

Multiomics Analysis Reveals Role of ncRNA in Hypoxia of Mouse Brain Microvascular Endothelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
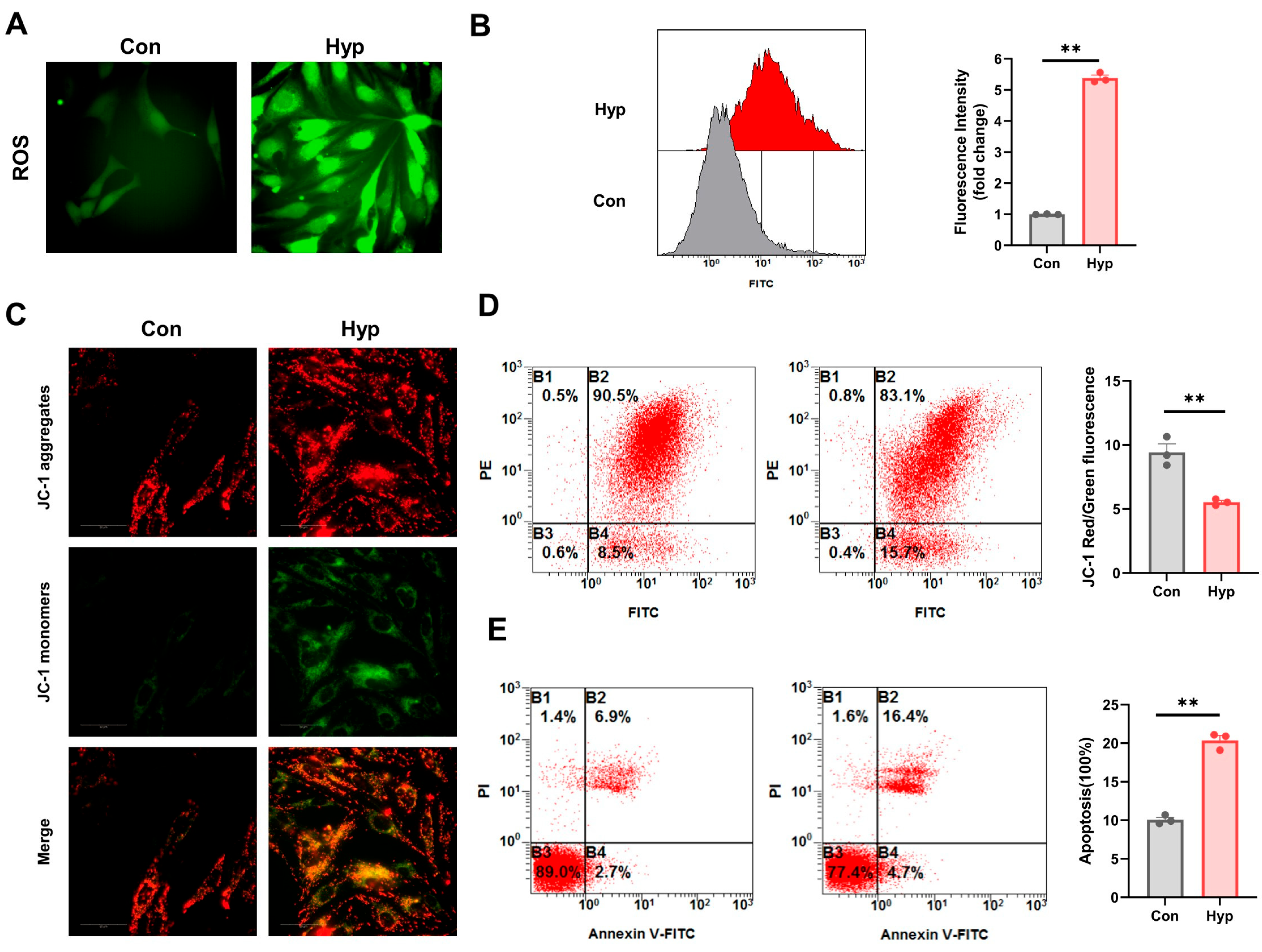
2.1. Hypoxia Impairs BMEC Mitochondrial Function and Increases ROS and Apoptosis Levels
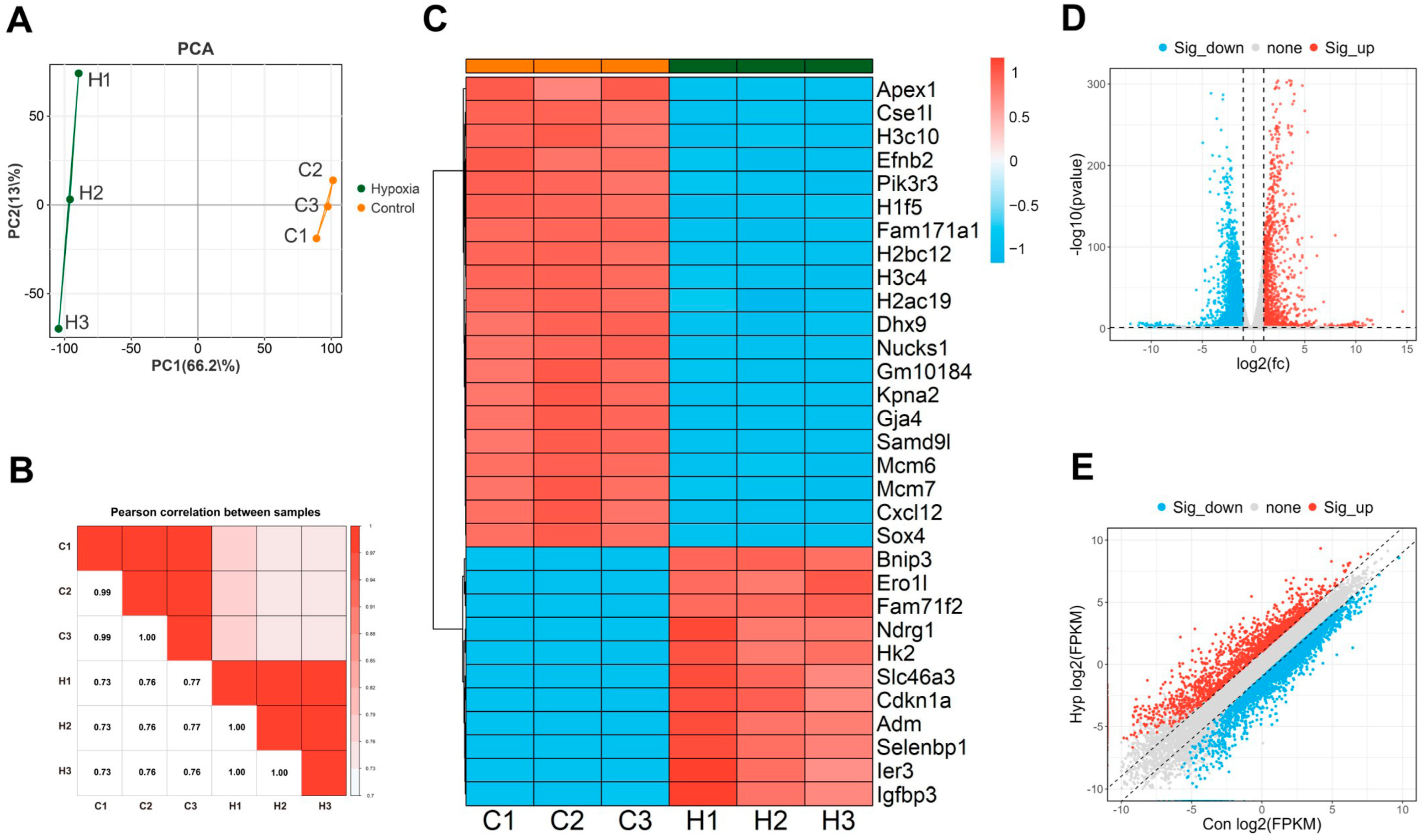
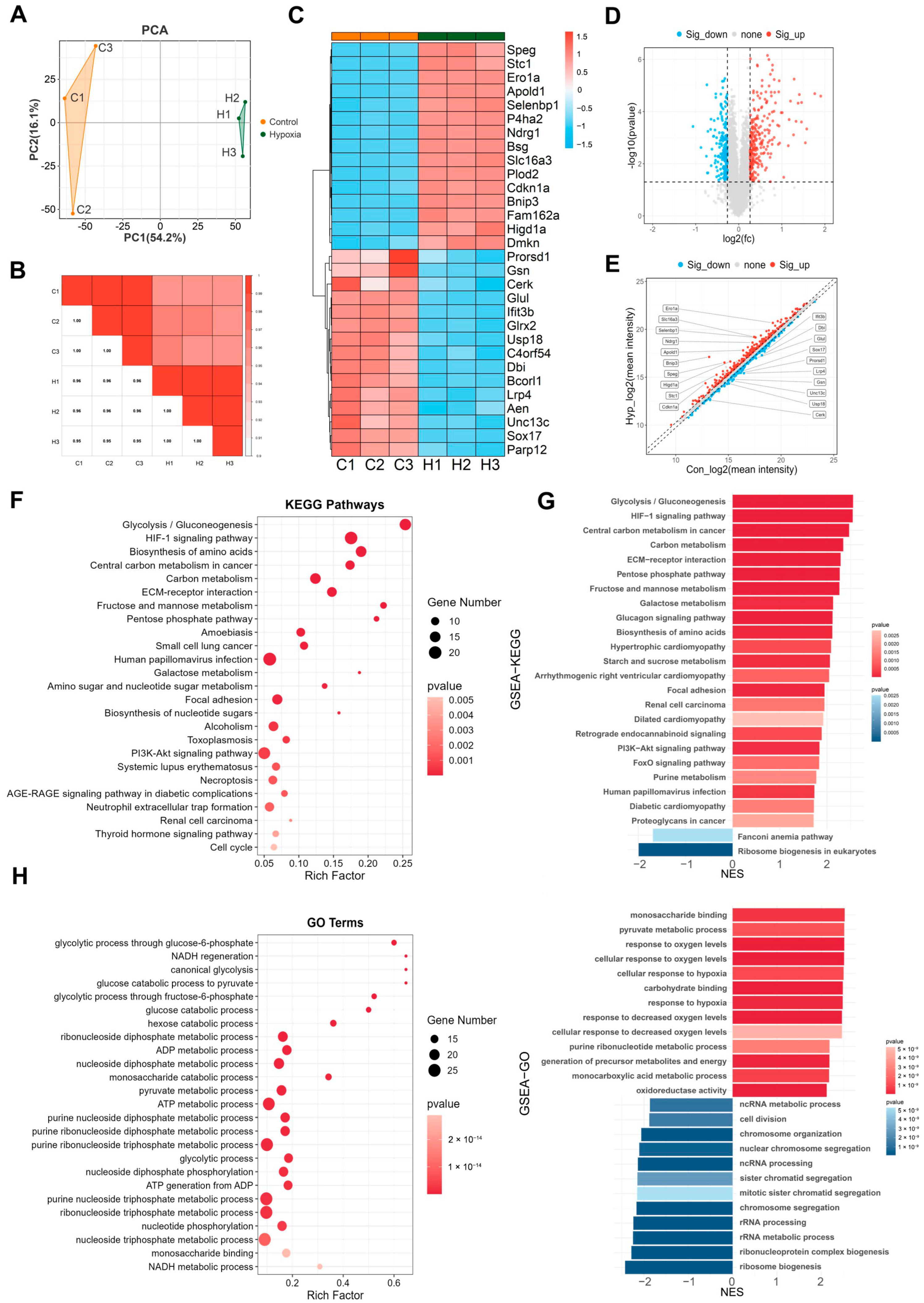
2.2. Differentially Expressed Genes (DEGs) in bEnd.3 Cells Under Hypoxic Conditions
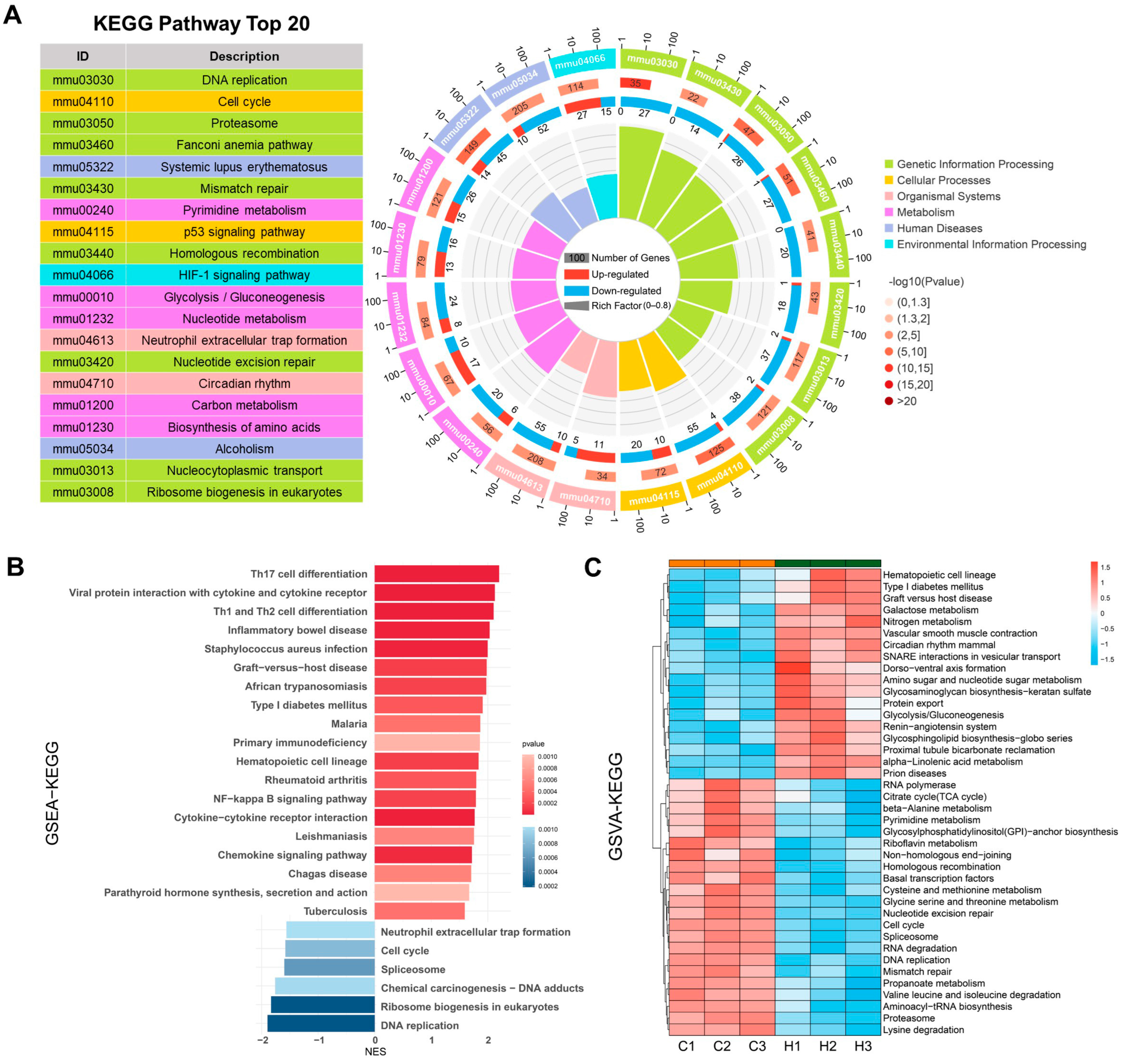
2.3. KEGG Analysis Reveals DEG Enrichment in DNA Replication, HIF-1 Signaling, and Glycolysis/Gluconeogenesis
2.4. GO Analysis Reveals DEGs Enrichment in ncRNA Processing
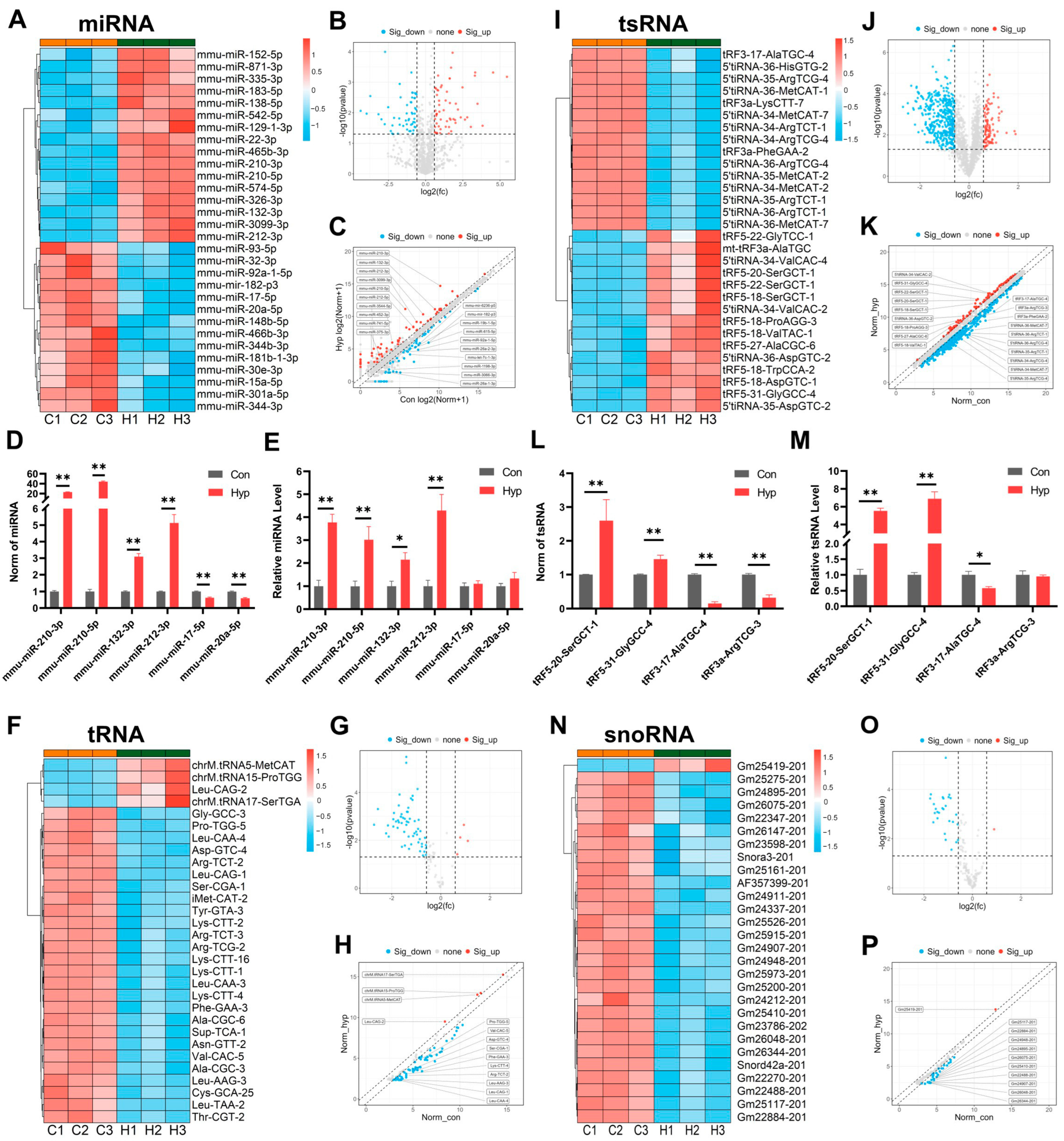
2.5. Hypoxia Alters Small Noncoding RNA Expression in BMECs
2.6. CircRNAs and lncRNAs Regulate Gene Expression Through ceRNA Mechanisms During Hypoxia
2.7. Proteomic GSEA Reveals ncRNA Processing Was Significantly Downregulated
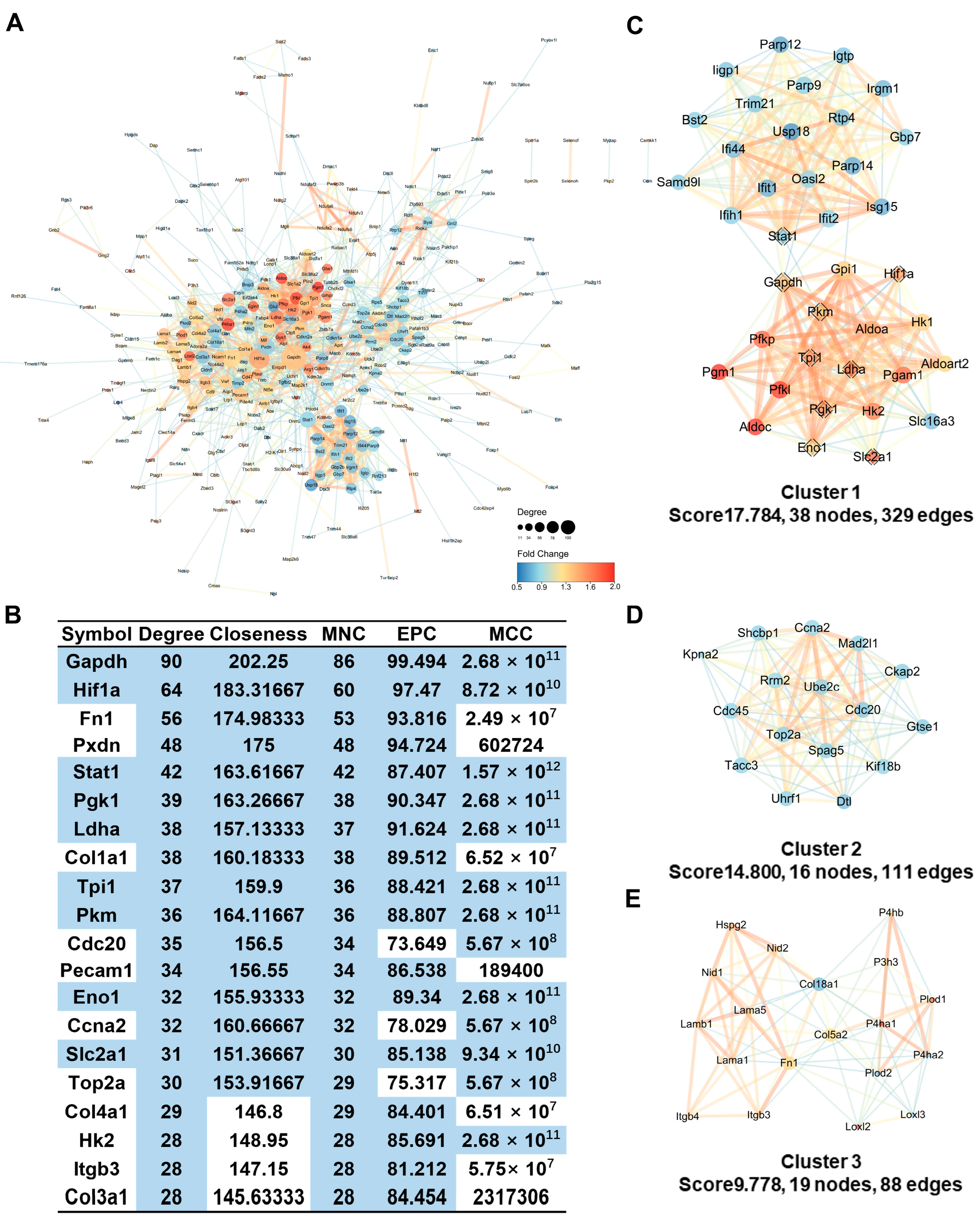
2.8. Protein-Protein Interaction Analysis Identifies Hub Genes Involved in Glycolytic Processes
2.9. Integrative Transcriptomic and Proteomic Analysis
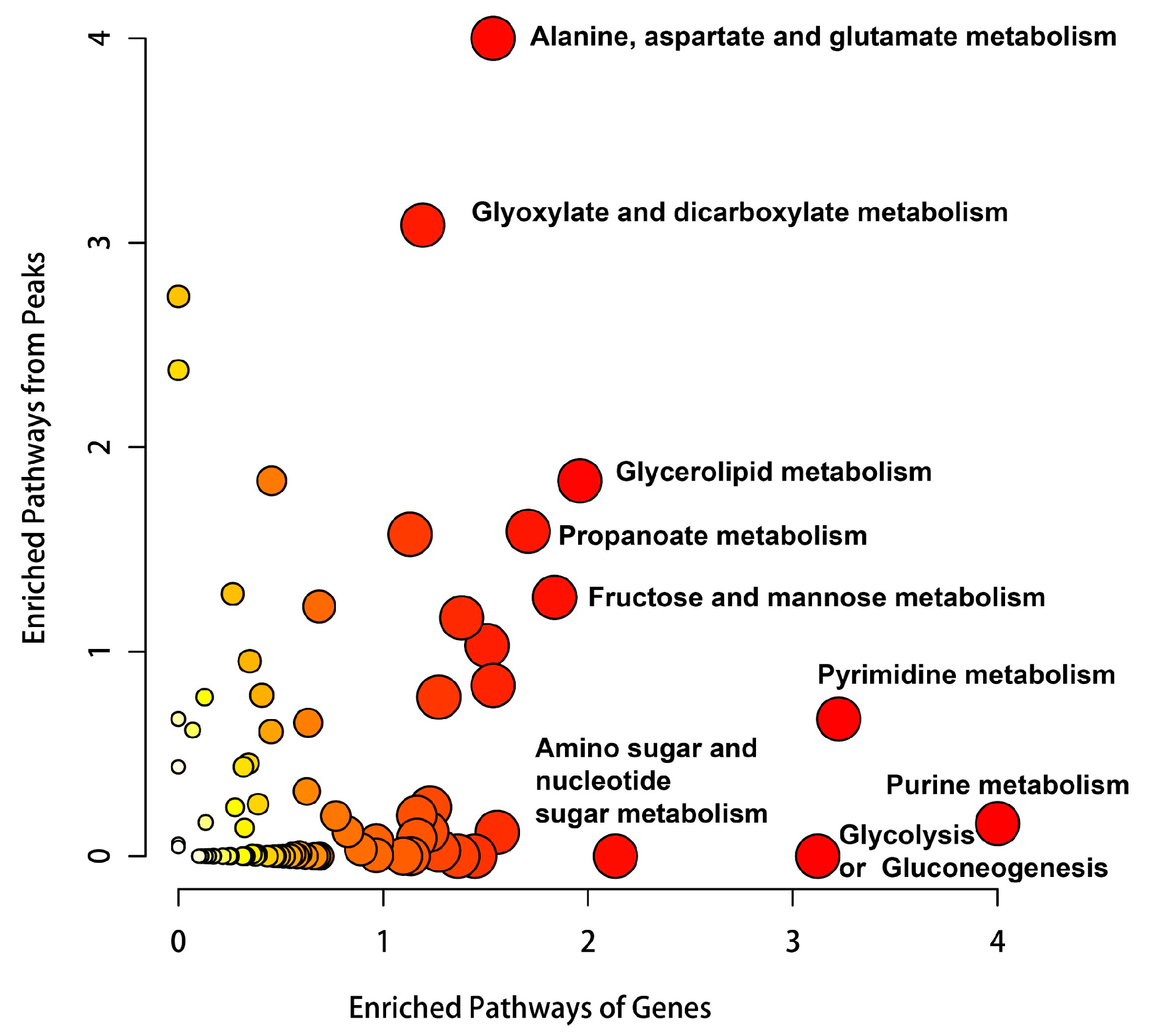
2.10. Metabolomic Analysis Shows Hypoxia Alters Purine Metabolism and Pyrimidine Metabolism
