
Peptide-Engineered Seliciclib Nanomedicine for Brain-Targeted Delivery and Neuroprotection
Abstract
1. Introduction
2. Results
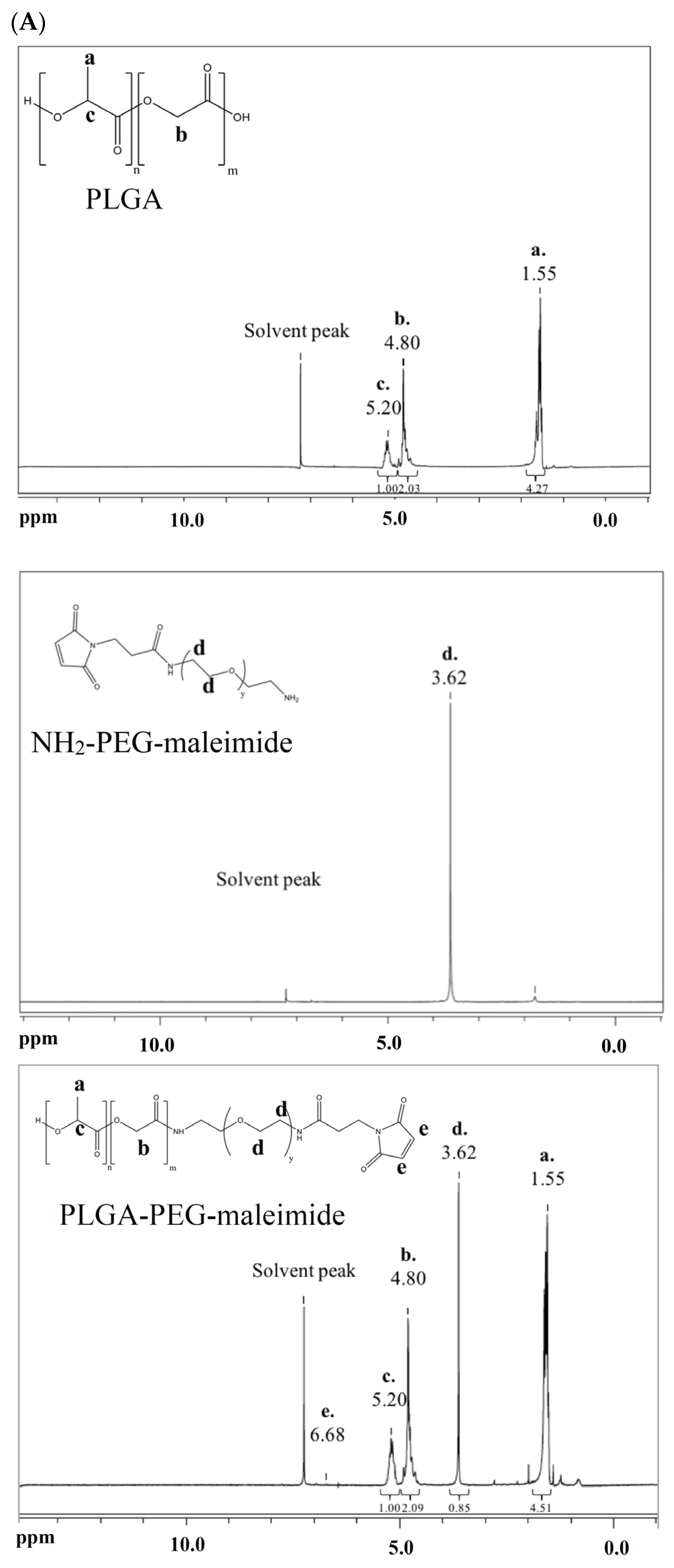
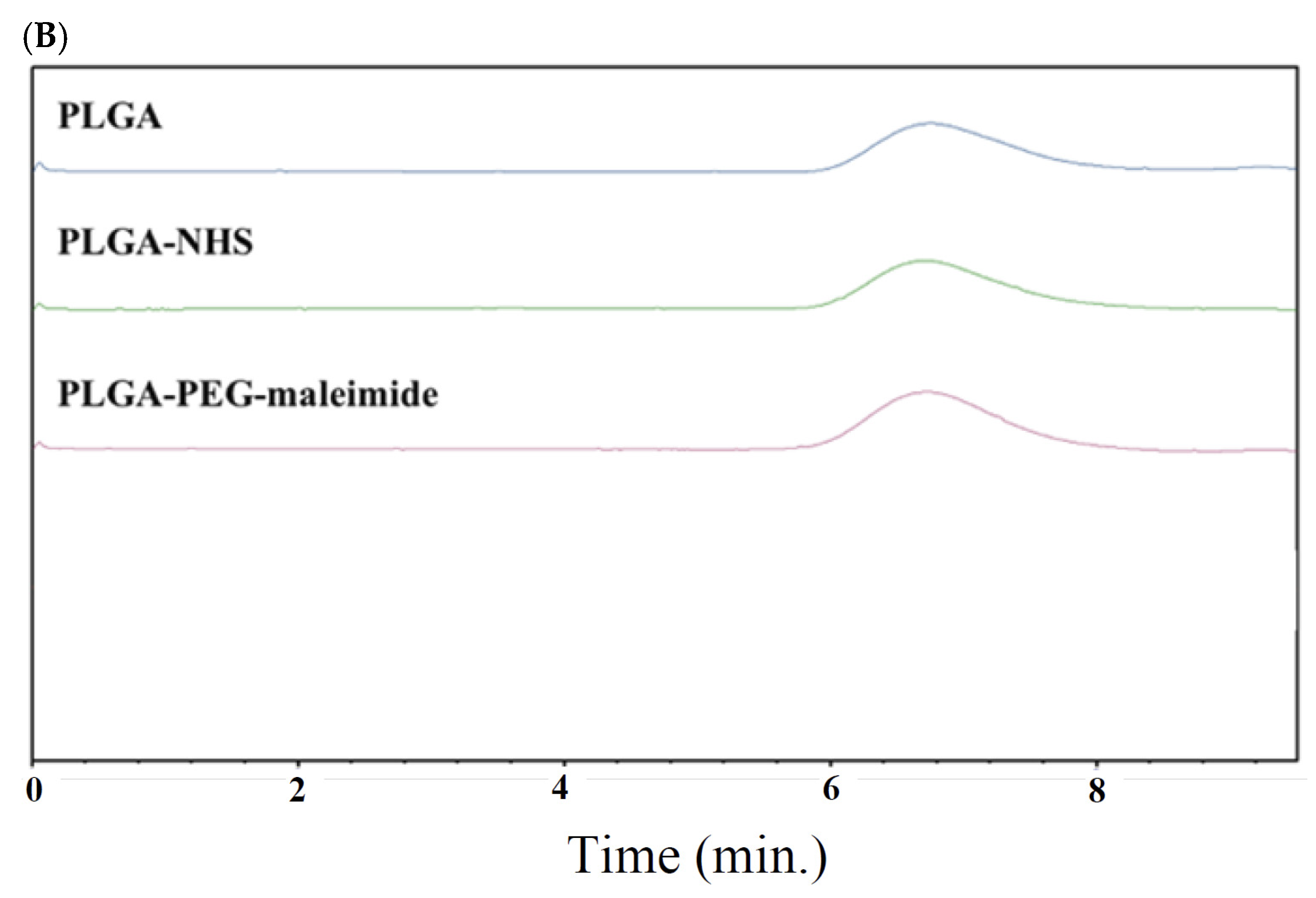
2.1. Characterization of PLGA-PEG-Maleimide Copolymer
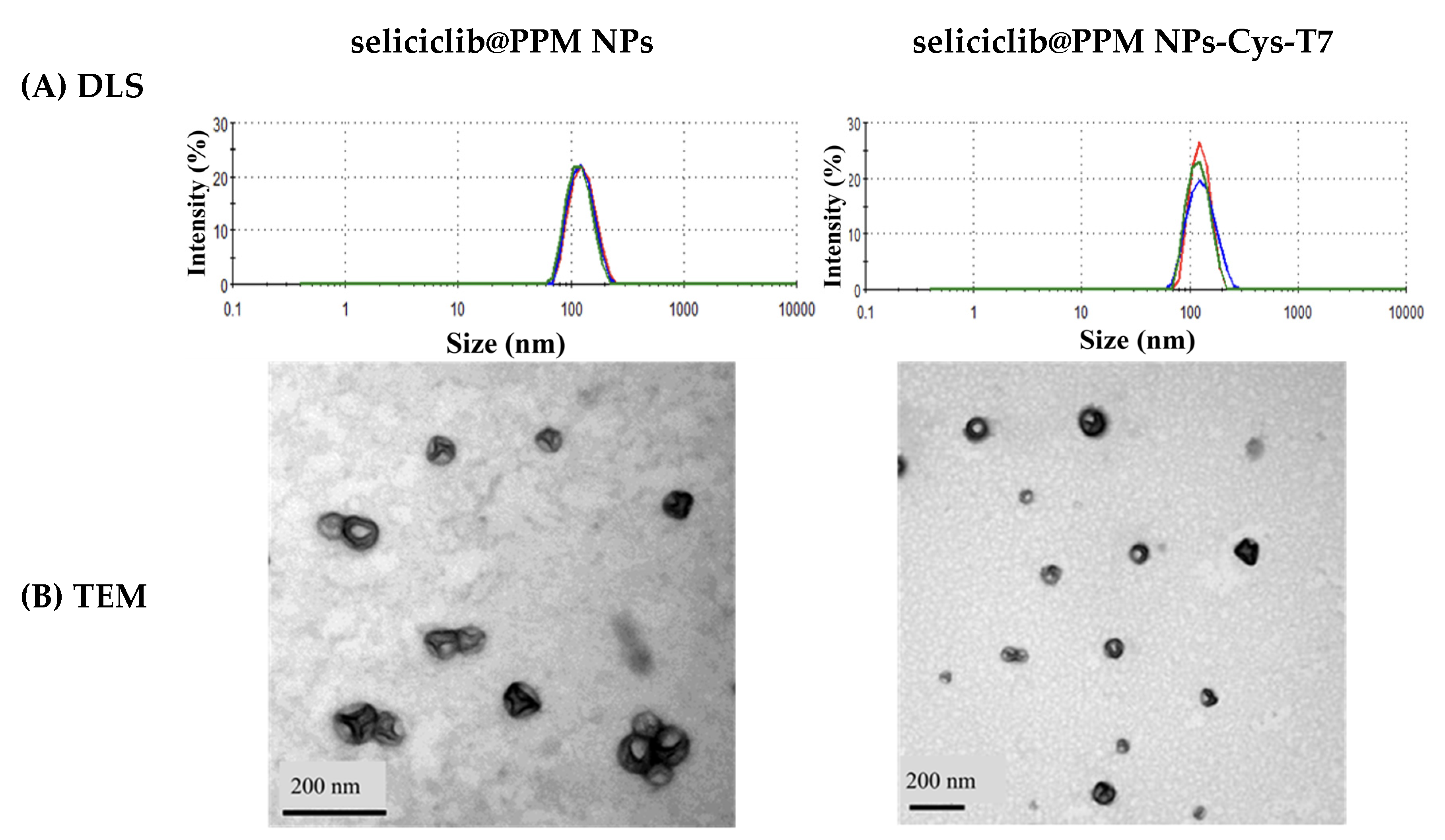
2.2. Characterization of Seliciclib@NPs
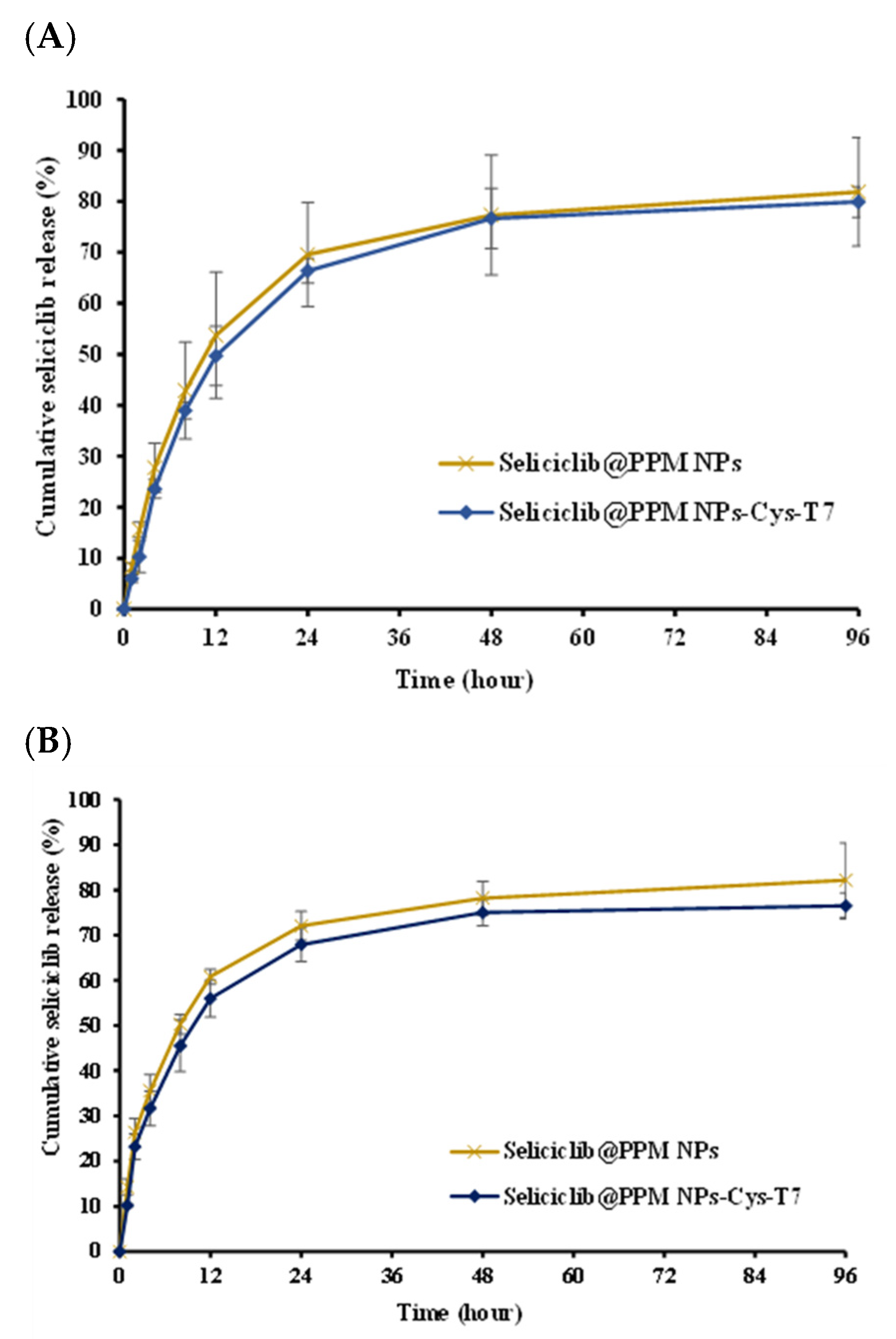
2.3. In Vitro Release
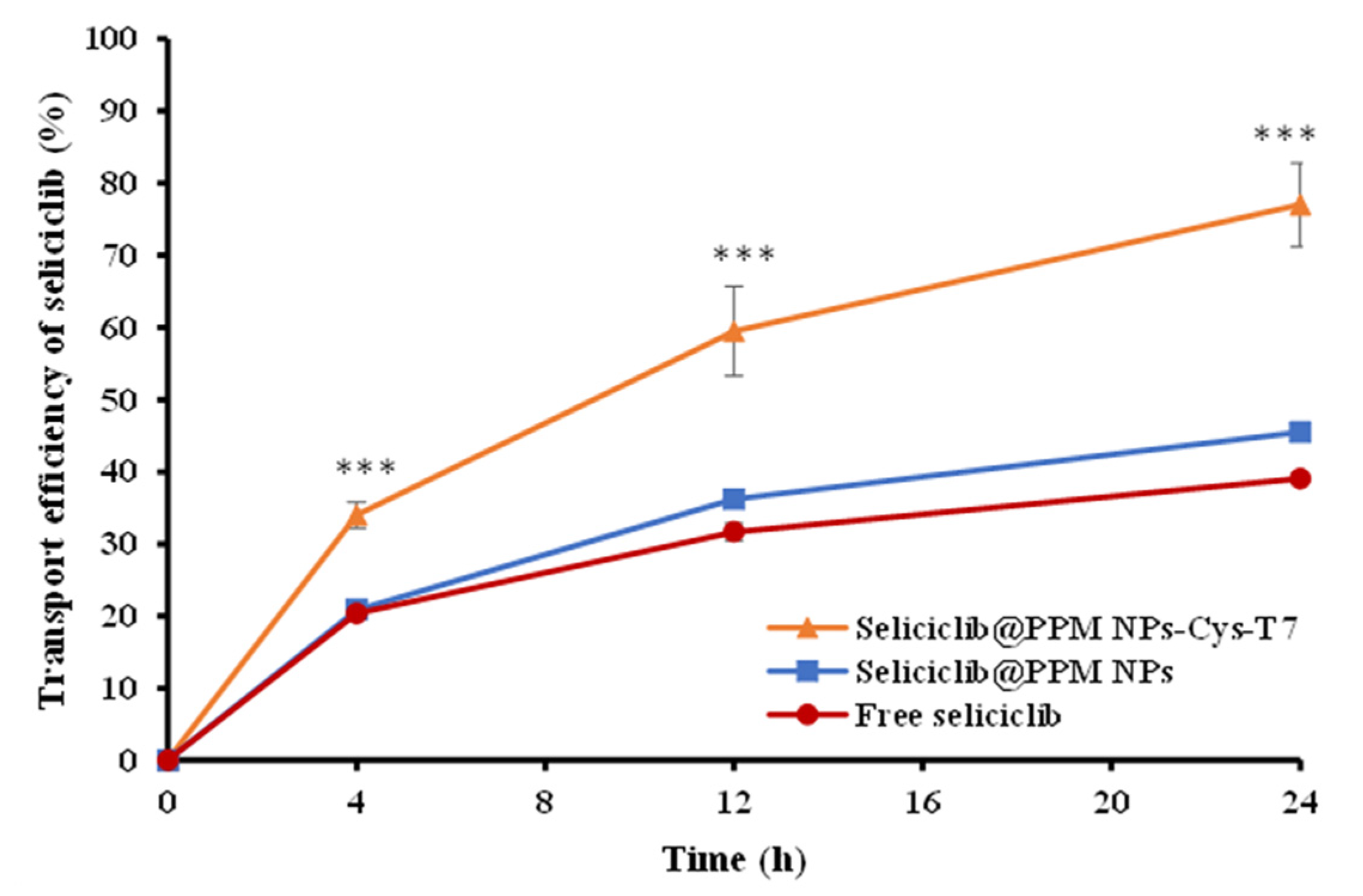
2.4. Transport of Seliciclib@NPs Across BBB Cell Model
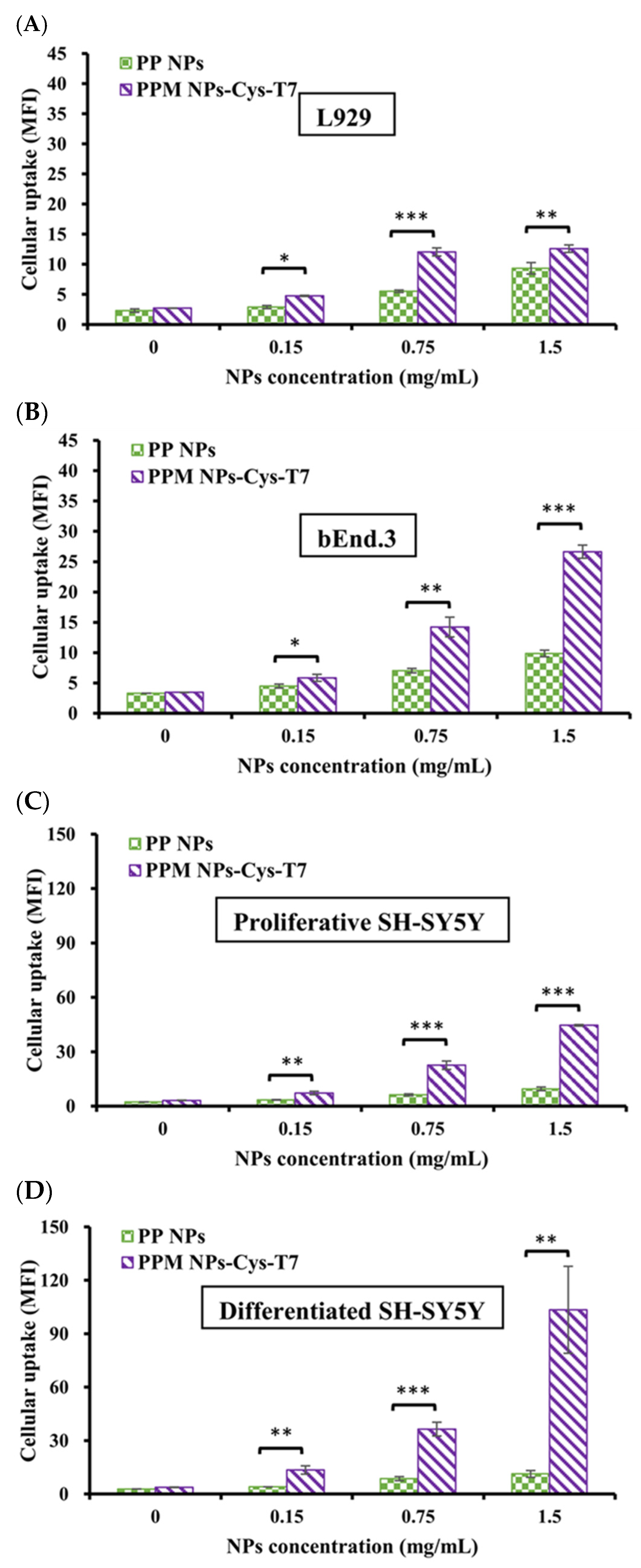
2.5. Cellular Uptake of Nanoparticles
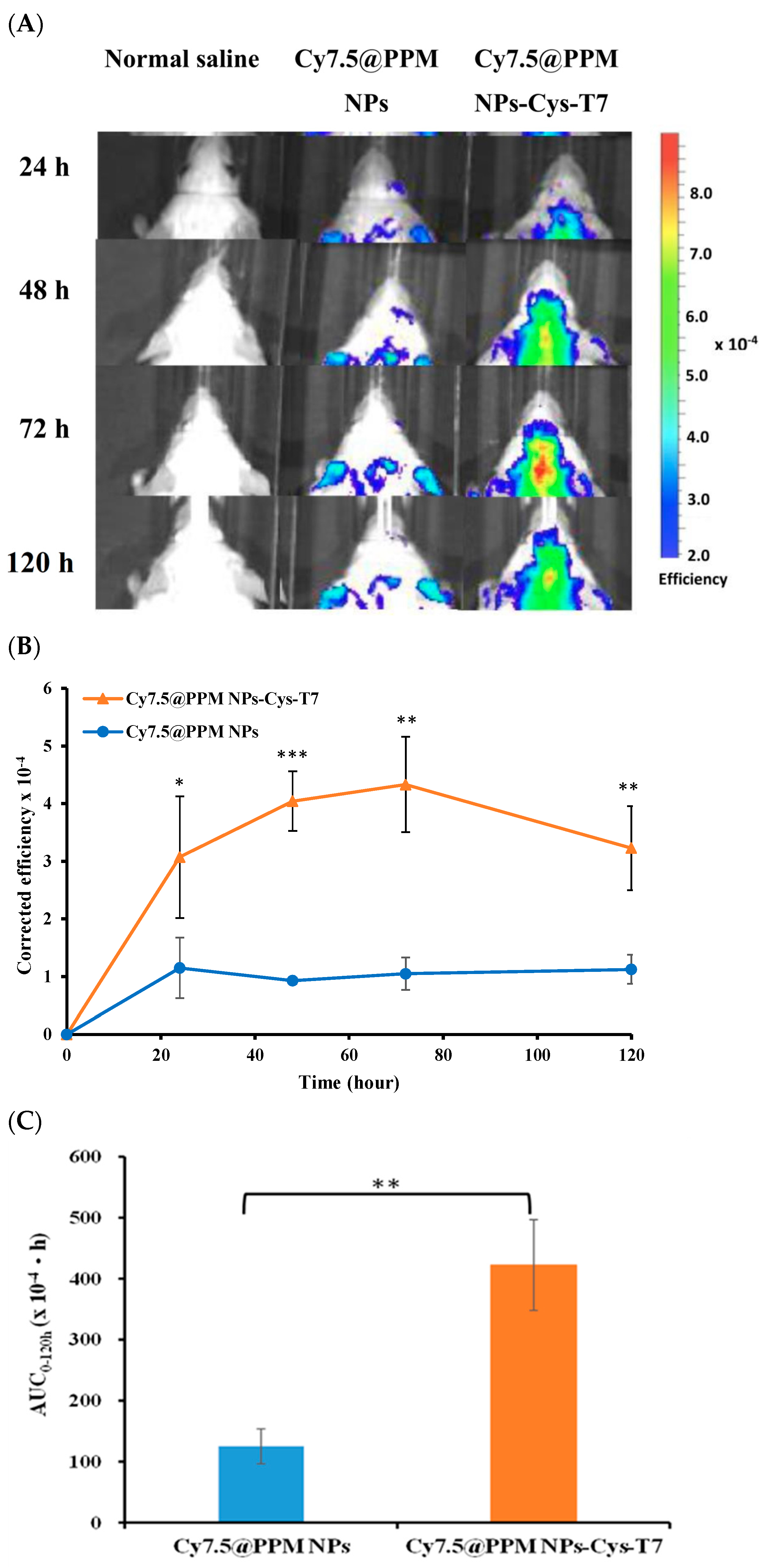
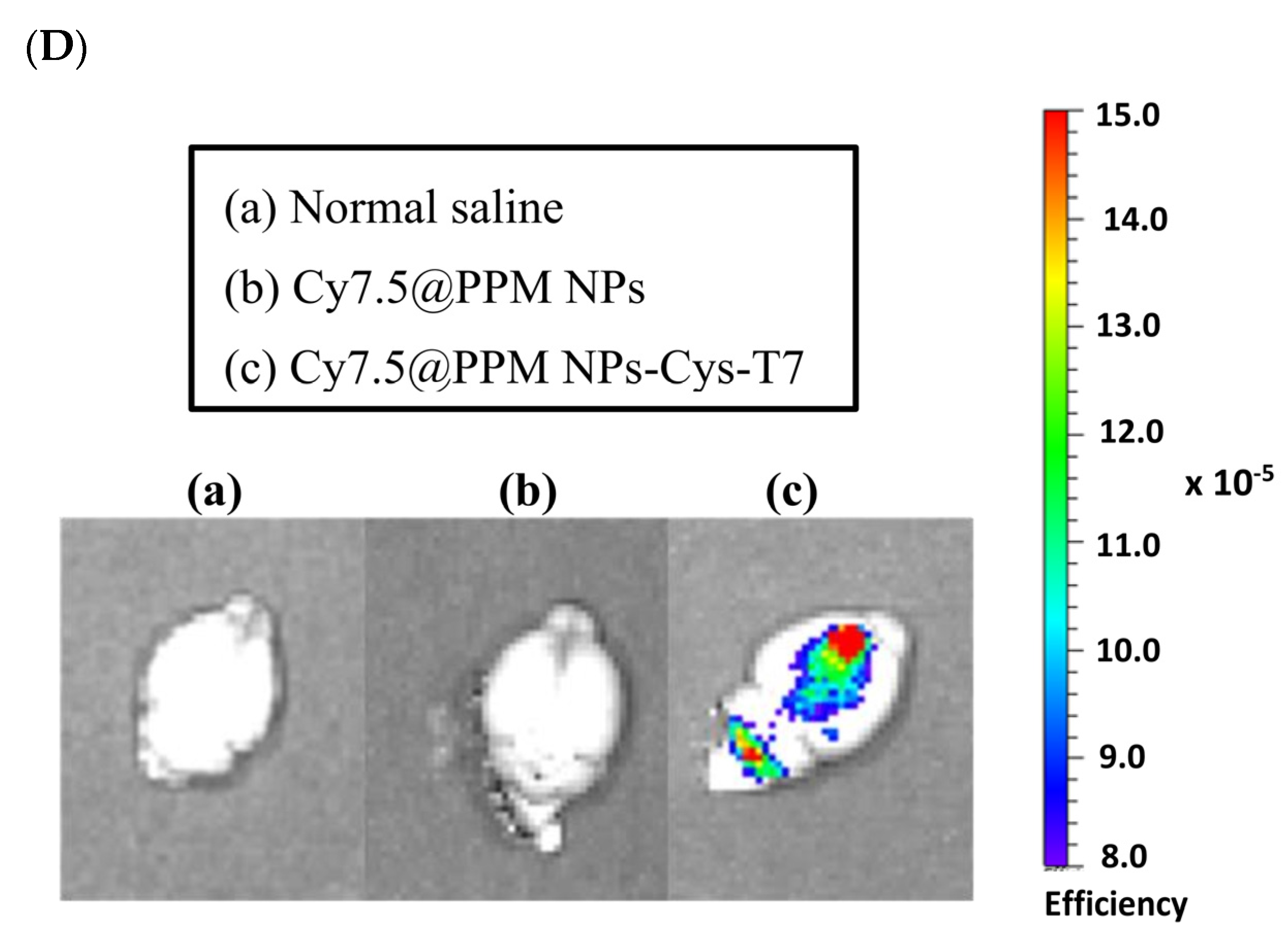
2.6. Brain Biodistribution of Nanoparticles by IVIS
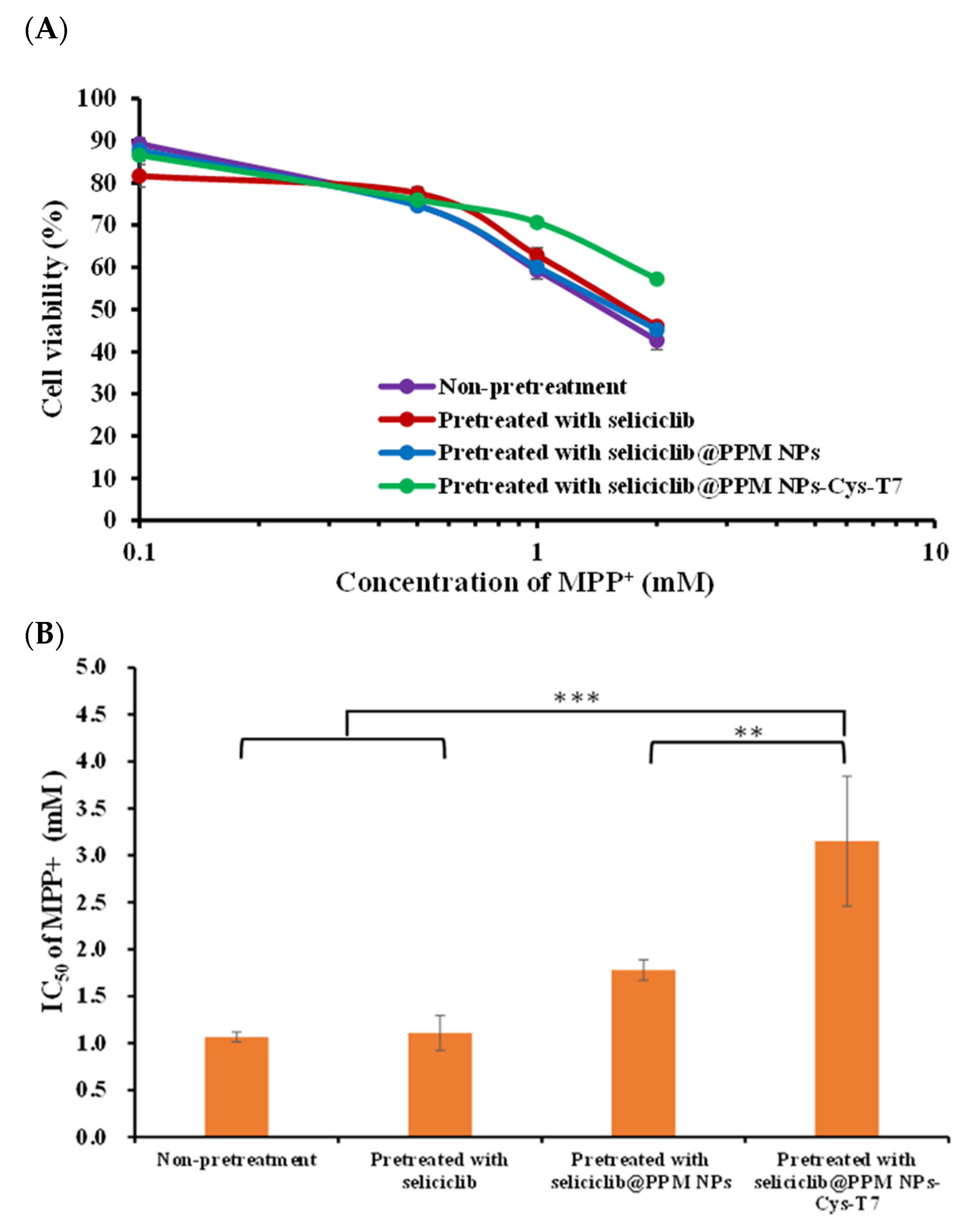
2.7. Neuroprotection Effect in MPP+-Induced Parkinsonian SH-SY5Y Cell Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis and Characterization of Copolymers
4.3. Preparation and Characterization of Nanoparticles
4.4. Transport of Seliciclib@NPs Across BBB Cell Model
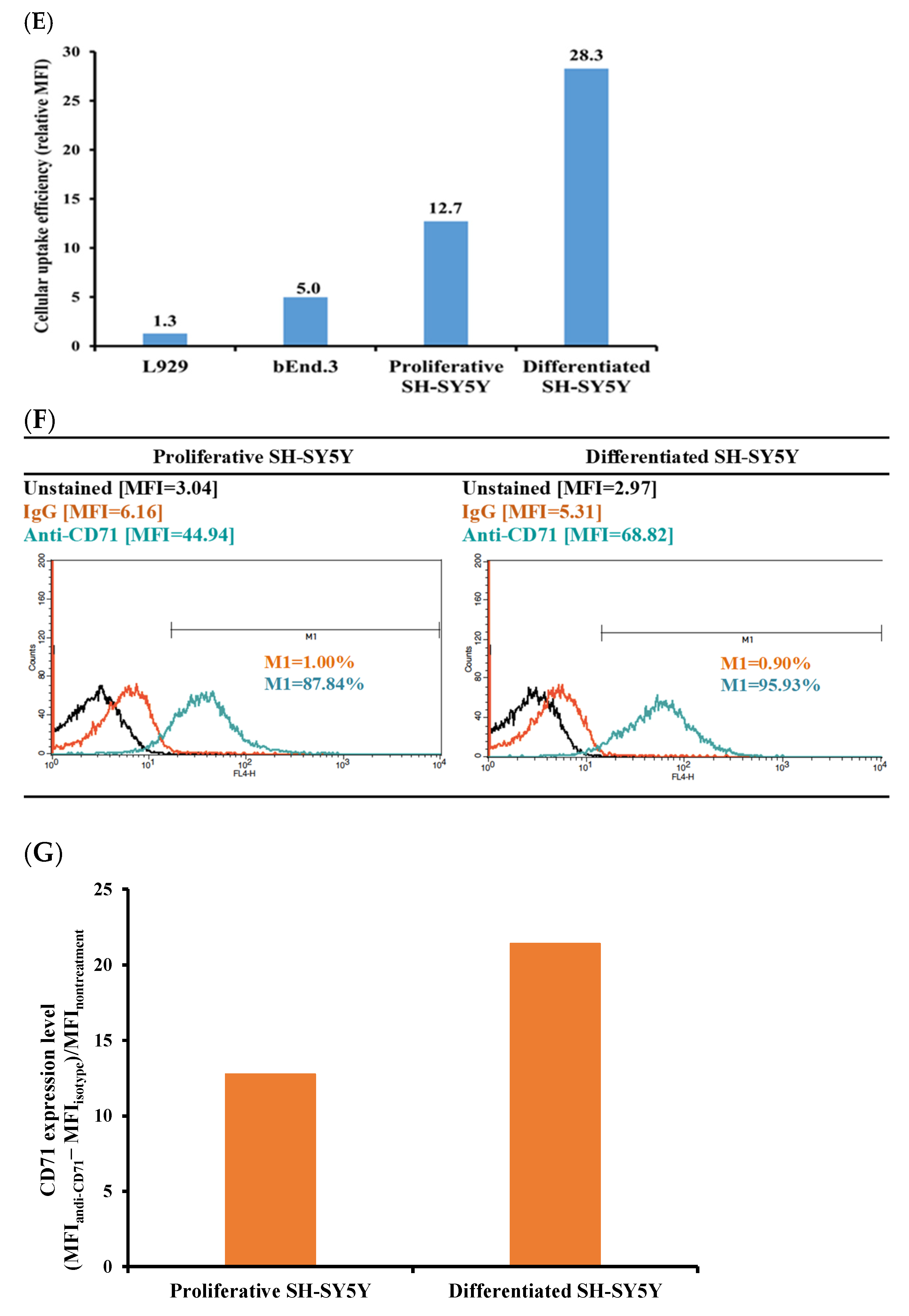
4.5. Identification of CD71 Expression Level
4.6. In Vitro Cellular Uptake of Nanoparticles
4.7. Brain Biodistribution of Nanoparticles by IVIS
4.8. Neuroprotective Effect in MPP+-Induced Parkinsonian SH-SY5Y Cell Model
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pao, P.C.; Tsai, L.H. Three decades of Cdk5. J. Biomed. Sci. 2021, 28, 79. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, W.; Mao, Z. Cdk5: Mediator of neuronal development, death and the response to DNA damage. Mech. Ageing Dev. 2011, 132, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Allnutt, A.B.; Waters, A.K.; Kesari, S.; Yenugond, V.M. Physiological and pathological roles of CDK5: Potential directions for therapeutic targeting in neurodegenerative diseases. ACS Chem. Neurosci. 2020, 11, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Skuntz, S.; Prochazkova, M.; Kesavapany, S.; Amin, N.D.; Shukla, V.; Grant, P.; Kulkarni, A.B.; Pant, H.C. Overex-pression of the Cdk5 inhibitory peptide in motor neurons rescue of amyotrophic lateral sclerosis phenotype in a mouse model. Hum. Mol. Genet. 2019, 28, 3175–3187. [Google Scholar]
