
Clinical and Allelic Heterogeneity in a Small Cohort of Patients with Inherited Epidermolysis Bullosa

 , , , , , , , , and
, , , , , , , , and 
Abstract
1. Introduction
2. Cases Presentation
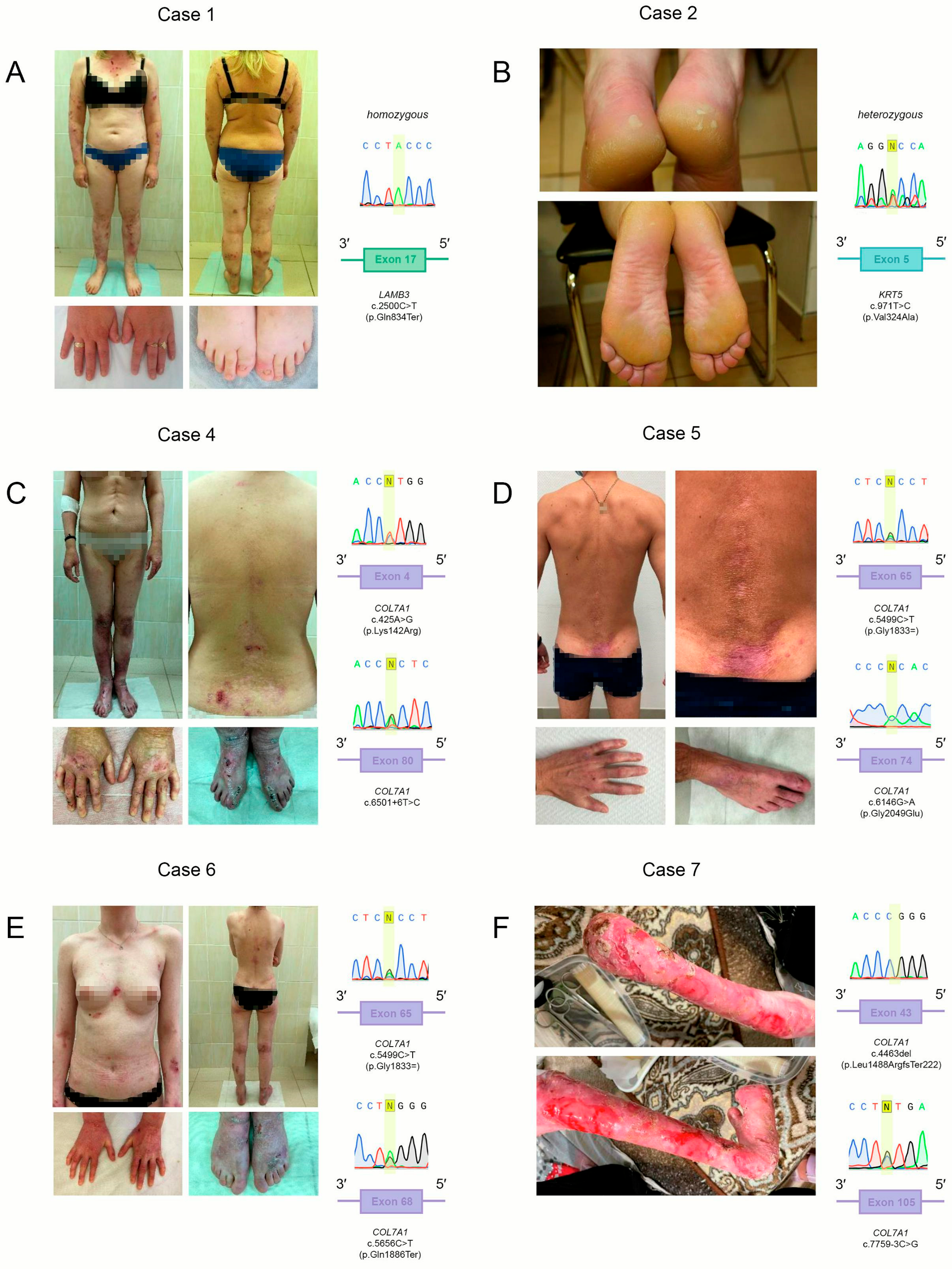
2.1. Case 1
2.2. Case 2 and Case 3
2.3. Case 4
2.4. Case 5
2.5. Case 6
2.6. Case 7
3. Discussion
3.1. Case 1
3.2. Case 2 and Case 3
3.3. Case 4
3.4. Case 5
3.5. Case 6
3.6. Case 7
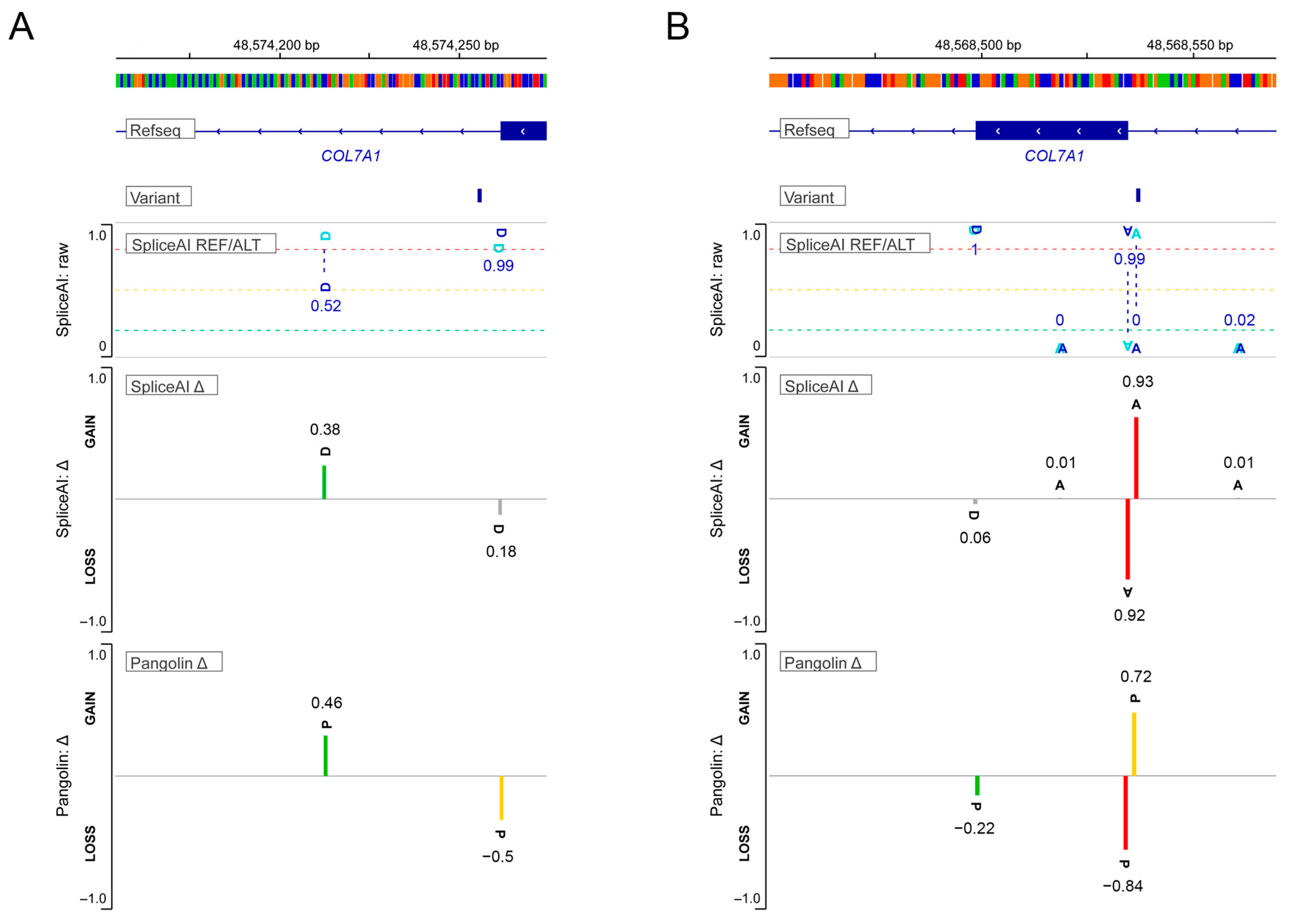
3.7. Intronic Sequence Variants of COL7A1 Associated with Aberrant Splicing
4. Materials and Methods
4.1. WES
4.2. IHC
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bardhan, A.; Bruckner-Tuderman, L.; Chapple, I.L.C.; Fine, J.-D.; Harper, N.; Has, C.; Magin, T.M.; Marinkovich, M.P.; Marshall, J.F.; McGrath, J.A.; et al. Epidermolysis Bullosa. Nat. Rev. Dis. Primers 2020, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- King, A.D.; Deirawan, H.; Klein, P.A.; Dasgeb, B.; Dumur, C.I.; Mehregan, D.R. Next-Generation Sequencing in Dermatology. Front. Med. 2023, 10, 1218404. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A.; Ashton, G.H.S.; Mellerio, J.E.; McMillan, J.R.; Eady, R.A.J.; Salas-Alanis, J.C.; Swensson, O. Moderation of Phenotypic Severity in Dystrophic and Junctional Forms of Epidermolysis Bullosa Through In-Frame Skipping of Exons Containing Non-Sense or Frameshift Mutations. J. Investig. Dermatol. 1999, 113, 314–321. [Google Scholar] [CrossRef]
- Chiang, Y.-Y.; Chao, S.-C.; Chen, W.-Y.; Lee, W.-R.; Wang, K.-H. Weber-Cockayne Type of Epidermolysis Bullosa Simplex Associated with a Novel Mutation in Keratin 5 and Amyloid Deposits. Br. J. Dermatol. 2008, 159, 1370–1372. [Google Scholar] [CrossRef]
- Csikos, M.; Szocs, H.I.; Laszik, A.; Mecklenbeck, S.; Horvath, A.; Karpati, S.; Bruckner-Tuderman, L. High Frequency of the 425AG Splice-Site Mutation and Novel Mutations of the COL7A1 Gene in Central Europe: Significance for Future Mutation Detection Strategies in Dystrophic Epidermolysis Bullosa. Br. J. Dermatol. 2005, 152, 879–886. [Google Scholar] [CrossRef]
- Kern, J.S.; Grüninger, G.; Imsak, R.; Müller, M.L.; Schumann, H.; Kiritsi, D.; Emmert, S.; Borozdin, W.; Kohlhase, J.; Bruckner-Tuderman, L.; et al. Forty-Two Novel COL7A1 Mutations and the Role of a Frequent Single Nucleotide Polymorphism in the MMP1 Promoter in Modulation of Disease Severity in a Large European Dystrophic Epidermolysis Bullosa Cohort. Br. J. Dermatol. 2009, 161, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Kopečková, L.; Bučková, H.; Kýrová, J.; Gaillyová, R.; Němečková, J.; Jeřábková, B.; Veselý, K.; Stehlíková, K.; Fajkusová, L. Ten Years of DNA Diagnostics of Epidermolysis Bullosa in the Czech Republic. Br. J. Dermatol. 2016, 174, 1388–1391. [Google Scholar] [CrossRef]