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Reactive Oxygen Species Assay
4.3. Mitochondrial Membrane Potential Assay
4.4. Apoptosis Detection
4.5. Whole-Transcriptome Sequencing Assay
4.6. Small RNA Microarray
4.7. Quantitative Reverse Transcription PCR (RT-qPCR)
4.8. TMT-Based Proteomic Quantification
4.9. Untargeted Metabolomics
4.10. Visualization of Results
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daneman, R.; Ransohoff, R.M.; Obermeier, B. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Gu, C.; Ayloo, S.; Langen, U.H. Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef]
- Shah, Y.M.; Solanki, S. Hypoxia-Induced Signaling in Gut and Liver Pathobiology. Annu. Rev. Pathol. Mech. Dis. 2024, 19, 291–317. [Google Scholar] [CrossRef]
- Verdikt, R.; Thienpont, B. Epigenetic remodelling under hypoxia. Semin. Cancer Biol. 2023, 98, 1–10. [Google Scholar] [CrossRef]
- Burtscher, M.; Millet, G.P.; Burtscher, J.; Mallet, R.T. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef] [PubMed]
- Jurajda, M.; Jelinek, M.; Duris, K. Oxidative Stress in the Brain: Basic Concepts and Treatment Strategies in Stroke. Antioxidants 2021, 10, 1886. [Google Scholar] [CrossRef]
