2004, 78, 243–256. [Google Scholar] [CrossRef]
- Luibl, V.; Isas, J.M.; Kayed, R.; Glabe, C.G.; Langen, R.; Chen, J. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J. Clin. Investig. 2006, 116, 378–385. [Google Scholar] [CrossRef]
- Kivinen, N.; Felszeghy, S.; Kinnunen, A.I.; Setälä, N.; Aikio, M.; Kinnunen, K.; Sironen, R.; Pihlajaniemi, T.; Kauppinen, A.; Kaarniranta, K. Absence of collagen XVIII in mice causes age-related insufficiency in retinal pigment epithelium proteostasis. Biogerontology 2016, 17, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and exosomes in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PLoS ONE 2009, 4, e4160. [Google Scholar] [CrossRef]
- La Cunza, N.; Tan, L.X.; Thamban, T.; Germer, C.J.; Rathnasamy, G.; Toops, K.A.; Lakkaraju, A. Mitochondria-dependent phase separation of disease-relevant proteins drivespathological features of age-related macular degeneration. JCI Insight 2021, 6, e142254. [Google Scholar] [CrossRef]
- Ren, C.; Cui, H.; Bao, X.; Huang, L.; He, S.; Fong, H.K.W.; Zhao, M. Proteopathy Linked to Exon-Skipping Isoform of RGR-Opsin Contributes to the Pathogenesis of Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2023, 64, 41. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, J.; Usategui-Martin, R.; Sanabria, M.R.; Fernandez-Perez, E.; Telleria, J.J.; Coco-Martin, R.M. Pathophysiology of Age-Related Macular Degeneration: Implications for Treatment. Ophthalmic. Res. 2022, 65, 615–636. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Veréb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef]
- Dialynaki, D.; Klionsky, D.J. Identification of the mammalian VPS4A as a selective lipophagy receptor. Autophagy 2025, 21, 691–692. [Google Scholar] [CrossRef]
- Das, D.; Sharma, M.; Gahlot, D.; Nia, S.S.; Gain, C.; Mecklenburg, M.; Zhou, Z.H.; Bourdenx, M.; Thukral, L.; Martinez-Lopez, N.; et al. VPS4A is the selective receptor for lipophagy in mice and humans. Mol. Cell 2024, 84, 4436–4453.e8. [Google Scholar] [CrossRef]
- Zhang, C.; Lai, M.B.; Khandan, L.; Lee, L.A.; Chen, Z.; Junge, H.J. Norrin-induced Frizzled4 endocytosis and endo-lysosomal trafficking control retinal angiogenesis and barrier function. Nat. Commun. 2017, 8, 16050. [Google Scholar] [CrossRef]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, S.; Guan, W.; Xu, N.; Zhu, L.; Du, W.; Liu, Z.; Fong, H.K.W.; Huang, L.; Zhao, M. Retinal G-protein-coupled receptor deletion exacerbates AMD-like changes via the PINK1-parkin pathway under oxidative stress. FASEB J. 2024, 38, e70135. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Clark, M.E.; Rudolf, M.; Curcio, C.A. Distribution and composition of esterified and unesterified cholesterol in extra-macular drusen. Exp. Eye Res. 2007, 85, 192–201. [Google Scholar] [CrossRef]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jia, L.; Feathers, K.; Lin, C.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Zacks, D.N. Autophagy-mediated catabolism ofvisual transduction proteins prevents retinal degeneration. Autophagy 2016, 12, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Sinha, D.; Kaarniranta, K. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res. 2016, 51, 69–89. [Google Scholar] [CrossRef]
- Heckel, E.; Cagnone, G.; Agnihotri, T.; Cakir, B.; Das, A.; Kim, J.S.; Kim, N.; Lavoie, G.; Situ, A.; Pundir, S.; et al. Triglyceride-derived fatty acids reduce autophagy in a model of retinal angiomatous proliferation. JCI Insight 2022, 7, e154174. [Google Scholar] [CrossRef]
- Tzankov, A. Retinoic acid-induced Golgi apparatus disruption in F2000 fibroblasts: A model for enhanced intracellular retrograde transport. J. Biochem. Mol. Biol. 2003, 36, 265–268. [Google Scholar] [CrossRef]
- Tokarz, P.; Piastowska-Ciesielska, A.W.; Kaarniranta, K.; Blasiak, J. All-Trans Retinoic Acid Modulates DNA Damage Response and the Expression of the VEGF-A and MKI67 Genes in ARPE-19 Cells Subjected to Oxidative Stress. Int. J. Mol. Sci. 2016, 17, 898. [Google Scholar] [CrossRef]
- Lazzeri, G.; Lenzi, P.; Signorini, G.; Raffaelli, S.; Giammattei, E.; Natale, G.; Ruffoli, R.; Fornai, F.; Ferrucci, M. Retinoic Acid Promotes Neuronal Differentiation While Increasing Proteins and Organelles Related to Autophagy. Int. J. Mol. Sci. 2025, 26, 1691. [Google Scholar] [CrossRef]
- Cao, J.J.; Han, F.F.; Kou, Z.Y.; Zhuang, T.T.; Dong, L.J.; Zhang, H.; Teng, H. Effect of SB431542 on autophagy and epithelial mesenchymal transition in retinal pigment epithelial cells induced by high glucose. Zhonghua Yan Ke Za Zhi 2025, 61, 202–210. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, C.; Zhang, J.; Xu, G.T.; Zhang, J. Molecular pathogenesis of subretinal fibrosis in neovascular AMD focusing on epithelial-mesenchymal transformation of retinal pigment epithelium. Neurobiol. Dis. 2023, 185, 106250. [Google Scholar] [CrossRef] [PubMed]