- Jain, S.V.; Harris, A.G.; Su, J.C.; Orchard, D.; Warren, L.J.; McManus, H.; Murrell, D.F. The Epidermolysis Bullosa Disease Activity and Scarring Index ( EBDASI ): Grading Disease Severity and Assessing Responsiveness to Clinical Change in Epidermolysis Bullosa. Acad. Dermatol. Venereol. 2017, 31, 692–698. [Google Scholar] [CrossRef]
- Woodley, D.T.; Hao, M.; Kwong, A.; Levian, B.; Cogan, J.; Hou, Y.; Mosallaei, D.; Kleinman, E.; Zheng, K.; Chung, C.; et al. Intravenous Gentamicin Therapy Induces Functional Type VII Collagen in Patients with Recessive Dystrophic Epidermolysis Bullosa: An Open-Label Clinical Trial. Br. J. Dermatol. 2024, 191, 267–274. [Google Scholar] [CrossRef]
- Gupta, D.; Jayashankar, C.; Srinivas, M.; Baraka Vishwanathan, G.; Reddy, K.R.; Kubba, A.; Batrani, M.; Hiremagalore, R. Clinical and Allelic Heterogeneity in Dystrophic Epidermolysis Bullosa- Lessons from an Indian Cohort. PLoS ONE 2023, 18, e0289558. [Google Scholar] [CrossRef]
- Posteraro, P.; Sorvillo, S.; Gagnoux-Palacios, L.; Angelo, C.; Paradisi, M.; Meneguzzi, G.; Castiglia, D.; Zambruno, G. Compound Heterozygosity for an Out-of-Frame Deletion and a Splice Site Mutation in the LAMB3 Gene Causes Nonlethal Junctional Epidermolysis Bullosa. Biochem. Biophys. Res. Commun. 1998, 243, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Pulkkinen, L.; Jonkman, M.F.; McGrath, J.A.; Kuijpers, A.; Paller, A.S.; Uitto, J. LAMB3 Mutations in Generalized Atrophic Benign Epidermolysis Bullosa: Consequences at the mRNA and Protein Levels. Lab. Investig. 1998, 78, 859–867. [Google Scholar] [CrossRef]
- Swensson, O.; Christophers, E. Generalized Atrophic Benign Epidermolysis Bullosa in 2 Siblings Complicated by Multiple Squamous Cell Carcinomas. Arch. Dermatol. 1998, 134, 199. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, Z.; Mi, Z.; Sun, L.; Fu, X.; Yu, G.; Pang, Z.; Liu, H.; Zhang, F. Epidermolysis Bullosa in Chinese Patients: Genetic Analysis and Mutation Landscape in 57 Pedigrees and Sporadic Cases. Acta Derm. Venereol. 2021, 101, adv00503. [Google Scholar] [CrossRef] [PubMed]
