
Molecular Mechanisms of L-Type Calcium Channel Dysregulation in Heart Failure

 , and
, and
Abstract
1. Introduction
2. Methods
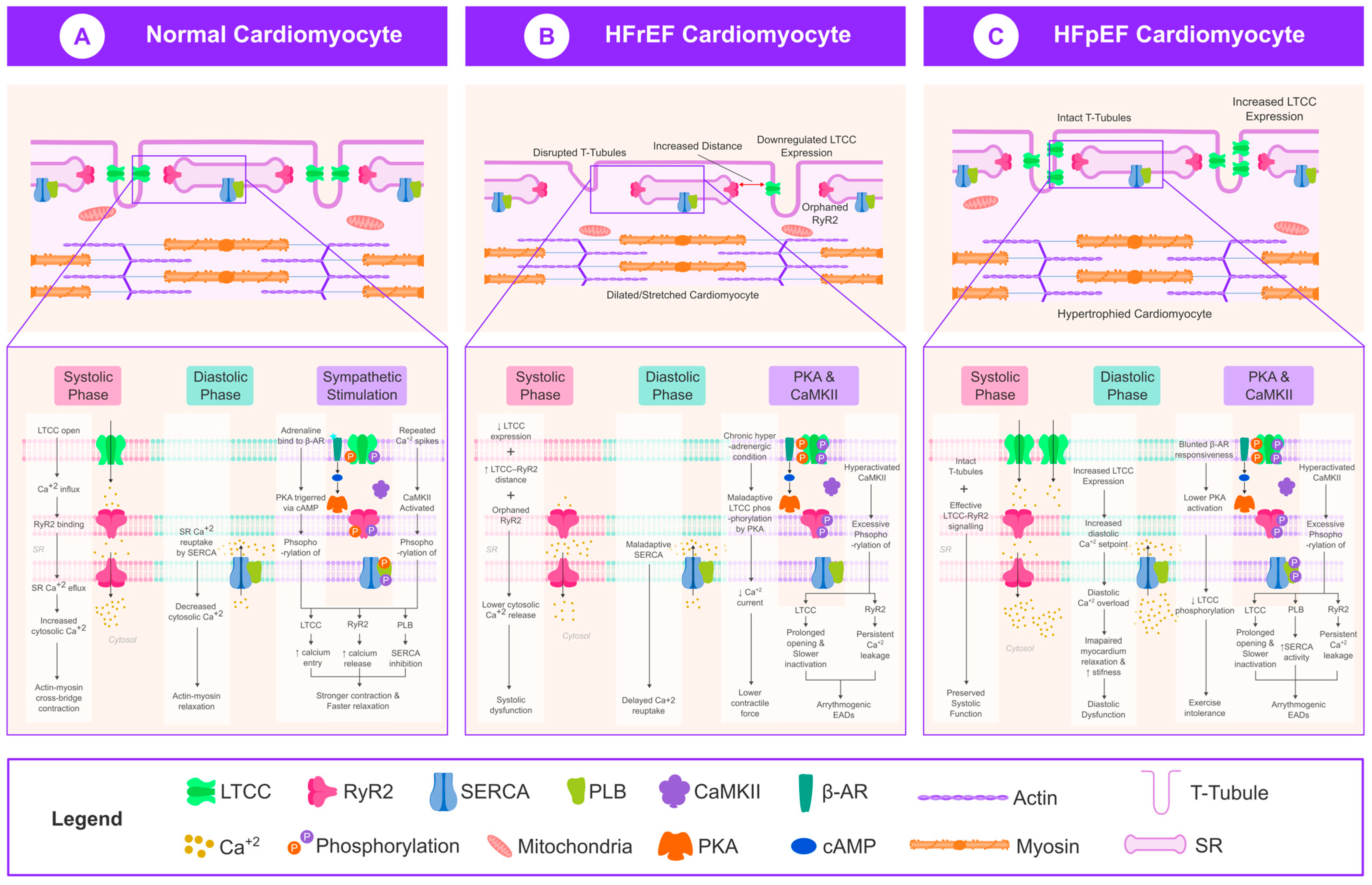
3. Discussion
3.1. LTCC Dysregulation in HFrEF
3.2. LTCC Dysregulation in HFpEF
3.3. Role of CaMKII in LTCC Regulation
3.4. Emerging Therapeutic Targets
3.4.1. CaMKII Inhibition
3.4.2. LTCC Modulation
3.4.3. RyR2 Stabilizers and Ca2+ Cycling Enhancers
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hersel, J.; Jung, S.; Mohacsi, P.; Hullin, R. Expression of the L-Type Calcium Channel in Human Heart Failure. Basic Res. Cardiol. 2002, 97 (Suppl. 1), I4–I10. [Google Scholar] [CrossRef]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol. 2018, 1067, 373–385. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium Cycling Proteins and Heart Failure: Mechanisms and Therapeutics. J. Clin. Invest. 2013, 123, 46–52. [Google Scholar] [CrossRef]
- Kilfoil, P.J.; Lotteau, S.; Zhang, R.; Yue, X.; Aynaszyan, S.; Solymani, R.E.; Cingolani, E.; Marbán, E.; Goldhaber, J.I. Distinct Features of Calcium Handling and Β-adrenergic Sensitivity in Heart Failure with Preserved versus Reduced Ejection Fraction. J. Physiol. 2020, 598, 5091–5108. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.; Le, C.; Shen, X.; Røe, Å.T.; Hou, Y.; Manfra, O.; Silva, G.J.J.; van Hout, I.; Norden, E.S.; Aronsen, J.M.; et al. Etiology-Dependent Impairment of Diastolic Cardiomyocyte Calcium Homeostasis in Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2021, 77, 405–419. [Google Scholar] [CrossRef]
- Alcaide, P.; Kallikourdis, M.; Emig, R.; Prabhu, S.D. Myocardial Inflammation in Heart Failure with Reduced and Preserved Ejection Fraction. Circ. Res. 2024, 134, 1752–1766. [Google Scholar] [CrossRef]
- van de Bovenkamp, A.A.; Nassiri, S.; Bakermans, A.J.; Burchell, G.L.; de Man, F.S.; van Loon, R.B.; Handoko, M.L. Long-Term Hemodynamic Responses and Reverse Remodeling after Pharmacotherapy in HFpEF versus HFrEF: A Systematic Review and Meta-Analysis. Am. J. Physiol. Heart Circ. Physiol. 2025, 328, H419–H432. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. CaMKII: Linking Heart Failure and Arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef] [PubMed]
- Beauverger, P.; Ozoux, M.-L.; Bégis, G.; Glénat, V.; Briand, V.; Philippo, M.-C.; Daveu, C.; Tavares, G.; Roy, S.; Corbier, A.; et al. Reversion of Cardiac Dysfunction by a Novel Orally Available Calcium/Calmodulin-Dependent Protein Kinase II Inhibitor, RA306, in a Genetic Model of Dilated Cardiomyopathy. Cardiovasc. Res. 2020, 116, 329–338. [Google Scholar] [CrossRef]