- Wang, D.; Wang, X.; Lu, Y.; Luo, Q.; Li, M.; Xue, Y.; Zhu, L.; Wan, B.; Cheng, K.; Wang, D. Caveolin-1 accelerates hypoxia-induced endothelial dysfunction in high-altitude cerebral edema. Cell Commun. Signal. 2022, 20, 1–16. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Zhang, W.; Lin, P.; Wu, S.; Huang, C.; Chen, B.; Xiao, W.; Feng, D.; Zhang, J. Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5). Autophagy 2020, 17, 3048–3067. [Google Scholar] [CrossRef]
- Baek, S.-H.; Majid, A.; Kim, E.-H.; Shin, Y.-J.; Kim, J.-H.; Kim, K.-A.; Akram, M.; Kim, D.; Bae, O.-N. Autophagy-mediated occludin degradation contributes to blood–brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Thum, T.; Woitzik, J.; Fiedler, J.; Förster, C.Y.; Bohnert, M.; König, A.; Blecharz-Lang, K.G.; Lang, M.; Oerter, S.; Roewer, N.; et al. Hypoxia-Induced MicroRNA-212/132 Alter Blood-Brain Barrier Integrity Through Inhibition of Tight Junction-Associated Proteins in Human and Mouse Brain Microvascular Endothelial Cells. Transl. Stroke Res. 2019, 10, 672–683. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Y.; Li, Y.; Cheng, Y.; Quan, X.; Zhao, T.; Han, Y.; Shen, X.; Zheng, Y.; Zhao, Y. Comparative study of extracellular vesicles derived from mesenchymal stem cells and brain endothelial cells attenuating blood–brain barrier permeability via regulating Caveolin-1-dependent ZO-1 and Claudin-5 endocytosis in acute ischemic stroke. J. Nanobiotechnol. 