- He, F.; Qi, G.; Cai, H.; Li, T.; Li, M.; Zhang, Q.; Chen, J.; Ming, J.; Tian, B.; Zhang, P. Quantitative phosphoproteomic analysis in alpha-synuclein transgenic mice reveals the involvement of aberrant p25/Cdk5 signaling in early-stage Parkinson’s disease. Cell Mol. Neurobiol. 2020, 40, 897–909. [Google Scholar] [CrossRef]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The role of Cdk5 in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Tran, J.; Taloy, S.K.; Gupta, A.; Amin, N.; Pant, H.; Gupta, B.P.; Mishra, R.K. Therapeutic effects of TP5, a Cdk/p25 inhibitor, in vitro and in vivo models of Parkinsons’s disease. Curr. Res. Neurobiol. 2021, 2, 100006. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zolkovic, B.V. Blood-brain barrier breakdown in Alzheimer’s disease and other neurodegen-erative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Kaya, S.; Callan, B.; Hawthorne, S. Non-invasive targeted nanoparticle-mediated drug delivery across a novel human BBB model. Pharmaceutics 2023, 15, 1382. [Google Scholar] [CrossRef]
- Moura, R.P.; Martins, C.; Pinto, S.; Sousa, F.; Sarmento, B. Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert Opin. Drug Deliv. 2019, 16, 271–285. [Google Scholar] [CrossRef]
- Ru, L.; Jaspers, T.; Degors, I.M.S.; Noppen, S.; Schols, D.; Strooper, B.D. Dewilde Novel human/non-human primate cross-reactive anti-transferrin receptor nanobodies for brain delivery of biologics. Pharmaceutics 2023, 15, 1748. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Parca, C.; Ferreira, R.; Santos, T. Nanoparticles-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Rel. 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Burkhart, A.; Melander, F.; Kempen, P.J.; Vejlebo, J.B.; Siupka, P.; Nielsen, M.S.; Andresen, T.L.; Moos, T. Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci. Rep. 2017, 7, 10396. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.R.; Albuquerque, T.; Faria, R.; Gonçalves, A.M.; Santos, C.; Vivès, E.; Boisguérin, P.; Passarinha, L.A.; Sousa, Â.; Costa, D. Development of WRAP5 peptide complexes for targeted drug/gene co-delivery toward glioblastoma therapy. Pharmaceutics 2022, 14, 2213. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Johnsen, K.B.; Kucharz, K.; Lauritzen, M.; Moos, T. Blood-brain barrier transport of transferrin receptor-targeted nanoparticles. Pharmaceutics 2022, 14, 2237. [Google Scholar] [CrossRef]
- Braatz, D.; Cherri, M.; Tully, M.; Dimde, M.; Ma, G.; Mohammadifar, E.; Reisbeck, F.; Ahmadi, V.; Schirner, M.; Haag, R. Chemical approaches to synthetic drug delivery systems for systemic applications. Angew. Chem. Int. Ed. 2022, 61, e202203942. [Google Scholar] [CrossRef]
- Ray, P.; Ferraro, M.; Haag, R.; Quadir, M. Dendritic polyglycerol-derived nano-architectures as delivery platforms of gemcitabine for pancreatic cancer. Macromol. Biosci. 2019, 19, 1900073. [Google Scholar] [CrossRef]
- Marzo, I.; Naval, J. Antimitotic drugs in cancer chemotherapy: Promises and pitfalls. Biochem. Pharma. 2013, 86, 703–710. [Google Scholar] [CrossRef]
- Molinsky, J.; Klanova, M.; Koc, M.; Beranova, L.; Andera, L.; Ludvikova, Z.; Bohmova, M.; Gasova, Z.; Strnad, M.; Ivanek, R.; et al. Roscovitine sensitizes leukemia and lymphoma cells to tumor necrosis fac-tor-related apoptosis-inducing ligand-induced apoptosis. Leuk. Lymphoma 2013, 54, 372–380. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Suys, E.J.A.; Lee, J.S.; Nguyen, D.H.; Park, K.D.; Truong, N.P. Lipid-based nanoparticles in the clinic and clinical trials: From cancernanomedicine to COVID-19 vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef]
- Far, B.F.; Safaei, M.; Pourmolaei, A.; Adibamini, S.; Shirdel, S.; Shirdel, S.; Emadi, R.; Kaushik, A.K. Exploring curcumin-loaded lipid-based nanomedicine as efficient targeted therapy for Alzheimer’s diseases. ACS Appl. Bio. Mater. 2024, 7, 3535–3555. [Google Scholar] [CrossRef] [PubMed]