- Marks, A.R. Targeting Ryanodine Receptors to Treat Human Diseases. J. Clin. Invest. 2023, 133, e162891. [Google Scholar] [CrossRef]
- Zaveri, S.; Srivastava, U.; Qu, Y.S.; Chahine, M.; Boutjdir, M. Pathophysiology of Cav1.3 L-Type Calcium Channels in the Heart. Front. Physiol. 2023, 14, 1144069. [Google Scholar] [CrossRef] [PubMed]
- Setterberg, I.E.; Le, C.; Frisk, M.; Perdreau-Dahl, H.; Li, J.; Louch, W.E. The Physiology and Pathophysiology of T-Tubules in the Heart. Front. Physiol. 2021, 12, 718404. [Google Scholar] [CrossRef]
- Høydal, M.A.; Kirkeby-Garstad, I.; Karevold, A.; Wiseth, R.; Haaverstad, R.; Wahba, A.; Stølen, T.L.; Contu, R.; Condorelli, G.; Ellingsen, Ø.; et al. Human Cardiomyocyte Calcium Handling and Transverse Tubules in Mid-Stage of Post-Myocardial-Infarction Heart Failure. ESC Heart Fail. 2018, 5, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Huo, Y. Changes of Calcium Cycling in HFrEF and HFpEF. Mechanobiol. Med. 2023, 1, 100001. [Google Scholar] [CrossRef] [PubMed]
- Kamp, T.J.; He, J.-Q. L-Type Ca2+ Channels Gaining Respect in Heart Failure. Circ. Res. 2002, 91, 451–453. [Google Scholar] [CrossRef]
- Schwinger, R.H.G. Pathophysiology of Heart Failure. Cardiovasc. Diagn. Ther. 2021, 11, 26376. [Google Scholar] [CrossRef]
- Bryant, S.M.; Kong, C.H.T.; Cannell, M.B.; Orchard, C.H.; James, A.F. Loss of Caveolin-3-Dependent Regulation of ICa in Rat Ventricular Myocytes in Heart Failure. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H521–H529. [Google Scholar] [CrossRef]
- Frisk, M.; Ruud, M.; Espe, E.K.S.; Aronsen, J.M.; Røe, Å.T.; Zhang, L.; Norseng, P.A.; Sejersted, O.M.; Christensen, G.A.; Sjaastad, I.; et al. Elevated Ventricular Wall Stress Disrupts Cardiomyocyte T-Tubule Structure and Calcium Homeostasis. Cardiovasc. Res. 2016, 112, 443–451. [Google Scholar] [CrossRef]
- Schröder, F.; Handrock, R.; Beuckelmann, D.J.; Hirt, S.; Hullin, R.; Priebe, L.; Schwinger, R.H.G.; Weil, J.; Herzig, S. Increased Availability and Open Probability of Single L-Type Calcium Channels From Failing Compared with Nonfailing Human Ventricle. Circulation 1998, 98, 969–976. [Google Scholar] [CrossRef]
- Sanchez-Alonso, J.L.; Loucks, A.; Schobesberger, S.; van Cromvoirt, A.M.; Poulet, C.; Chowdhury, R.A.; Trayanova, N.; Gorelik, J. Nanoscale Regulation of L-Type Calcium Channels Differentiates between Ischemic and Dilated Cardiomyopathies. EBioMedicine 2020, 57, 102845. [Google Scholar] [CrossRef]