2023, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Wang, Y.; Kai, G.; Ruan, W.; Gao, C.-L.; Zhao, S.; Pang, T.; Li, F. Medioresinol as a novel PGC-1α activator prevents pyroptosis of endothelial cells in ischemic stroke through PPARα-GOT1 axis. Pharmacol. Res. 2021, 169, 105640. [Google Scholar] [CrossRef]
- Zheng, Z.-J.; Hu, C.-L.; Zhu, L.-Z.; Qiu, H.; Huang, J.-R.; Chen, S.-H.; You, P.-T.; Zheng, W.-Y.; Zhou, Y.-J. Neferine inhibits BMECs pyroptosis and maintains blood–brain barrier integrity in ischemic stroke by triggering a cascade reaction of PGC-1α. Sci. Rep. 2024, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, S.; Bhawalkar, J.; Yadav, D.; Patil-Takbhate, B.; Khandagale, A.; Khopkar-Kale, P. Next-Generation sequencing transforming clinical practice and precision medicine. Clin. Chim. Acta 2023, 551, 117568. [Google Scholar] [CrossRef]
- Shi, H.; He, X.; Zuo, F.; Liu, X.; Jing, J. Artificial intelligence-based multi-omics analysis fuels cancer precision medicine. Semin. Cancer Biol. 2022, 88, 187–200. [Google Scholar] [CrossRef]
- Baysoy, A.; Satija, R.; Fan, R.; Bai, Z. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 2023, 24, 695–713. [Google Scholar] [CrossRef]