- Klingeborn, M.; Arora, V.; Chung, C.; Donchenko, P.; Wright, R.N.; Reese, E.D.; Gunn, T.M. Desmosome and Hemidesmosome Release via Exosomes from Retinal Pigmented Epithelium—A Precursor to Epithelial-Mesenchymal Transition in Early AMD? Curr. Eye Res. 2025, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gabrielle, P.H. Lipid metabolism and retinal diseases. Acta Ophthalmol. 2022, 100, 3–43. [Google Scholar] [CrossRef] [PubMed]
- Jarocki, M.; Turek, K.; Saczko, J.; Tarek, M.; Kulbacka, J. Lipids associated with autophagy: Mechanisms and therapeutic targets. Cell Death Discov. 2024, 10, 460. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef]
- Zhang, J.L.; Wang, X.F.; Li, J.L.; Duan, C.; Wang, J.F. The cholesterol metabolite 25-hydroxycholesterol suppresses porcine deltacoronavirus via lipophagy inhibition and mTORC1 modulation. Vet. Res. 2025, 56, 23. [Google Scholar] [CrossRef]
- Vessey, K.A.; Jobling, A.I.; Tran, M.X.; Wang, A.Y.; Greferath, U.; Fletcher, E.L. Treatments targeting autophagy ameliorate the age-related macular degeneration phenotype in mice lacking APOE (apolipoprotein E). Autophagy 2022, 18, 2368–2384. [Google Scholar] [CrossRef]
- Landowski, M.; Bowes Rickman, C. Targeting lipid metabolism for the treatment of age-related macular degeneration: Insights from preclinical mouse models. J. Ocul. Pharmacol. Ther. 2022, 38, 3–32. [Google Scholar] [CrossRef]
- George, A.A.; Hayden, S.; Stanton, G.R.; Brockerhoff, S.E. Arf6 and the 5′phosphataseof synaptojanin 1 regulate autophagy in cone photoreceptors. Bioessays 2016, 38, S119–S135. [Google Scholar] [CrossRef]
- Izumi, Y.; Ishikawa, M.; Nakazawa, T.; Kunikata, H.; Sato, K.; Covey, D.F.; Zorumski, C.F. Neurosteroids as stress modulators and neurotherapeutics: Lessons from the retina. Neural. Regen Res. 2023, 18, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, S.; Qin, X.; Hou, W.; Dong, H.; Yao, L.; Xiong, L. The pleiotropic roles of sphingolipid signaling in autophagy. Cell Death Dis. 2014, 5, e1245. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Domenech-Bendaña, A.; Avila-Portillo, N.; Rowan, S.; Edirisinghe, S.; Taylor, A. Glycative stress as a cause of macular degeneration. Prog. Retin. Eye Res. 2024, 101, 101260. [Google Scholar] [CrossRef] [PubMed]
- Hansman, D.S.; Du, J.; Casson, R.J.; Peet, D.J. Eye on the horizon: The metabolic landscape of the RPE in aging and disease. Prog. Retin. Eye Res. 2025, 104, 101306. [Google Scholar] [CrossRef]
- Ahmed, N.; Thornalley, P.J.; Dawczynski, J.; Franke, S.; Strobel, J.; Stein, G.; Haik, G.M. Methylglyoxalderived hydroimidazolone advanced glycation end-products of human lens proteins. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5287–5292. [Google Scholar] [CrossRef]
- Aragonès, G.; Dasuri, K.; Olukorede, O.; Francisco, S.G.; Renneburg, C.; Kumsta, C.; Hansen, M.; Kageyama, S.; Komatsu, M.; Rowan, S.; et al. Autophagic receptor p62 protects against glycation-derived toxicity and enhances viability. Aging Cell 2020, 19, e13257. [Google Scholar] [CrossRef]
- Aragonès, G.; Rowan, S.G.; Francisco, S.; Yang, W.; Weinberg, J.; Taylor, A.; Bejarano, E. Glyoxalase system as a therapeutic target against diabetic retinopathy. Antioxidants 2020, 9, 1062. [Google Scholar] [CrossRef]
- Saeed, M.; Kausar, M.A.; Singh, R.; Siddiqui, A.J.; Akhter, A. The role of glyoxalase in glycation and carbonyl stress induced metabolic disorders. Curr. Protein Pept. Sci. 2020, 21, 846–859. [Google Scholar] [CrossRef]
- Schutt, F.; Bergmann, M.; Holz, F.G.; Kopitz, J. Proteins modified bymalondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3663–3668. [Google Scholar] [CrossRef]
- Zhang, S.M.; Fan, B.; Li, Y.L.; Zuo, Z.Y.; Li, G.Y. Oxidative Stress-Involved Mitophagy of Retinal Pigment Epithelium and Retinal Degenerative Diseases. Cell Mol Neurobiol. 2023, 43, 3265–3276. [Google Scholar] [CrossRef]
- Koller, A.; Lamina, C.; Brandl, C.; Zimmermann, M.E.; Stark, K.J.; Weissensteiner, H.; Würzner, R.; Heid, I.M.; Kronenberg, F. Systemic Evidence for Mitochondrial Dysfunction in Age-Related Macular Degeneration as Revealed by mtDNA Copy Number Measurements in Peripheral Blood. Int. J. Mol. Sci. 2023, 24, 16406. [Google Scholar] [CrossRef] [PubMed]
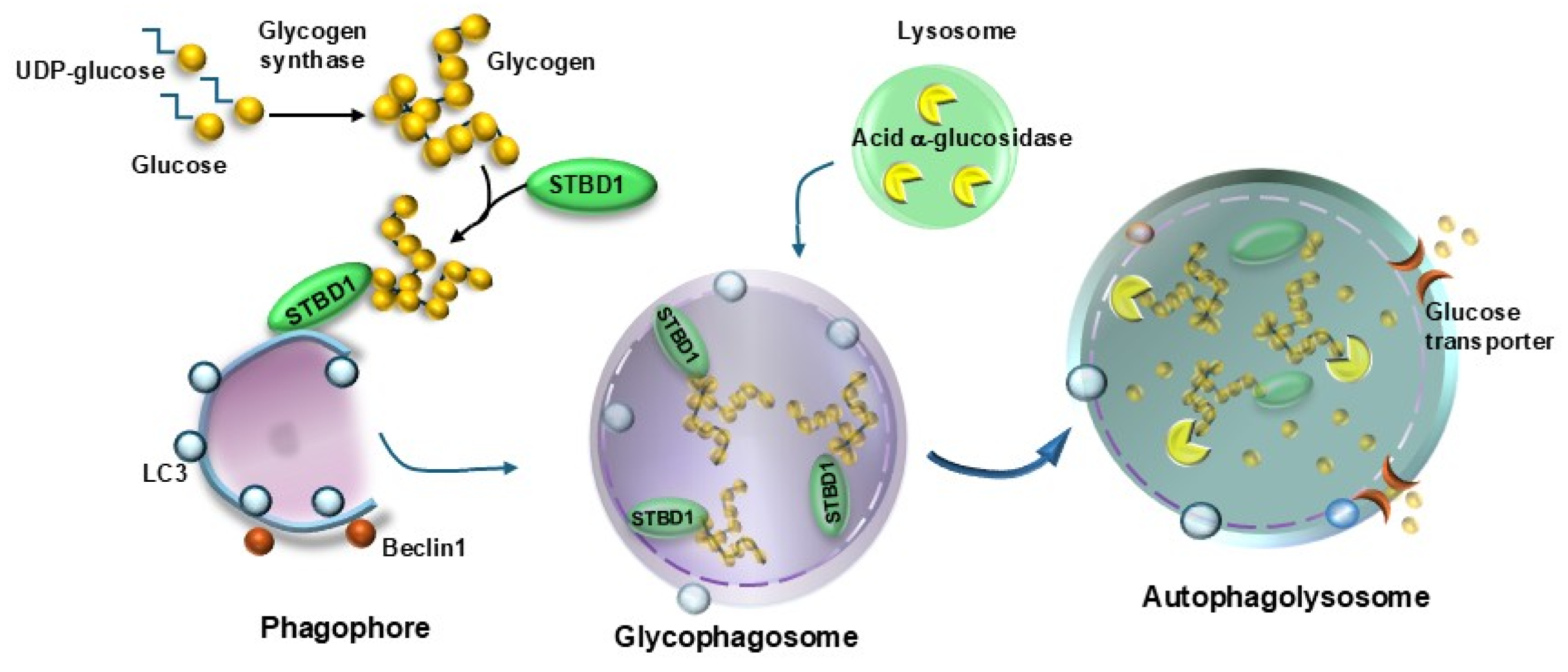
- Koutsifeli, P.; Varma, U.; Daniels, L.J.; Annandale, M.; Li, X.; Neale, J.P.H.; Hayes, S.; Weeks, K.L.; James, S.; Delbridge, L.M.D.; et al. Glycogen-autophagy: Molecular machinery and cellular mechanisms of glycophagy. J. Biol. Chem. 2022, 298, 102093. [Google Scholar] [CrossRef]
- Mancini, M.C.; Noland, R.C.; Collier, J.J.; Burke, S.J.; Stadler, K.; Heden, T.D. Lysosomalglucose sensing and glycophagy in metabolism. Trends Endocrinol. Metab. 2023, 34, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Laqtom, N.N.; Dong, W.; Medoh, U.N.; Cangelosi, A.L.; Dharamdasani, V.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Heinze, I.; Tang, R.; et al. CLN3 is required for the clearance of glycerophosphodiesters from lysosomes. Nature 2022, 609, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Canibano-Fraile, R.; Harlaar, L.; Dos Santos, C.A.; Hoogeveen-Westerveld, M.; Demmers, J.A.A.; Snijders, T.; Lijnzaad, P.; Verdijk, R.M.; van der Beek, N.A.M.E.; van Doorn, P.A.; et al. Lysosomal glycogen accumulation in Pompe disease results in disturbed cytoplasmic glycogen metabolism. J. Inherit. Metab. 2023, 46, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.X.; Xu, Y.C.; Pantopoulos, K.; Tan, X.Y.; Wei, X.L.; Zheng, H.; Luo, Z. Glycophagy mediated glucose-induced changes of hepatic glycogen metabolism via OGT1-AKT1-FOXO1(Ser238) pathway. J. Nutr. Biochem. 2023, 117, 109337. [Google Scholar] [CrossRef]