- Toader, C.; Dumitru, A.V.; Eva, L.; Serban, M.; Covache-Busuioc, R.A.; Ciurea, A.V. Nanoparticle strategies for treating CNS disorders: A comprehensive review of drug delivery and theranostic applications. Int. J. Mol. Sci. 2024, 25, 13302. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Liu, Z.; Yan, Z.; Wu, J.; Li, Z.; Gao, S.; Zheng, C.; Guo, S.; Pan, Y.; Chen, X.; et al. Minocycline nanoplatform penetrates the BBB and enables the targeted treatment of Parkinson’s disease with cognitive impairment. J. Control. Release 2025, 377, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-penetrating peptides in diagnosis and treatment of human diseases: From preclinical research to clinical application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Liu, C.W.; Lin, W.J. Polymeric nanoparticles conjugate a novel heptapeptide as an epidermal growth factor receptor-active targeting ligand for doxorubicin. Int. J. Nanomed. 2012, 7, 4749–4767. [Google Scholar]
- Liu, C.W.; Lin, W.J. Systemic co-delivery of doxorubicin and siRNA using nanoparticles conjugated with EGFR specific targeting peptide to enhance chemotherapy in ovarian tumor bearing mice. J. Nanopart. Res. 2013, 15, 1956–1969. [Google Scholar] [CrossRef]
- Lo, Y.C.; Lin, W.J. Benefit of a short chain peptide as a targeting ligand of nanocarriers for a brain-driven purpose. Pharmaceutics 2021, 13, 1249. [Google Scholar] [CrossRef]
- Lo, Y.C.; Lin, W.J. Improve BBB penetration and cytotoxicity of palbociclib in U87-MG glioblastoma cells delivered by dual peptide functionalized nanoparticles. Pharmaceutics 2023, 15, 2429. [Google Scholar] [CrossRef]
- Han, L.; Huang, R.; Liu, S.; Huang, S.; Jiang, C. Peptide-conjugated PAMAM for targeted soxorubicin delivery to transferrin receptor overexpressed tumors. Mol. Pharmaceutics 2010, 7, 2156–2165. [Google Scholar] [CrossRef]
- Lee, J.H.; Engler, J.A.; Collawn, J.F.; Moore, B.A. Receptor mediated uptake of peptides that bind the human transferrin receptor. Eur. J. Biochem. 2001, 268, 2004–2012. [Google Scholar] [CrossRef]