- Na Ayutthaya, W.D.; Mo, J.; Ngamratanapaiboon, S.; Yambangyang, P.; Srikornvit, N.; Hongthawonsiri, P.; Pornchokchai, K.; Wongpitoonmanachai, S.; Pholkla, P. Exploring the mechanisms of clozapine-induced blood–brain barrier dysfunction using untargeted metabolomics and cellular metabolism analysis. Environ. Toxicol. Pharmacol. 2023, 102, 104219. [Google Scholar] [CrossRef]
- Couraud, P.-O.; Guillonneau, F.; Bouzinba-Segard, H.; Bourdoulous, S.; Borderie, D.; Parmentier, Y.; Chafey, P.; Moreau, A.; Nakib, S.; Le Gall, M.; et al. Exposure of human cerebral microvascular endothelial cells hCMEC/D3 to laminar shear stress induces vascular protective responses. Fluids Barriers CNS 2022, 19, 1–24. [Google Scholar] [CrossRef]
- Mysiorek, C.; Sevin, E.; Kowalska, A.; Duban-Deweer, S.; Shimizu, F.; Hachani, J.; Rizzi, E.; Loiola, R.A.; Mazurek, M.; Kanda, T.; et al. Secretome of brain microvascular endothelial cells promotes endothelial barrier tightness and protects against hypoxia-induced vascular leakage. Mol. Med. 2024, 30, 1–23. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, Y.-G.; Zhang, H.-X.; Chen, H.-W.; Lv, P.; Su, J.; Chen, Y.-R.; Fu, Z.-F. Type I/type III IFN and related factors regulate JEV infection and BBB endothelial integrity. J. Neuroinflammation 2023, 20, 1–19. [Google Scholar] [CrossRef]