- Banevicius, M.; Vilkeviciute, A.; Kriauciuniene, L.; Liutkeviciene, R.; Deltuva, V.P. The Association Between Variants of Receptor for Advanced Glycation End Products (RAGE) Gene Polymorphisms and Age-Related Macular Degeneration. Med. Sci. Monit. 2018, 24, 190–199. [Google Scholar] [CrossRef]
- Roddy, G.W. Metabolic syndrome and the aging retina. Curr. Opin. Ophthalmol. 2021, 32, 280–287. [Google Scholar] [CrossRef]
- Roehlecke, C.; Valtink, M.; Frenzel, A.; Goetze, D.; Knels, L.; Morawietz, H.; Funk, R.H. Stress responses of human retinal pigment epithelial cells to glyoxal. Graefes. Arch. Clin. Exp. Ophthalmol. 2016, 254, 2361–2372. [Google Scholar] [CrossRef]
- Glenn, J.V.; Mahaffy, H.; Wu, K.; Smith, G.; Nagai, R.; Simpson, D.A.; Boulton, M.E.; Stitt, A.W. Advanced glycation end product (AGE) accumulation on Bruch’s membrane: Links to age-related RPE dysfunction. Investig. Ophthalmol. Vis. Sci. 2009, 50, 441–451. [Google Scholar] [CrossRef]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD pathobiology: A bioenergetic crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhang, Z.; Wang, S. Role of mitochondria in retinal pigment epithelial aging and degeneration. Front. Aging 2022, 3, 926627. [Google Scholar] [CrossRef]
- Ramirez-Pardo, I.; Villarejo-Zori, B.; Jimenez-Loygorri, J.I.; Sierra-Filardi, E.; Alonso-Gil, S.; Mariño, G.; de la Villa, P.; Fitze, P.S.; Fuentes, J.M.; García-Escudero, R.; et al. Ambra1 haploinsufficiency in CD1 mice results in metabolic alterations and exacerbates age-associated retinal degeneration. Autophagy 2023, 19, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.R.; Shaaeli, A.A.; Ebeling, M.C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondrial fission, fusion, and autophagy in retinal pigment epithelium from donors with age-related macular degeneration. Sci. Rep. 2022, 12, 21725. [Google Scholar] [CrossRef]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [PubMed]
- Mohtashami, Z.; Schneider, K.; Azimi, R.; Atilano, S.; Chwa, M.; Kenney, M.C.; Singh, M.K. Exploring the therapeutic potential of MOTS-c in age-related macular degeneration: From cellular responses to patient-derived cybrids. Hum. Cell 2025, 38, 57. [Google Scholar] [CrossRef]
- Taskintuna, K.; Bhat, M.A.; Shaikh, T.; Hum, J.; Golestaneh, N. Sex-dependent regulation of retinal pigment epithelium and retinal function by Pgc-1α. Front. Cell Neurosci. 2024, 18, 1442079. [Google Scholar] [CrossRef]
- Shu, D.Y.; Butcher, E.R.; Saint-Geniez, M. Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4701. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coupling mitogenesis and mitophagy for longevity. Autophagy 2015, 11, 1428–1430. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mitophagy: In sickness and in health. Mol. Cell Oncol. 2015, 3, e1056332. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharm. Acool Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Markaki, M.; Palikaras, K.; Tavernarakis, N. Novel Insights Into the Anti-aging Role of Mitophagy. Int. Rev. Cell Mol. Biol. 2018, 340, 169–208. [Google Scholar] [CrossRef]
- Zaninello, M.; Palikaras, K.; Naon, D.; Iwata, K.; Herkenne, S.; Quintana-Cabrera, R.; Semenzato, M.; Grespi, F.; Ross-Cisneros, F.N.; Carelli, V.; et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 2020, 11, 4029. [Google Scholar] [CrossRef]
- Bell, K.; Rosignol, I.; Sierra-Filardi, E.; Rodriguez-Muela, N.; Schmelter, C.; Cecconi, F.; Grus, F.; Boya, P. Age related retinal Ganglion cell susceptibility in context of autophagy deficiency. Cell Death Discov. 2020, 6, 21. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef]
- Sepe, S.; Nardacci, R.; Fanelli, F.; Rosso, P.; Bernardi, C.; Cecconi, F.; Mastroberardino, P.G.; Piacentini, M.; Moreno, S. Expression of Ambra1 in mouse brain during physiological and Alzheimer type aging. Neurobiol. Aging 2014, 35, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhao, Y.; Li, Z.; Zhu, M.; Wang, Z.; Li, Y.; Xu, T.; Feng, D.; Zhang, S.; Tang, F.; et al. miR-103a-3p regulates mitophagy in Parkinson’s disease through Parkin/Ambra1 signaling. Pharmacol. Res. 2020, 160, 105197. [Google Scholar] [CrossRef]
- Zhang, M.; Schekman, R. Cell biology. Unconventional secretion, unconventional solutions. Science 2013, 340, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Spraul, C.W.; Lang, G.E.; Grossniklaus, H.E.; Lang, G.K. Histologic and morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv. Ophthalmol. 1999, 44, S10–S32. [Google Scholar] [CrossRef] [PubMed]