- Budde, H.; Hassoun, R.; Mügge, A.; Kovács, Á.; Hamdani, N. Current Understanding of Molecular Pathophysiology of Heart Failure with Preserved Ejection Fraction. Front. Physiol. 2022, 13, 928232. [Google Scholar] [CrossRef] [PubMed]
- Zhazykbayeva, S.; Pabel, S.; Mügge, A.; Sossalla, S.; Hamdani, N. The Molecular Mechanisms Associated with the Physiological Responses to Inflammation and Oxidative Stress in Cardiovascular Diseases. Biophys. Rev. 2020, 12, 947–968. [Google Scholar] [CrossRef]
- Zhang, P. CaMKII: The Molecular Villain That Aggravates Cardiovascular Disease (Review). Exp. Ther. Med. 2017, 13, 815–820. [Google Scholar] [CrossRef]
- Bers, D.M.; Grandi, E. CaMKII Regulation of Cardiac Ion Channels. J. Cardiovasc. Pharmacol. 2009, 54, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.; Nickel, L.; Estrada, M.; Backs, J.; van den Hoogenhof, M.M.G. CaMKIIδ Splice Variants in the Healthy and Diseased Heart. Front. Cell Dev. Biol. 2021, 9, 644630. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-M.; Huang, J.; Shioda, N.; Fukunaga, K.; Shirasaki, Y.; Li, X.-M.; Han, F. CaMKIIδB Mediates Aberrant NCX1 Expression and the Imbalance of NCX1/SERCA in Transverse Aortic Constriction-Induced Failing Heart. PLoS ONE 2011, 6, e24724. [Google Scholar] [CrossRef]
- Nassal, D.; Gratz, D.; Hund, T.J. Challenges and Opportunities for Therapeutic Targeting of Calmodulin Kinase II in Heart. Front. Pharmacol. 2020, 11, 35. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, R.; Wang, K.; Zhang, W.; Zhang, M.; Jin, L.; Xie, P.; Zheng, W.; Shang, H.; Hu, Q.; et al. Novel CaMKII-δ Inhibitor Hesperadin Exerts Dual Functions to Ameliorate Cardiac Ischemia/Reperfusion Injury and Inhibit Tumor Growth. Circulation 2022, 145, 1154–1168. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Sharma, K.; Kass, D.A. Heart Failure with Preserved Ejection Fraction: Mechanisms, Clinical Features, and Therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef]
- Ortner, N.J.; Striessnig, J. L-Type Calcium Channels as Drug Targets in CNS Disorders. Channels 2016, 10, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.L.; Yatani, A.; Hawkes, M.J.; Redding, K.; Brown, A.M. Atrotoxin: A Specific Agonist for Calcium Currents in Heart. Science 1985, 229, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Xi, D.; Van Dolah, F.M.; Ramsdell, J.S. Maitotoxin Induces a Calcium-Dependent Membrane Depolarization in GH4C1 Pituitary Cells via Activation of Type L Voltage-Dependent Calcium Channels. J. Biol. Chem. 1992, 267, 25025–25031. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.L.; Perez, M. Toxins That Affect Voltage-Dependent Calcium Channels. Biochem. Pharmacol. 1987, 36, 3325–3329. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, G.; Wu, J.; Wu, Q.; Gao, S.; Yan, Z.; Lei, J.; Yan, N. Molecular Basis for Ligand Modulation of a Mammalian Voltage-Gated Ca2⁺ Channel. Cell 2019, 177, 1495–1506.e12. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Mechanisms of Dihydropyridine Agonists and Antagonists in View of Cryo-EM Structures of Calcium and Sodium Channels. J. Gen. Physiol. 2023, 155, e202313418. [Google Scholar] [CrossRef]
- Cardurion Pharmaceuticals, Inc. Cardurion Pharmaceuticals to Provide Update on CaMKII Inhibitor Program at the American Heart Association’s Scientific Sessions 2023. Business Wire. 7 November 2023. Available online: https://www.businesswire.com/news/home/20231106993542/en/Cardurion-Pharmaceuticals-to-Provide-Update-on-CaMKII-Inhibitor-Program-at-the-American-Heart-Associations-Scientific-Sessions-2023 (accessed on 3 June 2025).
- Li, J.; Balmaceda, P.; Ha, T.; Visker, J.R.; Maalouf, N.; Kwan, E.; Hoareau, G.L.; Accad, M.; Ranjan, R.; Selzman, C.H.; et al. Cardiac Bridging Integrator 1 Gene Therapy Rescues Chronic Non-Ischemic Heart Failure in Minipigs. NPJ Regen. Med. 2024, 9, 36. [Google Scholar] [CrossRef]
- Lebek, S.; Chemello, F.; Caravia, X.M.; Tan, W.; Li, H.; Chen, K.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Ablation of CaMKIIδ Oxidation by CRISPR-Cas9 Base Editing as a Therapy for Cardiac Disease. Science 2023, 379, 179–185. [Google Scholar] [CrossRef]
- Lebek, S.; Caravia, X.M.; Chemello, F.; Tan, W.; McAnally, J.R.; Chen, K.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Elimination of CaMKIIδ Autophosphorylation by CRISPR-Cas9 Base Editing Improves Survival and Cardiac Function in Heart Failure in Mice. Circulation 2023, 148, 1490–1504. [Google Scholar] [CrossRef]
- Medera Inc. Medera Presented Updated Results from First-In-Human Gene Therapy Trial for Heart Failure with Preserved Ejection Fraction at 2025 HFpEF Summit. GlobeNewswire. 1 April 2025. Available online: https://www.globenewswire.com/news-release/2025/04/01/3053271/0/en/Medera-Presented-Updated-Results-from-First-In-Human-Gene-Therapy-Trial-for-Heart-Failure-with-Preserved-Ejection-Fraction-at-2025-HFpEF-Summit.html (accessed on 3 June 2025).