- Meng, F.; Hamblin, M.; Yuan, L.; Zhu, T.; Li, Y.; Chen, Y.; Zhang, J.; Zhang, X.; Yin, K. Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Exp. Neurol. 2016, 277, 162–170. [Google Scholar] [CrossRef]
- Huang, X.; Yin, M.; Ji, Y.; Ma, Y.; Tan, X.; Fang, C.; Chen, Y.; Gao, Q.; Wang, Y.; Yu, M.; et al. Integrated transcriptomic and proteomic profiling reveals the key molecular signatures of brain endothelial reperfusion injury. CNS Neurosci. Ther. 2023, 30. [Google Scholar] [CrossRef]
- Senavirathne, G.; Shang, R.; Lai, E.C.; Lee, S. microRNAs in action: Biogenesis, function and regulation. Nat. Rev. Genet. 2023, 24, 816–833. [Google Scholar] [CrossRef]
- Ma, J.; Li, S.; Lyu, Q.; Zhang, S.; Bai, Y.; Shi, Q. Hypoxia Inhibits Cell Cycle Progression and Cell Proliferation in Brain Microvascular Endothelial Cells via the miR-212-3p/MCM2 Axis. Int. J. Mol. Sci. 2023, 24, 2788. [Google Scholar] [CrossRef]
- Gaetano, C.; Voellenkle, C.; Greco, S.; Martelli, F.; Zaccagnini, G. miR-210 hypoxamiR in Angiogenesis and Diabetes. Antioxidants Redox Signal. 2022, 36, 685–706. [Google Scholar] [CrossRef]
- Siegal, E.; Orellana, E.A.; Gregory, R.I. tRNA dysregulation and disease. Nat. Rev. Genet. 2022, 23, 651–664. [Google Scholar] [CrossRef]