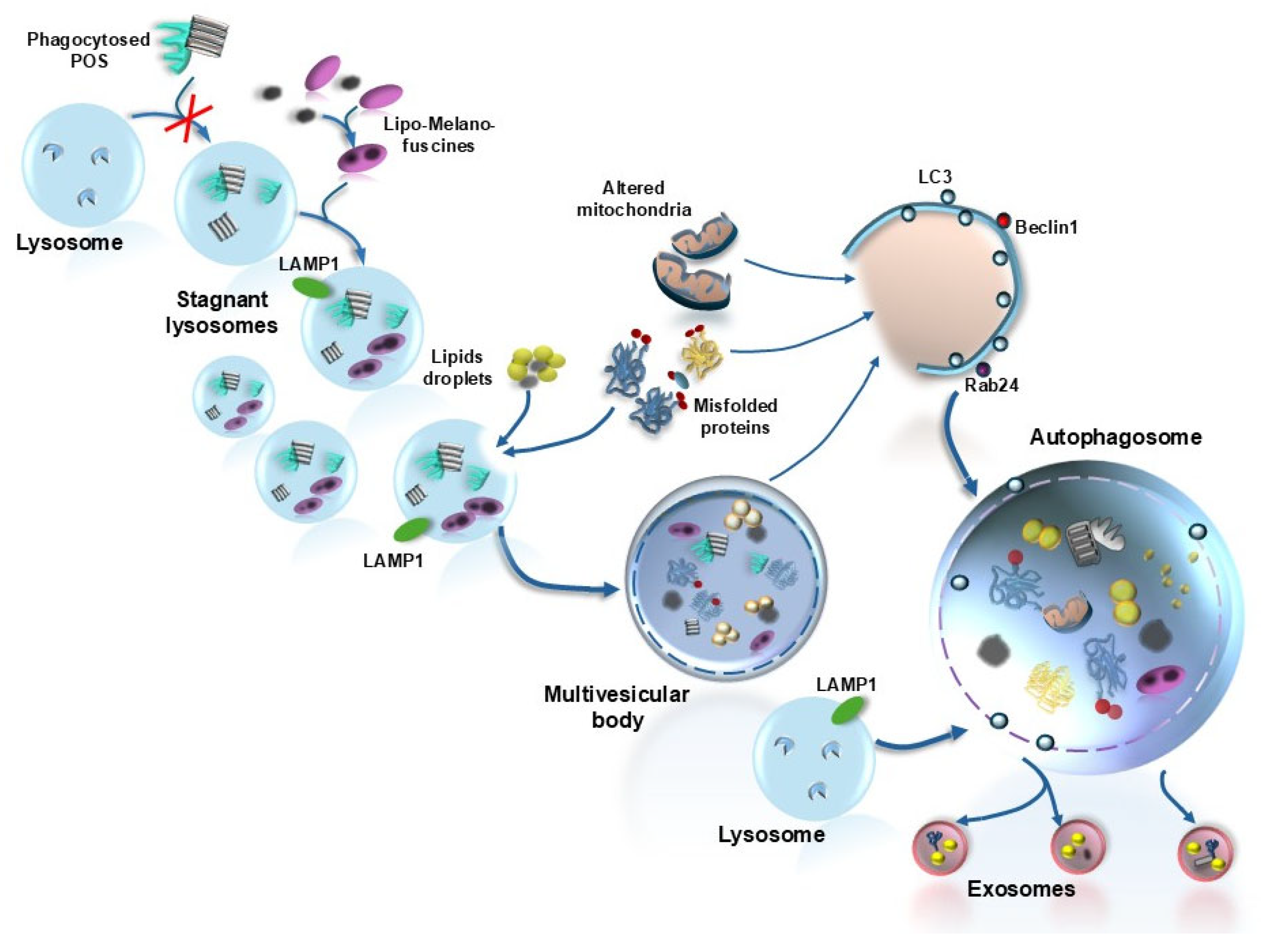
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Hyttinen, J.; Ryhänen, T.; Viiri, J.; Paimela, T.; Toropainen, E.; Sorri, I.; Salminen, A. Mechanism of protein aggregation on the retinal pigment epitelial cells. Front. Biosci. 2010, 2, 1374–1384. [Google Scholar] [CrossRef]
- Boyer, N.P.; Tang, P.H.; Higbee, D.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. Lipofuscin and A2E accumulate with age in the retinal pigment epithelium of Nrl-/- mice. Photochem. Photobiol. 2012, 88, 1373–1377. [Google Scholar] [CrossRef]
- Grimes, W.N.; Berson, D.M.; Sabnis, A.; Hoon, M.; Sinha, R.; Tian, H.; Diamond, J.S. Layer-specific anatomical and physiological features of the retina’s neurovascular unit. Curr. Biol. 2025, 35, 109–120.e4. [Google Scholar] [CrossRef]
- Yang, L.; Yao, Y.; Zheng, W.; Zheng, X.; Xie, M.; Huang, L. Nitric oxide mediates negative feedback on the TXNIP/NLRP3 inflammasome pathway to prevent retinal neurovascular unit dysfunction in early diabetic retinopathy. Free Radic. Biol. Med. 2025, 233, 279–291. [Google Scholar] [CrossRef]
- Udom, G.J.; Oritsemuelebi, B.; Frazzoli, C.; Bocca, B.; Ruggieri, F.; Orisakwe, O.E. Tyazhelyemetallyinarusheniyauglevodnogoobmenaprizabolevaniyakh organa zreniya: Sistematicheskiiobzor [Heavy metals and derangement in carbohydrate metabolism in eye diseases: A systematic review]. Vestn. Oftalmol. 2025, 141, 89–100. (In Russian) [Google Scholar] [CrossRef]
- Qambari, H.; Hein, M.; Balaratnasingam, C.; Yu, P.; Yu, D.Y. Enabling visualization of GFAP-positive retinal glial cells, neurons and microvasculature in three-dimensions. Exp. Eye Res. 2025, 28, 110410. [Google Scholar] [CrossRef]
- Villeneuve, J.; Bassaganyas, L.; Lepreux, S.; Chiritoiu, M.; Costet, P.; Ripoche, J.; Malhotra, V.; Schekman, R. Unconventional secretion of FABP4 by endosomes and secretory lysosomes. J. Cell Biol. 2018, 217, 649–665. [Google Scholar] [CrossRef]
- McKechnie, N.M.; King, B.C.; Fletcher, E.; Braun, G. Fas-ligand is stored in secretory lysosomes of ocular barrier epithelia and released with microvesicles. Exp. Eye Res. 2006, 83, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact endothelial autophagy is required to maintain vascular lipidhomeostasis. Aging Cell 2016, 15, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Gupta, V.; Bae, S.; Sharma, S. Metamorphopsia and vision-related quality of life among patients with age-related macular degeneration. Can. J. Ophthalmol. 2018, 53, 168–172. [Google Scholar] [CrossRef]
- Zhang, S.; Ren, J.; Chai, R.; Yuan, S.; Hao, Y. Global burden of low vision and blindness due to age-related macular degeneration from 1990 to 2021 and projections for 2050. BMC Public Health 2024, 24, 3510. [Google Scholar] [CrossRef] [PubMed]
- Sverdlichenko, I.; Mandelcorn, M.S.; IssasharLeibovitzh, G.; Mandelcorn, E.D.; Markowitz, S.N.; Tarita-Nistor, L. Binocular visual function and fixational control in patients with macular disease: A review. Ophthalmic Physiol. Opt. 2022, 42, 258–271. [Google Scholar] [CrossRef]
- Altınbay, D.; İdil, Ş.A. Fixation Stability and Preferred Retinal Locus in Advanced Age-Related Macular Degeneration. Turk J. Ophthalmol. 2022, 52, 23–29. [Google Scholar] [CrossRef]
- Guadron, L.; Titchener, S.A.; Abbott, C.J.; Ayton, L.N.; van Opstal, J.; Petoe, M.A.; Goossens, J. The Saccade Main Sequence in Patients With Retinitis Pigmentosa and Advanced Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2023, 64, 1. [Google Scholar] [CrossRef]
- Hanumunthadu, D.; Lescrauwaet, B.; Jaffe, M.; Sadda, S.; Wiecek, E.; Hubschman, J.P.; Patel, P.J. Clinical Update on Metamorphopsia: Epidemiology, Diagnosis and Imaging. Curr. Eye Res. 2021, 46, 1777–1791. [Google Scholar] [CrossRef]
- Nomoto, H.; Matsumoto, C.; Arimura, E.; Okuyama, S.; Takada, S.; Hashimoto, S.; Shimomura, Y. Quantification of changes in metamorphopsia and retinal contraction in eyes with spontaneous separation of idiopathic epiretinal membrane. Eye 2013, 27, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Birner, K.; Reiter, G.S.; Steiner, I.; Deák, G.; Mohamed, H.; Schürer-Waldheim, S.; Gumpinger, M.; Bogunović, H.; Schmidt-Erfurth, U. Topographic and quantitative correlation of structure and function using deep learning in subclinical biomarkers of intermediate age-related macular degeneration. Sci. Rep. 2024, 14, 28165. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.X.; Pickel, L.; Berger, A.R.; Sivachandran, N. Improvement in Dry Age-Related Macular Degeneration with Photobiomodulation. Case Rep. Ophthalmol. 2025, 16, 155–162. [Google Scholar] [CrossRef]
- Bennett, C.; Romano, F.; Vingopoulos, F.; Garcia, M.; Ding, X.; Bannerman, A.; Ploumi, I.; Ntentakis, D.; Stettler, I.; Overbey, K.; et al. Associations Between Contrast Sensitivity, Optical Coherence Tomography Features and Progression From Intermediate to Late Age-related Macular Degeneration. Am. J. Ophthalmol. 2025, 271, 175–187. [Google Scholar] [CrossRef]