- Varki, R.; Sadowski, S.; Uitto, J.; Pfendner, E. Epidermolysis Bullosa. II. Type VII Collagen Mutations and Phenotype-Genotype Correlations in the Dystrophic Subtypes. J. Med. Genet. 2006, 44, 181–192. [Google Scholar] [CrossRef]
- Wertheim-Tysarowska, K.; Sobczyńska-Tomaszewska, A.; Kowalewski, C.; Kutkowska-Kaźmierczak, A.; Woźniak, K.; Niepokój, K.; Klausegger, A.; Sypniewska-Jutkiewicz, J.; Stępień, A.; Bal, J. Novel and Recurrent COL7A1 Mutation in a Polish Population. Eur. J. Dermatol. 2012, 22, 23–28. [Google Scholar] [CrossRef]
- Beilin, A.K.; Evtushenko, N.A.; Lukyanov, D.K.; Murashkin, N.N.; Ambarchian, E.T.; Pushkov, A.A.; Savostyanov, K.V.; Fisenko, A.P.; Rogovaya, O.S.; Vasiliev, A.V.; et al. Signatures of Dermal Fibroblasts from RDEB Pediatric Patients. Int. J. Mol. Sci. 2021, 22, 1792. [Google Scholar] [CrossRef]
- Savostyanov, K.; Murashkin, N.; Pushkov, A.; Zhanin, I.; Suleymanov, E.; Akhkiamova, M.; Shchagina, O.; Balanovska, E.; Epishev, R.; Polyakov, A.; et al. Targeted NGS in Diagnostics of Genodermatosis Characterized by the Epidermolysis Bullosa Symptom Complex in 268 Russian Children. Int. J. Mol. Sci. 2022, 23, 14343. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Wagner, R.N.; Klausegger, A.; Guttmann-Gruber, C.; Hainzl, S.; Bauer, J.W.; Reichelt, J.; Koller, U. Improved Double-Nicking Strategies for COL7A1-Editing by Homologous Recombination. Mol. Ther. Nucleic Acids 2019, 18, 496–507. [Google Scholar] [CrossRef]
- Chong, S.C.; Hon, K.L.; Yuen, L.Y.P.; Choi, P.C.L.; Ng, W.G.G.; Chiu, T.W. Neonatal Epidermolysis Bullosa: Lessons to Learn about Genetic Counseling. J. Dermatol. Treat. 2021, 32, 29–32. [Google Scholar] [CrossRef]
- Rashidghamat, E.; McGrath, J.A. Novel and Emerging Therapies in the Treatment of Recessive Dystrophic Epidermolysis Bullosa. Intractable Rare Dis. Res. 2017, 6, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Zingman, L.V.; Park, S.; Olson, T.M.; Alekseev, A.E.; Terzic, A. Aminoglycoside-Induced Translational Read-through in Disease: Overcoming Nonsense Mutations by Pharmacogenetic Therapy. Clin. Pharmacol. Ther. 2007, 81, 99–103. [Google Scholar] [CrossRef]