- Ruan, Y.; Yu, X.; Xie, Y.; Yao, L.; Li, Z.; Guo, J. Action mechanisms and research methods of tRNA-derived small RNAs. Signal Transduct. Target. Ther. 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Wajahat, M.; Bracken, C.P.; Orang, A. Emerging Functions for snoRNAs and snoRNA-Derived Fragments. Int. J. Mol. Sci. 2021, 22, 10193. [Google Scholar] [CrossRef]
- Liu, C.-X.; Chen, L.-L. Circular RNAs: Characterization, cellular roles, and applications. Cell 2022, 185, 2016–2034. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.-L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nature reviews Molecular cell biology 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Pyysalo, S.; Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; von Mering, C.; Doncheva, N.T.; Mehryary, F.; Jensen, L.J.; Nastou, K.; Hachilif, R.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Bono, H.; Hirota, K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines 2020, 8, 10. [Google Scholar] [CrossRef]
- Chen, R.; Yang, X.; Li, Y.; Chen, Y.; Chen, J.; Nie, X.; Zhang, J.; Bian, J.-S.; Tan, J.; Deng, L. ErbB3 Governs Endothelial Dysfunction in Hypoxia-Induced Pulmonary Hypertension. Circulation 2024, 150, 1533–1553. [Google Scholar] [CrossRef]