- Oh, S.; Kim, B.J.; Singh, N.P.; Lai, H.; Sasaki, T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. 2009, 274, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Gao, C.; Wang, Y.; Gong, W.; Fu, S.; Cui, L.; Zhou, Z.; Chu, X.; Zhang, Y.; Liu, Q.; et al. Enhanced blood-brain-barrier penetration and glioma therapy mediated by T7 pep-tide-modified low-density lipoprotein particles. Drug Deliv. 2018, 25, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J.; Xu, Y.; Guo, G.; Cai, L.; Wu, H.; Zhao, Y.; Zhang, X. Roscovitine, a CDK5 inhibitor, alleviates sevoflu-rane-induced cognitive dysfunction via regulation Tau/GSK3β and ERK/PPARγ/CREB signaling. Cell Physiol. Biochem. 2017, 44, 423–435. [Google Scholar] [CrossRef] [PubMed]
- He, G.Z.; Lin, W.J. Peptide-functionalized nanoparticles-encapsulated cyclin-dependent kinases inhibitor seliciclib in transferrin receptor overexpressed cancer cells. Nanomaterials 2021, 11, 772. [Google Scholar] [CrossRef]
- Chagniel, L.; Robitaille, L.M.; Cyr, M. Striatal inhibition of calpains prevents levodopa-induced neurochemical changes and abnormal involuntary movements in the hemiparkinsonian rat model. Neurobiol. Dis. 2012, 45, 645–655. [Google Scholar] [CrossRef]
- Do, P.A.; Lee, C.H. The role of CDK5 in tumours and tumour microenvironments. Cancers 2021, 13, 101. [Google Scholar] [CrossRef]
- Gao, L.; Xiao, H.; Ai, L.Q.Y.; Chen, C.; Lin, S.; Zhou, Y.; Ye, J.; Liu, W. Vps35 deficiency impairs CDK5/p35 degradation and promotes the hyperphosphorylation of tau protein in retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 1. [Google Scholar] [CrossRef]
- Wilkaniec, A.; Gąssowska-Dobrowolska, M.; Strawski, M.; Adamczyk, A.; Czapski, G.A. Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signaling in murine model of amyloid beta toxicity. J. Neuroinflammation 2018, 15, 1. [Google Scholar] [CrossRef]
- Erdő, F.; Nagy, I.; Tóth, B.; Bui, A.; Molnár, É.; Tímár, Z.; Magnan, R.; Krajcsi, P. Abcb1a (p-glycoprotein) limits brain exposure of the anticancer drug candidate seliciclib in vivo in adult mice. Brain Res. Bull. 2017, 132, 232–236. [Google Scholar] [CrossRef]