- Küçük, E.; Çoban Karataş, M. Comparison of choroidal thickness and vascularity in patients with subretinal drusenoid deposits and large drusen using swept-source optical coherence tomography. Int. Ophthalmol. 2025, 45, 94. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, R.; Bertelli, M.; Scaffidi, E.; Fulceri, F.; Busceti, C.L.; Biagioni, F.; Fornai, F. Measurement of drusen and their correlation with visual symptoms in patients affected by age-related macular degeneration. Arch. Ital. Biol. 2020, 158, 82–104. [Google Scholar] [CrossRef]
- Medical Advisory Secretariat. Optical coherence tomography for age-related macular degeneration and diabetic macular edema: An evidence-based analysis. Ont. Health Technol. Assess. Ser. 2009, 9, 1–22. [Google Scholar]
- Ahmed, H.; Sierpina, D.I.; Khazaeni, L. Retinal Pattern Dystrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gurubaran, I.S. Mitochondrial damage and clearance in retinal pigment epithelial cells. Acta Ophthalmol. 2024, 102, 3–53. [Google Scholar] [CrossRef]
- Blasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef]
- Huang, P.; Sun, J.; Wang, F.; Luo, X.; Feng, J.; Gu, Q.; Liu, T.; Sun, X. MicroRNA Expression Patterns Involved in Amyloid Beta-Induced Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1726–1735. [Google Scholar] [CrossRef]
- Ferguson, T.A.; Green, D.R. Autophagy and phagocytosis converge for better vision. Autophagy 2014, 10, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Tucker, J.; Marc, R.E. Retinal Remodeling and Metabolic Alterations in Human AMD. Front. Cell Neurosci. 2016, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Naso, F.; Intartaglia, D.; Falanga, D.; Soldati, C.; Polishchuk, E.; Giamundo, G.; Tiberi, P.; Marrocco, E.; Scudieri, P.; Di Malta, C.; et al. Light-responsive microRNA miR-211 targets Ezrin to modulate lysosomal biogenesis and retinal cell clearance. EMBO J. 2020, 39, e102468. [Google Scholar] [CrossRef]
- Intartaglia, D.; Giamundo, G.; Naso, F.; Nusco, E.; Di Giulio, S.; Salierno, F.G.; Polishchuk, E.; Conte, I. Induction of Autophagy Promotes Clearance of RHOP23H Aggregates and Protects From Retinal Degeneration. Front. Aging Neurosci. 2022, 14, 878958. [Google Scholar] [CrossRef]
- Intartaglia, D.; Giamundo, G.; Conte, I. Autophagy in the retinal pigment epithelium: A new vision and future challenges. FEBS J. 2022, 289, 7199–7212. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Satyanarayana, G.; Liu, T.; Wang, L.; Wang, J.; Cheng, J.; Itoh, K.; Sharma, A.; Bhutto, I.; et al. Mitophagy initiates retrograde mitochondrial-nuclear signaling to guide retinal pigment cell heterogeneity. Autophagy 2023, 19, 966–983. [Google Scholar] [CrossRef]
- Choi, M.S.; Kim, H.J.; Ham, M.; Choi, D.H.; Lee, T.R.; Shin, D.W. Amber Light (590 nm) Induces the Breakdown of Lipid Droplets through Autophagy-Related Lysosomal Degradation in Differentiated Adipocytes. Sci. Rep. 2016, 6, 28476. [Google Scholar] [CrossRef]
- Wen, R.H.; Stanar, P.; Tam, B.; Moritz, O.L. Autophagy in Xenopus laevis rod photoreceptors is independently regulated by phototransduction and misfolded RHOP23H. Autophagy 2019, 15, 1970–1989. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wei, L.; Liu, D.; Zhang, Q.; Xia, X.; Ding, L.; Xiong, S. Identification and Validation of Autophagy-Related Genes in Diabetic Retinopathy. Front. Endocrinol. 2022, 13, 867600. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Błasiak, J.; Niittykoski, M.; Kinnunen, K.; Kauppinen, A.; Salminen, A.; Kaarniranta, K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2017, 36, 64–77. [Google Scholar] [CrossRef]
- Santo, M.; Conte, I. Emerging Lysosomal Functions for Photoreceptor Cell Homeostasis and Survival. Cells 2021, 11, 60. [Google Scholar] [CrossRef]
- Ramachandra Rao, S.; Fliesler, S.J. Monitoring basal autophagy in the retina utilizing CAG-mRFP-EGFP-MAP1LC3B reporter mouse: Technical and biological considerations. Autophagy 2022, 18, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.; Freeman, S.A. Phagocytosis by the Retinal Pigment Epithelium: Recognition, Resolution, Recycling. Front. Immunol. 2020, 11, 604205. [Google Scholar] [CrossRef]
- Frost, L.S.; Lopes, V.S.; Bragin, A.; Reyes-Reveles, J.; Brancato, J.; Cohen, A.; Mitchell, C.H.; Williams, D.S.; Boesze-Battaglia, K. The Contribution of Melanoregulin to Microtubule-Associated Protein 1 Light Chain 3 (LC3) Associated Phagocytosis in Retinal Pigment Epithelium. Mol. Neurobiol. 2015, 52, 1135–1151. [Google Scholar] [CrossRef] [PubMed]
- Muniz-Feliciano, L.; Doggett, T.A.; Zhou, Z.; Ferguson, T.A. RUBCN/rubicon and EGFR regulate lysosomal degradative processes in the retinal pigment epithelium (RPE) of the eye. Autophagy 2017, 13, 2072–2085. [Google Scholar] [CrossRef]
- Archibald, N.K.; Clarke, M.P.; Mosimann, U.P.; Burn, D.J. The retina in Parkinson’s disease. Brain 2009, 132, 1128–1145. [Google Scholar] [CrossRef]