- Cogan, J.; Weinstein, J.; Wang, X.; Hou, Y.; Martin, S.; South, A.P.; Woodley, D.T.; Chen, M. Aminoglycosides Restore Full-Length Type VII Collagen by Overcoming Premature Termination Codons: Therapeutic Implications for Dystrophic Epidermolysis Bullosa. Mol. Ther. 2014, 22, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Woodley, D.T.; Cogan, J.; Hou, Y.; Lyu, C.; Marinkovich, M.P.; Keene, D.; Chen, M. Gentamicin Induces Functional Type VII Collagen in Recessive Dystrophic Epidermolysis Bullosa Patients. J. Clin. Investig. 2017, 127, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Maquat, L.E. A Rule for Termination-Codon Position within Intron-Containing Genes: When Nonsense Affects RNA Abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Levian, B.; Hou, Y.; Tang, X.; Bainvoll, L.; Zheng, K.; Badarinarayana, V.; Aghamohammadzadeh, S.; Chen, M. Novel readthrough agent suppresses nonsense mutations and restores functional type VII collagen and laminin 332 in epidermolysis bullosa. Mol. Ther. Nucleic Acids 2024, 35, 102334. [Google Scholar] [CrossRef]
- Rivas, M.A.; Pirinen, M.; Conrad, D.F.; Lek, M.; Tsang, E.K.; Karczewski, K.J.; Maller, J.B.; Kukurba, K.R.; DeLuca, D.S.; Fromer, M.; et al. Effect of Predicted Protein-Truncating Genetic Variants on the Human Transcriptome. Science 2015, 348, 666–669. [Google Scholar] [CrossRef]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J.; et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef]
- Vermeer, F.; Bremer, J.; Sietsma, R.; Sandilands, A.; Hickerson, R.; Bolling, M.; Pasmooij, A.; Lemmink, H.; Swertz, M.; Knoers, N.; et al. Therapeutic Prospects of Exon Skipping for Epidermolysis Bullosa. Int. J. Mol. Sci. 2021, 22, 12222. [Google Scholar] [CrossRef]
- Gardella, R.; Castiglia, D.; Posteraro, P.; Bernardini, S.; Zoppi, N.; Paradisi, M.; Tadini, G.; Barlati, S.; McGrath, J.A.; Zambruno, G.; et al. Genotype–Phenotype Correlation in Italian Patients with Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2002, 119, 1456–1462. [Google Scholar] [CrossRef]
- Von Bartenwerffer, W.; Has, C.; Arin, M.J.; Tantcheva-Poór, I.; Kreuter, A.; Kremer, K.; Arshah, T.; Hoffmann, M.; Eming, S.A.; Kohlhase, J.; et al. Mild Recessive Dystrophic Epidermolysis Bullosa Associated with Two Compound Heterozygous COL7A1 Mutations. Eur. J. Dermatol. 2011, 21, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Pironon, N.; Bourrat, E.; Prost, C.; Chen, M.; Woodley, D.T.; Titeux, M.; Hovnanian, A. Splice Modulation Strategy Applied to Deep Intronic Variants in COL7A1 Causing Recessive Dystrophic Epidermolysis Bullosa. Proc. Natl. Acad. Sci. USA 2024, 121, e2401781121. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.; Santos, J.I.; Coutinho, M.F.; Matos, L.; Alves, S. Development of Engineered-U1 snRNA Therapies: Current Status. Int. J. Mol. Sci. 2023, 24, 14617. [Google Scholar] [CrossRef] [PubMed]