- Bourassa, P.; Alata, W.; Tremblay, C.; Paris-Robidas, S.; Calon, F. Transferrin receptor-mediated uptake at the blood-brain barrier is not impaired by Alzheimer’s disease neuropathology. Mol. Pharm. 2019, 16, 583–594. [Google Scholar] [CrossRef]
- Fan, W.; Peng, H.; Yu, Z.; Wang, L.; He, H.; Ma, Y.; Qi, J.; Lu, Y.; Wu, W. The long-circulating effect of pegylated nanoparticles revisited via simultaneous monitoring of both the drug payloads and nanocarriers. Acta Pharm. Sin. B 2022, 12, 2479–2493. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, W.; Wu, G.; Kong, J.; Yuan, S.; Chen, L. A magnetic T7 peptide&AS1411 aptamer-modified microemulsion for triple glioma-targeted delivery of shikonin and docetaxel. J. Pharm. Sci. 2021, 110, 2946–2954. [Google Scholar] [PubMed]
- Li, C.; Guan, N.; Liu, F. T7 peptide-decorated exosome-based nanocarrier system for delivery of Galectin-9 siRNA to stimulate macrophage repolarization in glioblastoma. J. Neurooncol. 2023, 162, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.M.; Schröder, R.; da Frota, M.L.C., Jr.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F.; et al. Comparison between proliferative and neuron-like SH-SY5Y cells as an in vitro model for Parkinson disease studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef]
- Huot, P.; Parent, A. Dopaminergic neurons intrinsic to the striatum. J. Neurochem. 2007, 101, 1441–1447. [Google Scholar] [CrossRef]
- Presgraves, S.P.; Ahmed, T.; Borwege, S.; Joyce, J.N. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox. Res. 2004, 5, 579–598. [Google Scholar] [CrossRef]
- Krsek, A.; Jagodic, A.; Baticic, L. Nanomedicine in neuroprotection, neuroregeneration, and blood-brain barrier modulation: A narrative review. Medicina 2024, 60, 1384. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Ferro, M.M.; Bellissimo, M.I.; Anselmo-Franci, J.A.; Angellucci, M.E.M.; Canteras, N.S.; Cunha, C.D. Comparison of bilaterally 6-OHDA- and MPTP-lesioned rats as models of the early phase of Parkinson’s disease: Histological, neurochemical, motor and memory alterations. J. Neurosci. Meth. 2005, 148, 78–87. [Google Scholar] [CrossRef]