- Dwivedi, A.; Kumar, A.; Faruq, M.; Singh, V.K.; Dwivedi, N.; Singh, K.; Hussain, I.; Parida, S.; Jha, G.K.; Kumar, N.; et al. Co-occurrence of Parkinson’s disease and Retinitis Pigmentosa: A genetic and in silico analysis. Neuroscience 2025, 565, 519–526. [Google Scholar] [CrossRef]
- Lee, J.; Kang, S.H.; Koh, S.B. Postural instability and gait disturbance are associated with abnormal stereopsis in Parkinson’s disease. PLoS ONE 2025, 20, e0317935. [Google Scholar] [CrossRef]
- Cunha, L.P.; Martins, P.N.; Martins, L.C.; Almada, F.M.D.N.; Shigaeff, N.; Araújo, D.O.; Mello, L.G.M.; Monteiro, M.L.R.; Snyder, P.J.; Vale, T.C. Correlation between motor symptoms, cognitive function, and optical coherence tomography findings in Parkinson’s disease. Arq. Bras. Oftalmol. 2024, 88, e2024-0049. [Google Scholar] [CrossRef]
- Fornai, F.; Soldani, P.; Lazzeri, G.; di Poggio, A.B.; Biagioni, F.; Fulceri, F.; Batini, S.; Ruggieri, S.; Paparelli, A. Neuronal inclusions in degenerative disorders Do they represent static features or a key to understand the dynamics of the disease? Brain Res. Bull. 2005, 65, 275–290. [Google Scholar] [CrossRef]
- Lazzeri, G.; Lenzi, P.; Busceti, C.L.; Ferrucci, M.; Falleni, A.; Bruno, V.; Paparelli, A.; Fornai, F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007, 101, 1414–1427. [Google Scholar] [CrossRef]
- Lenzi, P.; Lazzeri, G.; Ferrucci, M.; Busceti, C.L.; Puglisi-Allegra, S.; Fornai, F. In situ stoichiometry amounts of p62 and poly-ubiquitin exceed the increase of alpha-synuclein during degeneration of catecholamine cells induced by autophagy inhibition in vitro. J. Neural. Transm. 2024, 131, 1397–1414. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Ferrucci, M.; Scotto, M.; Frati, A.; Puglisi-Allegra, S.; Busceti, C.L.; Fornai, F. Is There a Place for Lewy Bodies before and beyond Alpha-Synuclein Accumulation? Provocative Issues in Need of Solid Explanations. Int. J. Mol. Sci. 2024, 25, 3929. [Google Scholar] [CrossRef]
- Ono, K.; Yamada, M. Vitamin A potently destabilizes preformed alpha-synuclein fibrils in vitro: Implications for Lewy body diseases. Neurobiol. Dis. 2007, 25, 446–454. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J. Neural. Transm. 2015, 122, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Giorgi, F.S.; Alessandrì, M.G.; Giusiani, M.; Corsini, G.U. Effects of pretreatment with N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) on methamphetamine pharmacokinetics and striatal dopamine losses. J. Neurochem. 1999, 72, 777–784. [Google Scholar] [CrossRef]
- Battaglia, G.; Busceti, C.L.; Pontarelli, F.; Biagioni, F.; Fornai, F.; Paparelli, A.; Bruno, V.; Ruggieri, S.; Nicoletti, F. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neuropharmacology 2003, 45, 155–166. [Google Scholar] [CrossRef]
- Firmani, G.; Salducci, M.; Testa, F.; Covelli, G.P.; Sagnelli, P.; Lambiase, A. Ocular Biomarkers in Alzheimer’s Disease: Insights into Early Detection Through Eye-Based Diagnostics—A Literature Review. Clin. Ter. 2024, 175, 352–361. [Google Scholar] [CrossRef]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.M.; Clement, C.; Sambamurti, K.; Dua, P.; Lukiw, W.J. Beta-Amyloid Precursor Protein (βAPP) Processing in Alzheimer’s Disease (AD) and Age-Related Macular Degeneration (AMD). Mol. Neurobiol. 2015, 52, 533–544. [Google Scholar] [CrossRef]
- Donato, L.; Mordà, D.; Scimone, C.; Alibrandi, S.; D’Angelo, R.; Sidoti, A. Bridging Retinal and Cerebral Neurodegeneration: A Focus on Crosslinks between Alzheimer-Perusini’s Disease and Retinal Dystrophies. Biomedicines 2023, 11, 3258. [Google Scholar] [CrossRef]
- Rinaldi, M.; Pezone, A.; Quadrini, G.I.; Abbadessa, G.; Laezza, M.P.; Passaro, M.L.; Porcellini, A.; Costagliola, C. Targeting shared pathways in tauopathies and age-related macular degeneration: Implications for novel therapies. Front. Aging Neurosci. 2024, 16, 1371745. [Google Scholar] [CrossRef] [PubMed]
- Abyadeh, M.; Gupta, V.; Paulo, J.A.; Sheriff, S.; Shadfar, S.; Fitzhenry, M.; Amirkhani, A.; Gupta, V.; Salekdeh, G.H.; Haynes, P.A.; et al. Apolipoprotein ε in Brain and Retinal Neurodegenerative Diseases. Aging Dis. 2023, 14, 1311–1330. [Google Scholar] [CrossRef]
- Suimon, Y.; Nishimura, M.; Murata, M.; Yoshida, S.; Yokoi, K.; Dong, Z.; Kuno, N.; Fujii, S.; Tanei, Z.I.; Yabe, I.; et al. Leucine-Rich Repeat Kinase 2 Promotes Disintegration of Retinal Pigment Epithelial Cell: Implication in the Pathogenesis of Dry Age-Related Macular Degeneration. Am. J. Pathol. 2025, in press. [CrossRef]
- Meng, M.; Shen, X.; Xie, Y.; Wang, J.; Liu, J. The association between age-related macular degeneration and risk of Parkinson disease: A systematic review and meta-analysis. Medicine 2024, 103, e40524. [Google Scholar] [CrossRef]
- Khalatyan, A.S.; Yusef, Y.; Avetisov, K.S.; Plyukhova, A.A. Cognitive impairment and age-related eye pathology. Adv. Gerontol. 2024, 37, 559–565. [Google Scholar]