- Igreja, S.; Clarke, L.A.; Botelho, H.M.; Marques, L.; Amaral, M.D. Correction of a Cystic Fibrosis Splicing Mutation by Antisense Oligonucleotides. Hum. Mutat. 2016, 37, 209–215. [Google Scholar] [CrossRef]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Décha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef]
- Bremer, J.; Bornert, O.; Nyström, A.; Gostynski, A.; Jonkman, M.F.; Aartsma-Rus, A.; Van Den Akker, P.C.; Pasmooij, A.M. Antisense Oligonucleotide-Mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther.-Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef]
- Bornert, O.; Hogervorst, M.; Nauroy, P.; Bischof, J.; Swildens, J.; Athanasiou, I.; Tufa, S.F.; Keene, D.R.; Kiritsi, D.; Hainzl, S.; et al. QR-313, an Antisense Oligonucleotide, Shows Therapeutic Efficacy for Treatment of Dominant and Recessive Dystrophic Epidermolysis Bullosa: A Preclinical Study. J. Investig. Dermatol. 2021, 141, 883–893.e6. [Google Scholar] [CrossRef]
- Bornert, O.; Kühl, T.; Bremer, J.; Van Den Akker, P.C.; Pasmooij, A.M.; Nyström, A. Analysis of the Functional Consequences of Targeted Exon Deletion in COL7A1 Reveals Prospects for Dystrophic Epidermolysis Bullosa Therapy. Mol. Ther. 2016, 24, 1302–1311. [Google Scholar] [CrossRef]
- Bremer, J.; Van Der Heijden, E.H.; Eichhorn, D.S.; Meijer, R.; Lemmink, H.H.; Scheffer, H.; Sinke, R.J.; Jonkman, M.F.; Pasmooij, A.M.G.; Van Den Akker, P.C. Natural Exon Skipping Sets the Stage for Exon Skipping as Therapy for Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2019, 18, 465–475. [Google Scholar] [CrossRef]
- Masunaga, T.; Kubo, A.; Ishiko, A. Splice Site Mutation in COL 7A1 Resulting in Aberrant In-frame Transcripts Identified in a Case of Recessive Dystrophic Epidermolysis Bullosa, Pretibial. J. Dermatol. 2018, 45, 742–745. [Google Scholar] [CrossRef]
- Belova, V.; Pavlova, A.; Afasizhev, R.; Moskalenko, V.; Korzhanova, M.; Krivoy, A.; Cheranev, V.; Nikashin, B.; Bulusheva, I.; Rebrikov, D.; et al. System Analysis of the Sequencing Quality of Human Whole Exome Samples on BGI NGS Platform. Sci. Rep. 2022, 12, 609. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | cDNA Change | Protein Change | rs ID | ACMG Classification (According to VarSome) | ACMG Criteria | Previously Described |