- Shi, W.; Yuan, S.; Cheng, G.; Zhang, H.; Liu, K.J.; Ji, X.; Du, L.; Qi, Z. Blood brain barrier-targeted lipid nanoparticles improved the neuroprotection of Ferrostatin-1 against cerebral ischemic damage in an experimental stroke model. Exp. Neurol. 2024, 379, 114849. [Google Scholar] [CrossRef]
- Azouz, A.A.; El Komy, M.H.; Elmowafy, M.; Mahmoud, M.O.; Ali, F.E.M.; Aboud, H.M. Crafting cationic lecithmer nanocomposites as promising wagons for brain targeting of cinnamaldehyde: Accentuated neuroprotection via downregulation of Aβ1-42/p-tau crosstalk. J. Drug Deliv. Sci. Tec. 2025, 106, 106664. [Google Scholar] [CrossRef]
- Xu, J.; Wan, L.; Wang, X.; Wei, Y.; He, Y.; You, S.; Zhong, R.; Wang, C.; Li, H.; You, C.; et al. A combined strategy of brain neuroprotection and endogenous neuroregeneration for enhanced intracerebral hemorrhage treatment via an injectable biomimetic hydrogel with efficient ROS scavenging and therapeutics delivery. Chem. Eng. J. 2025, 503, 158069. [Google Scholar] [CrossRef]
- Lin, W.J.; Kao, L.T. Cytotoxic enhancement of hexapeptide-conjugated micelles in EGFR high-expressed cancer cells. Expert Opin. Drug Deliv. 2014, 11, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, A.; Vega, E.; Pérez, Y.; Gómara, M.J.; García, M.L.; Haro, I. Conjugation of cell-penetrating peptides with poly(lactic-co-glycolic acid)-polyethylene glycol nanoparticles improves ocular drug delivery. Int. J. Nanomed. 2015, 10, 609–631. [Google Scholar]
- Halevas, E.; Kokotidou, C.; Zaimai, E.; Moschona, A.; Lialiaris, E.; Mitraki, A.; Lialiaris, T.; Pantazaki, A. Evaluation of the hemocompatibility and anticancer potential of poly(ε-caprolactone) and poly(3-hydroxybutyrate) microcarriers with encapsulated chrysin. Pharmaceutics 2021, 13, 109. [Google Scholar] [CrossRef]