- Tao, J.X.; Zhou, W.C.; Zhu, X.G. Mitochondria as Potential Targets and Initiators of the Blue Light Hazard to the Retina. Oxid. Med. Cell Longev. 2019, 2019, 6435364. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Yang, C.M.; Yang, C.H. Protective Effect of Astaxanthin on Blue Light Light-Emitting Diode-Induced Retinal Cell Damage via Free Radical Scavenging and Activation of PI3K/Akt/Nrf2 Pathway in 661W Cell Model. Mar. Drugs 2020, 18, 387. [Google Scholar] [CrossRef]
- Lin, Y.H.; Sheu, S.J.; Liu, W.; Hsu, Y.T.; He, C.X.; Wu, C.Y.; Chen, K.J.; Lee, P.Y.; Chiu, C.C.; Cheng, K.C. Retinal protective effect of curcumin metabolite hexahydrocurcumin against blue light-induced RPE damage. Phytomedicine 2023, 110, 154606. [Google Scholar] [CrossRef]
- Cheng, K.C.; Hsu, Y.T.; Liu, W.; Huang, H.L.; Chen, L.Y.; He, C.X.; Sheu, S.J.; Chen, K.J.; Lee, P.Y.; Lin, Y.H.; et al. The Role of Oxidative Stress and Autophagy in Blue-Light-Induced Damage to the Retinal Pigment Epithelium in Zebrafish In Vitro and In Vivo. Int. J. Mol. Sci. 2021, 22, 1338. [Google Scholar] [CrossRef]
- Wang, Q.; He, F.; Wu, L. NLRX1 increases human retinal pigment epithelial autophagy and reduces H2O2-induced oxidative stress and inflammation by suppressing FUNDC1 phosphorylation and NLRP3 activation. Allergol. Immunopathol. 2023, 51, 177–186. [Google Scholar] [CrossRef]
- Domalpally, A.; Xing, B.; Pak, J.W.; Agrón, E.; Ferris, F.L., 3rd; Clemons, T.E.; Chew, E.Y. Extramacular Drusen and Progression of Age-Related Macular Degeneration: Age Related Eye Disease Study 2 Report 30. Ophthalmol. Retina 2023, 7, 111–117. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Asp. Med. 2012, 33, 399–417. [Google Scholar] [CrossRef]
- Matsumoto, B.; Defoe, D.M.; Besharse, J.C. Membrane turnover in rod photoreceptors: Ensheathment and phagocytosis of outer segment distal tips by pseudopodia of the retinal pigment epithelium. Proc. R Soc. Lond. B Biol. Sci. 1987, 230, 339–354. [Google Scholar]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implication for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, R.; Bertelli, M.; Scaffidi, E.; Busceti, C.L.; Biagioni, F.; Fornai, F. Exosomes and alpha-synuclein within retina from autophagy to protein spreading in neurodegeneration. Arch. Ital. Biol. 2021, 159, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, R.; Berti, C.; Scaffidi, E.; Lazzeri, G.; Bumah, V.V.; Ruffoli, R.; Biagioni, F.; Busceti, C.L.; Puglisi-Allegra, S.; Fornai, F. Combined pulses of light and sound in the retina with nutraceuticals may enhance the recovery of foveal holes. Arch. Ital. Biol. 2022, 160, 1–19. [Google Scholar] [CrossRef]
- Rojas, J.C.; Lee, J.; John, J.M.; Gonzalez-Lima, F. Neuroprotective effects of nearinfrared light in an in vivo model of mitochondrial optic neuropathy. J. Neurosci. 2008, 28, 13511–13521. [Google Scholar] [CrossRef]
- Tata, D.B.; Waynant, R.W. Laser therapy: A review of its mechanism of action and potential medical applications. Laser Photonics Rev. 2011, 5, 1–12. [Google Scholar] [CrossRef]
- Rojas, J.C.; Gonzalaz-Lima, F. Low level light therapy of the eye and brain. Eye Brain 2011, 3, 49–67. [Google Scholar]
- Stefenon, L.; Boasquevisque, M.; Garcez, A.S.; de Araújo, V.C.; Soares, A.B.; Santos-Silva, A.R.; Sperandio, F.; Brod, J.M.M.; Sperandio, M. Autophagy upregulation may explain inhibition of oral carcinoma in situ by photobiomodulation in vitro. J. Photochem. Photobiol. B 2021, 221, 112245. [Google Scholar] [CrossRef] [PubMed]
- Comerota, M.M.; Tumurbaatar, B.; Krishnan, B.; Kayed, R.; Taglialatela, G. Near Infrared Light Treatment Reduces Synaptic Levels of Toxic Tau Oligomers in Two Transgenic Mouse Models of Human Tauopathies. Mol. Neurobiol. 2019, 56, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.L.; Khoo, B.Y.; Ong, M.T.; Yoong, I.C.K.; Sreeramanan, S. In vitro anti-breast cancer studies of LED red light therapy through autophagy. Breast Cancer 2021, 28, 60–66. [Google Scholar] [CrossRef]
- Dong, L.; He, J.; Luo, L.; Wang, K. Targeting the Interplay of Autophagy and ROS for Cancer Therapy: An Updated Overview on Phytochemicals. Pharmaceuticals 2023, 16, 92. [Google Scholar] [CrossRef]
- Patra, S.; Mishra, S.R.; Behera, B.P.; Mahapatra, K.K.; Panigrahi, D.P.; Bhol, C.S.; Praharaj, P.P.; Sethi, G.; Patra, S.K.; Bhutia, S.K. Autophagy-modulating phytochemicals in cancer therapeutics: Current evidences and future perspectives. Semin Cancer Biol. 2022, 80, 205–217. [Google Scholar] [CrossRef]
- Shannar, A.; Sarwar, M.S.; Kong, A.T. A New Frontier in Studying Dietary Phytochemicals in Cancer and in Health: Metabolic and Epigenetic Reprogramming. Prev. Nutr. Food Sci. 2022, 27, 335–346. [Google Scholar] [CrossRef]
- Jafari-Nozad, A.M.; Jafari, A.; Zangooie, A.; Behdadfard, M.; Zangouei, A.S.; Aschner, M.; Farkhondeh, T.; Samarghandian, S. Curcumin Combats Against Gastrointestinal Cancer: A Review of Current Knowledge Regarding Epigenetics Mechanisms with a Focus on DNA Methylation. Curr. Med. Chem. 2023, 30, 4374–4388. [Google Scholar] [CrossRef]