- Wang, D.; Zhao, X.; Zhang, H.; Dang, L.; Huang, W.; Zhang, Z.; Song, J. Harnessing RNA Therapeutics: Novel Approaches and Emerging Strategies for Cardiovascular Disease Management. Front. Cardiovasc. Med. 2025, 12, 1546515. [Google Scholar] [CrossRef]
- Oh, J.; Kwon, O.-B.; Park, S.-W.; Kim, J.-W.; Lee, H.; Kim, Y.-K.; Choi, E.J.; Jung, H.; Choi, D.K.; Oh, B.J.; et al. Advancing Cardiovascular Drug Screening Using Human Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci. 2024, 25, 7971. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Feature | HFrEF | HFpEF |
|---|---|---|
| LTCC expression and peak L-type Ca2⁺ current | ↓ (~30–40% loss); diminished channel binding capacity | Normal or slightly ↑ |
| T-tubule architecture | Sparse, disorganized network → “orphaned” RyR2s | Preserved or mildly ↑ density |
| Post-translational state of LTCC/RyR2 | Chronic PKA + CaMKII hyper-phosphorylation → maladaptive gating | Limited PKA activity; CaMKII phosphorylation drives diastolic Ca2+ leak |
| LTCC–RyR2 coupling distance | Widened → lower excitation–contraction gain | Normal/narrow → efficient systolic trigger |
| Systolic Ca2+ release and contractility | Depressed Ca2+ transients → systolic dysfunction | Preserved Ca2+ transients → maintained EF |
| Diastolic Ca2+ handling | Normal-to-low resting Ca2+ | Elevated diastolic Ca2+, higher passive tension |
| β-adrenergic reserve | Hyper-adrenergic environment yet desensitized receptors | Blunted reserve (“ceiling effect”) |
| Predominant arrhythmogenic trigger | Early after-depolarizations (late Ca2⁺ entry + RyR2 leak) | Delayed after-depolarizations (diastolic Ca2⁺ overload) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, A.; Ahmed, A.-B.; Gill, R.; Shaikh, T.; Khorsandi, J.; Kia, A. Molecular Mechanisms of L-Type Calcium Channel Dysregulation in Heart Failure. Int. J. Mol. Sci. 2025, 26, 5738. https://doi.org/10.3390/ijms26125738
Khalid A, Ahmed A-B, Gill R, Shaikh T, Khorsandi J, Kia A. Molecular Mechanisms of L-Type Calcium Channel Dysregulation in Heart Failure. International Journal of Molecular Sciences. 2025; 26(12):5738. https://doi.org/10.3390/ijms26125738
Chicago/Turabian StyleKhalid, Arbab, Abu-Bakr Ahmed, Randeep Gill, Taha Shaikh, Joshua Khorsandi, and Ali Kia. 2025. "Molecular Mechanisms of L-Type Calcium Channel Dysregulation in Heart Failure" International Journal of Molecular Sciences 26, no. 12: 5738. https://doi.org/10.3390/ijms26125738
APA StyleKhalid, A., Ahmed, A.-B., Gill, R., Shaikh, T., Khorsandi, J., & Kia, A. (2025). Molecular Mechanisms of L-Type Calcium Channel Dysregulation in Heart Failure. International Journal of Molecular Sciences, 26(12), 5738. https://doi.org/10.3390/ijms26125738