- Byeon, H.J.; Thao, L.Q.; Lee, S.; Min, S.Y.; Lee, E.S.; Shin, B.S.; Choi, H.G.; Youn, Y.S. Doxorubicin-loaded nanoparticles consisted of cationic- and mannose-modified-albumins for dual-targeting in brain tumors. J. Control. Release 2016, 225, 301–313. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, M.; Zeng, F.; Jin, H.; Xu, Q.; Huang, Y. Dual-targeting magnetic PLGA nanoparticles for codelivery of paclitaxel and curcumin for brain tumor therapy. ACS Appl. Mater. Interfaces 2016, 8, 32159–32169. [Google Scholar] [CrossRef]
- Wei, L.; Guo, X.Y.; Yang, T.; Yu, M.Z.; Chen, D.W.; Wang, J.C. Brain tumor-targeted therapy by systemic delivery of siRNA with Transferrin receptor-mediated core-shell nanoparticles. Int. J. Pharm. 2016, 510, 394–405. [Google Scholar] [CrossRef]
- Zhukova, V.; Osipova, N.; Semyonkin, A.; Malinovskaya, J.; Melnikov, P.; Valikhov, M.; Porozov, Y.; Solovev, Y.; Kuliaev, P.; Zhang, E.; et al. Fluorescently labeled PLGA nanoparticles for visualization in vitro and in vivo: The importance of dye properties. Pharmaceutics 2021, 13, 1145. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Size (nm) | PDI | ZP (mV) | Yield (%) | EE (%) | DL (%) |
|---|---|---|---|---|---|---|
| Seliciclib@PPM NPs | 115.7 ± 5.5 | 0.11 ± 0.03 | −30.8 ± 9.2 | 72.5 ± 3.6 | 64.8 ± 3.7 | 14.9 ± 1.0 |
| Seliciclib@PPM NPs-Cys-T7 | 127.3 ± 0.7 | 0.19 ± 0.03 | −20.0 ± 4.2 | 81.3 ± 1.7 | 60.0 ± 1.2 | 12.3 ± 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, G.Z.; Lin, W.J. Peptide-Engineered Seliciclib Nanomedicine for Brain-Targeted Delivery and Neuroprotection. Int. J. Mol. Sci. 2025, 26, 5768. https://doi.org/10.3390/ijms26125768
He GZ, Lin WJ. Peptide-Engineered Seliciclib Nanomedicine for Brain-Targeted Delivery and Neuroprotection. International Journal of Molecular Sciences. 2025; 26(12):5768. https://doi.org/10.3390/ijms26125768
Chicago/Turabian StyleHe, Guan Zhen, and Wen Jen Lin. 2025. "Peptide-Engineered Seliciclib Nanomedicine for Brain-Targeted Delivery and Neuroprotection" International Journal of Molecular Sciences 26, no. 12: 5768. https://doi.org/10.3390/ijms26125768
APA StyleHe, G. Z., & Lin, W. J. (2025). Peptide-Engineered Seliciclib Nanomedicine for Brain-Targeted Delivery and Neuroprotection. International Journal of Molecular Sciences, 26(12), 5768. https://doi.org/10.3390/ijms26125768