- Dong, X.; Nao, J. Relationship between the therapeutic potential of various plant-derived bioactive compounds and their related microRNAs in neurological disorders. Phytomedicine 2023, 108, 154501. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Yu, P.; Luo, L.; Sun, J.; Tao, H.; Wang, X.; Meng, X. Berberis dictyophylla F. inhibits angiogenesis and apoptosis of diabetic retinopathy via suppressing HIF-1/VEGF/DLL-4/Notch-1 pathway. J. Ethnopharmacol. 2022, 296, 115453. [Google Scholar] [CrossRef]
- Kang, Q.; Dai, H.; Jiang, S.; Yu, L. Advanced glycation end products in diabetic retinopathy and phytochemical therapy. Front. Nutr. 2022, 9, 1037186. [Google Scholar] [CrossRef]
- Takkar, B.; Sheemar, A.; Jayasudha, R.; Soni, D.; Narayanan, R.; Venkatesh, P.; Shivaji, S.; Das, T. Unconventional avenues to decelerate diabetic retinopathy. Surv. Ophthalmol. 2022, 67, 1574–1592. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Morell, F.; Villagrasa, V.; Ortega, T.; Acero, N.; Muñoz-Mingarro, D.; González-Rosende, M.E.; Castillo, E.; Sanahuja, M.A.; Soriano, P.; Martínez-Solís, I. Medicinal plants and natural products as neuroprotective agents in age-related macular degeneration. Neural Regen. Res. 2020, 15, 2207–2216. [Google Scholar] [PubMed]
- Hyttinen, J.; Blasiak, J.; Tavi, P.; Kaarniranta, K. Therapeutic potential of PGC-1α in age-related macular degeneration (AMD)—The involvement of mitochondrial quality control, autophagy, and antioxidant response. Expert Opin. Ther. Targets 2021, 25, 773–785. [Google Scholar] [CrossRef]
- Lewis Luján, L.M.; McCarty, M.F.; Di Nicolantonio, J.J.; Gálvez Ruiz, J.C.; Rosas-Burgos, E.C.; Plascencia-Jatomea, M.; Iloki Assanga, S.B. Nutraceuticals/Drugs Promoting Mitophagy and Mitochondrial Biogenesis May Combat the Mitochondrial Dysfunction Driving Progression of Dry Age-Related Macular Degeneration. Nutrients 2022, 14, 1985. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, C.; Zhang, Q.; Lv, Y.; Lu, C.; Su, W.; Zhou, J.; Zhang, H.; Gong, H.; Liu, Y.; et al. Strategic delivery of rapamycin and ranibizumab with intravitreal hydrogel depot disrupts multipathway-driven angiogenesis loop for boosted wAMD therapy. J. Control. Release 2025, 377, 239–255. [Google Scholar] [CrossRef]
- Ren, Y.; Liang, H.; Xie, M.; Zhang, M. Natural plant medications for the treatment of retinal diseases: The blood-retinal barrier as a clue. Phytomedicine 2024, 130, 155568. [Google Scholar] [CrossRef]
- Mei, L.; Yu, M.; Liu, Y.; Weh, E.; Pawar, M.; Li, L.; Besirli, C.G.; Schwendeman, A.A. Synthetic high-density lipoprotein nanoparticles delivering rapamycin for the treatment of age-related macular degeneration. Nanomedicine 2022, 44, 102571. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Effects | References |
|---|---|---|
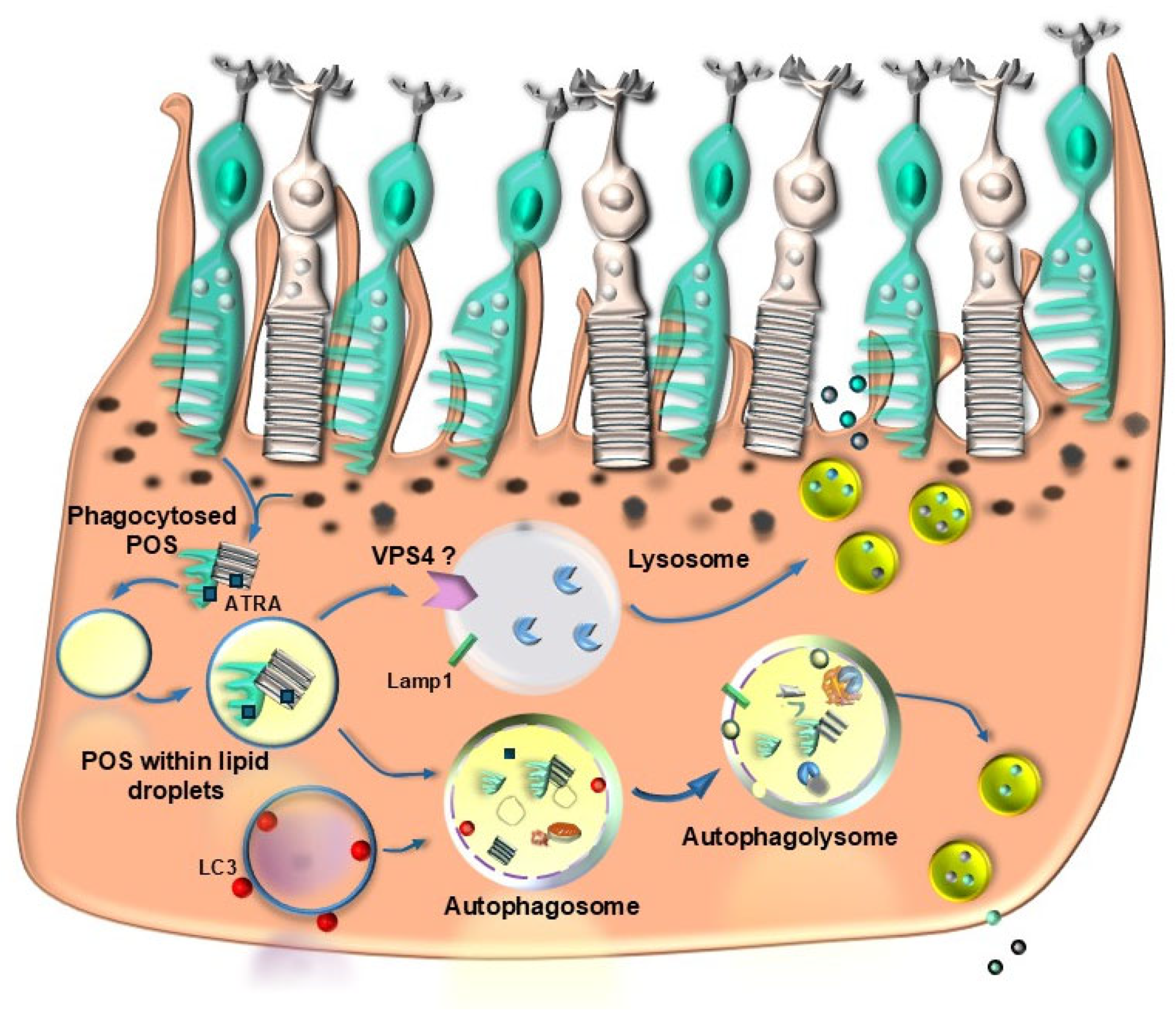
| Vps4 | Regulates transport within multivesicular bodies | [79] |
| MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3)-associated phagocytosis (LAP) | Recycles the actual platform for phototransduction | [83,84] |
| ATRA (all trans retinoic acid) | Directlystimulates autophagywithin RPE cells | [88] |
| Lipidated LC3 (LC3II) | Promoteselongation and membrane folding of the nascent autophagy vesicle | [46] |
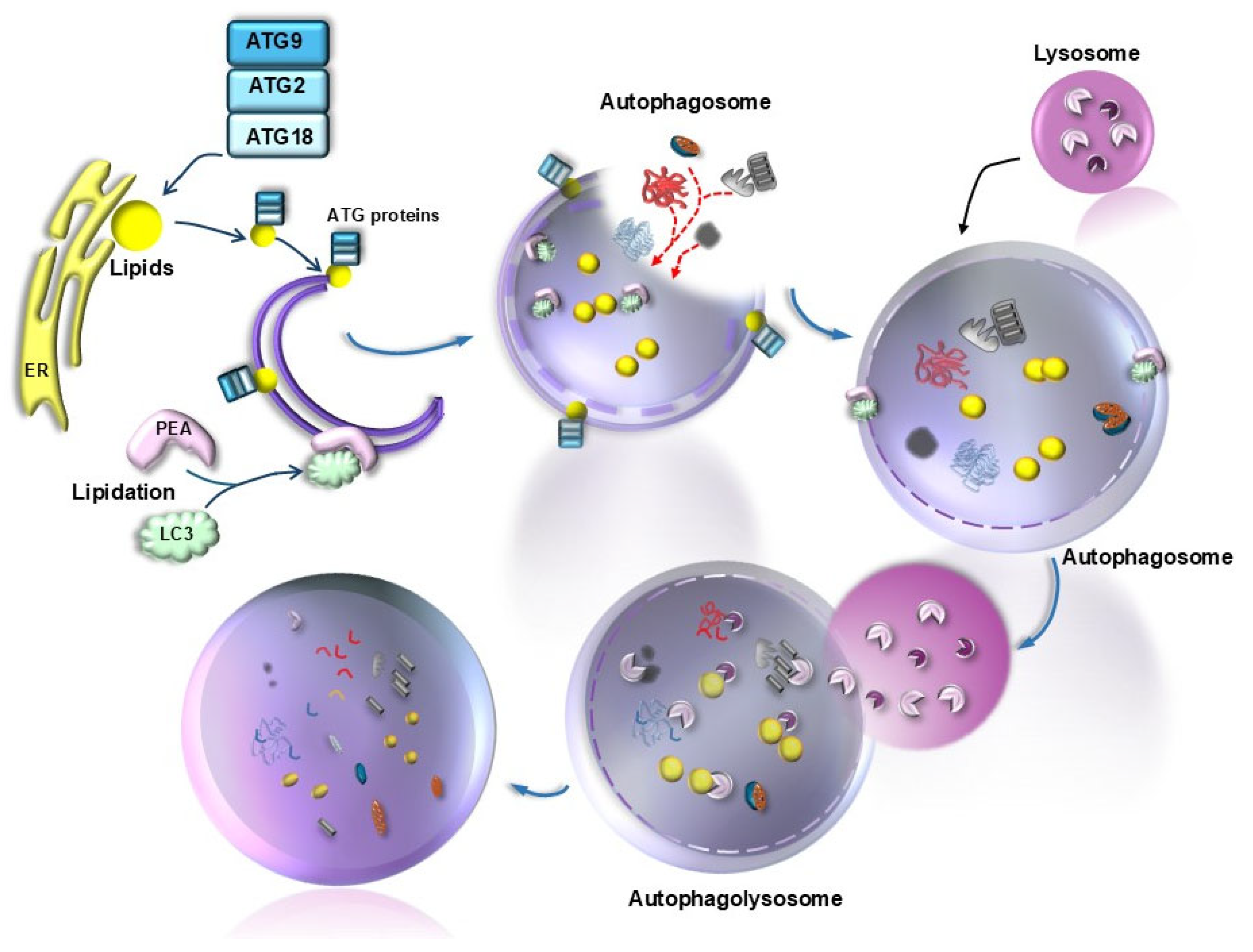
| Apo-E |
| [35,98,99] |
| Phosphoinositides/Oxysterols Synaptojanin 1 |
| [94,100] |
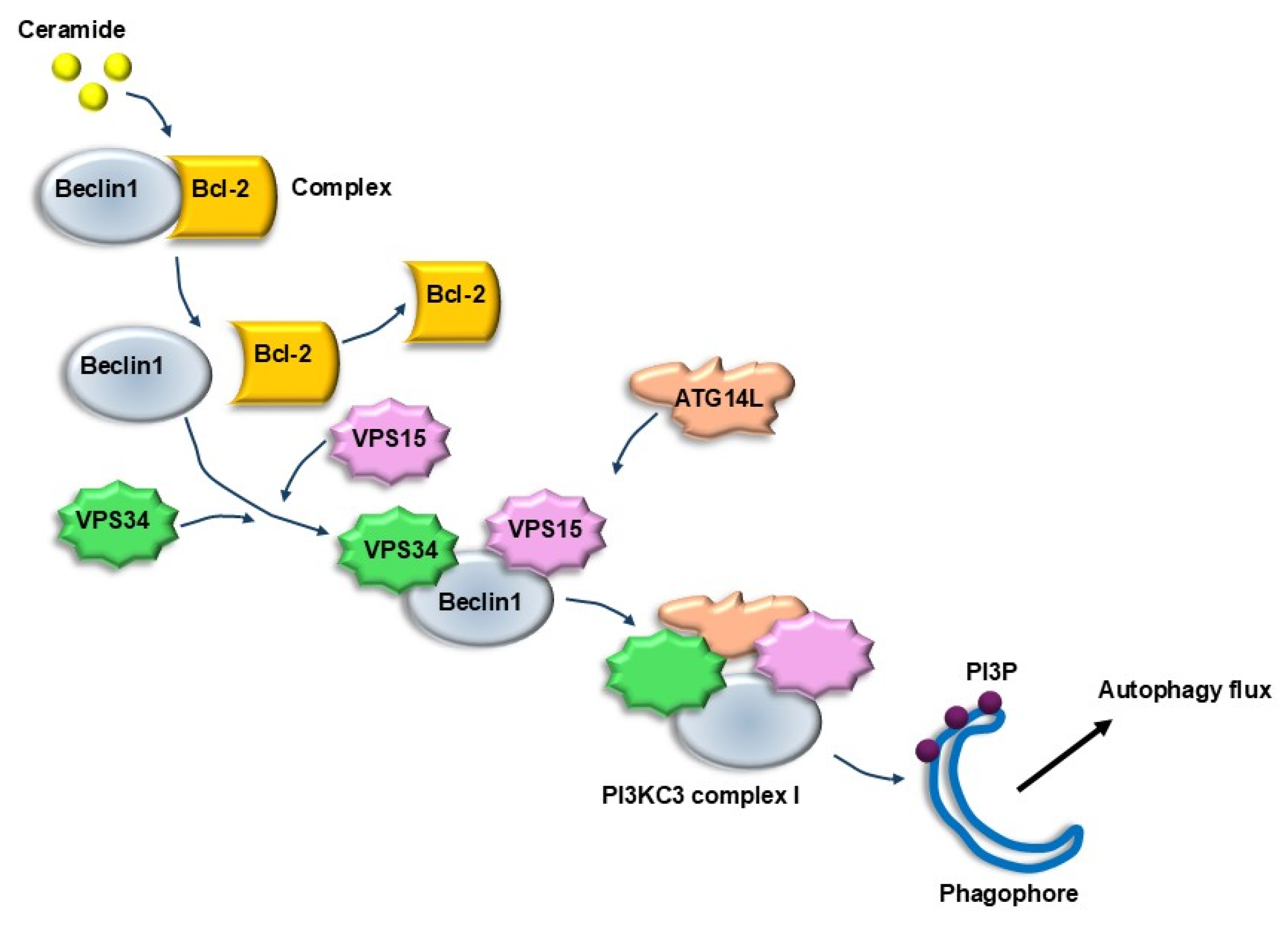
| Ceramide | Promotes the removal of Beclin1 from the Beclin1/Bcl-2 complex, and promotes the interaction of Beclin1/VPS34/VPS15 forming a complex which is a strong lipophagy stimulator | [102] |
| Total Mitochondria | % Healthy Mitochondria | % Altered Mitochondria | |
|---|---|---|---|
| Control (n = 30 cells) | 13.72 ± 0.08 | 74.84 ± 0.22 | 25.16 ± 0.40 |
| 3-MA, 10 mM (n = 30 cells) | 7.28 ± 0.02 * p < 0.0001 | 55.71 ± 0.32 * p < 0.0001 | 44.29 ± 0.32 * p < 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinelli, R.; Lazzeri, G.; Berti, C.; Biagioni, F.; Scaffidi, E.; Ferrucci, M.; Bumah, V.V.; Fornai, F. Retinal Autophagy for Sustaining Retinal Integrity as a Proof of Concept for Age-Related Macular Degeneration. Int. J. Mol. Sci. 2025, 26, 5773. https://doi.org/10.3390/ijms26125773
Pinelli R, Lazzeri G, Berti C, Biagioni F, Scaffidi E, Ferrucci M, Bumah VV, Fornai F. Retinal Autophagy for Sustaining Retinal Integrity as a Proof of Concept for Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2025; 26(12):5773. https://doi.org/10.3390/ijms26125773
Chicago/Turabian StylePinelli, Roberto, Gloria Lazzeri, Caterina Berti, Francesca Biagioni, Elena Scaffidi, Michela Ferrucci, Violet Vakunseh Bumah, and Francesco Fornai. 2025. "Retinal Autophagy for Sustaining Retinal Integrity as a Proof of Concept for Age-Related Macular Degeneration" International Journal of Molecular Sciences 26, no. 12: 5773. https://doi.org/10.3390/ijms26125773
APA StylePinelli, R., Lazzeri, G., Berti, C., Biagioni, F., Scaffidi, E., Ferrucci, M., Bumah, V. V., & Fornai, F. (2025). Retinal Autophagy for Sustaining Retinal Integrity as a Proof of Concept for Age-Related Macular Degeneration. International Journal of Molecular Sciences, 26(12), 5773. https://doi.org/10.3390/ijms26125773