|---|---|---|---|---|---|---|
| LAMB3 (NM_000228.3) | c.2500C>T | p.Gln834Ter | rs770868939 | LP | PVS1, PM2 | [3] |
| KRT5 (NM_000424.4) | c.971T>C | p.Val324Ala | rs59335325 | P | PM1, PM2, PM5, PP3 | [4] |
| COL7A1 (NM_000094.4) | c.425A>G | p.Lys142Arg (aberrant splicing) | rs121912856 | P | PM2, PP3, PP5 | [5] |
| c.4463del | p.Leu1488ArgfsTer222 | rs2044901784 | P | PVS1, PM2, PP5 | No | |
| c.5499C>T | p.Gly1833= (aberrant splicing) | rs758886532 | LP | PM2, PP5, BP4 | [6] | |
| c.5656C>T | p.Gln1886Ter | rs2044329211 | P | PVS1, PM2, PP5 | No | |
| c.6146G>A | p.Gly2049Glu | rs1410793870 | P | PM1, PM2, PM5, PP3, PP5 | [7] | |
| c.6501+6T>C | rs752070397 | VUS | PM2 | No | ||
| c.7759-3C>G | rs1030248456 | LP | PM2, PP3, PP5 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buianova, A.A.; Yagizarova, A.S.; Kosykh, A.V.; Kubanov, A.A.; Belova, V.A.; Shmitko, A.O.; Karamova, A.E.; Martynova, A.A.; Podmoskovnikov, G.S.; Nefedova, M.A.; et al. Clinical and Allelic Heterogeneity in a Small Cohort of Patients with Inherited Epidermolysis Bullosa. Int. J. Mol. Sci. 2025, 26, 5762. https://doi.org/10.3390/ijms26125762
Buianova AA, Yagizarova AS, Kosykh AV, Kubanov AA, Belova VA, Shmitko AO, Karamova AE, Martynova AA, Podmoskovnikov GS, Nefedova MA, et al. Clinical and Allelic Heterogeneity in a Small Cohort of Patients with Inherited Epidermolysis Bullosa. International Journal of Molecular Sciences. 2025; 26(12):5762. https://doi.org/10.3390/ijms26125762
Chicago/Turabian StyleBuianova, Anastasiia A., Anastasia S. Yagizarova, Anastasiya V. Kosykh, Alexey A. Kubanov, Vera A. Belova, Anna O. Shmitko, Arfenya E. Karamova, Aleksandra A. Martynova, Grigoriy S. Podmoskovnikov, Maria A. Nefedova, and et al. 2025. "Clinical and Allelic Heterogeneity in a Small Cohort of Patients with Inherited Epidermolysis Bullosa" International Journal of Molecular Sciences 26, no. 12: 5762. https://doi.org/10.3390/ijms26125762
APA StyleBuianova, A. A., Yagizarova, A. S., Kosykh, A. V., Kubanov, A. A., Belova, V. A., Shmitko, A. O., Karamova, A. E., Martynova, A. A., Podmoskovnikov, G. S., Nefedova, M. A., Monchakovskaya, E. S., Korostin, D. O., Gurskaya, N. G., & Rebrikov, D. V. (2025). Clinical and Allelic Heterogeneity in a Small Cohort of Patients with Inherited Epidermolysis Bullosa. International Journal of Molecular Sciences, 26(12), 5762. https://doi.org/10.3390/ijms26125762