- Mukherjee, C.; Islam, S. Molecular regulation of hypoxia through the lenses of noncoding RNAs and epitranscriptome. Wiley Interdiscip. Rev. RNA 2022, 14, e1750. [Google Scholar] [CrossRef]
- Wang, S.-M.; Chen, Y.-M.; Xia, Y.; He, X.-Z. δ-Opioid Receptors, microRNAs, and Neuroinflammation in Cerebral Ischemia/Hypoxia. Front. Immunol. 2020, 11, 421. [Google Scholar] [CrossRef]
- Pescador, N.; Sanchez-Gonzalez, L.; del Peso, L.; Ramos-Ruiz, R.; Puente-Santamaria, L.; Martinez-Costa, O. Formal Meta-Analysis of Hypoxic Gene Expression Profiles Reveals a Universal Gene Signature. Biomedicines 2022, 10, 2229. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.P.; Dzau, V.J. RNA Therapeutics for the Cardiovascular System. Circulation 2024, 149, 707–716. [Google Scholar] [CrossRef]
- Brüne, B.; Fuhrmann, D.C. A graphical journey through iron metabolism, microRNAs, and hypoxia in ferroptosis. Redox Biol. 2022, 54, 102365. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, L.; Liu, J.; Guo, X.; Song, S.; Lu, J.; Zhang, L.; Yang, C.; Zeng, B.; Fu, Q. Epigenetic regulation of programmed cell death in hypoxia-induced pulmonary arterial hypertension. Front. Immunol. 2023, 14, 1206452. [Google Scholar] [CrossRef]
- Doevendans, P.A.; De Windt, L.J.; el Azzouzi, H.; Leptidis, S. HypoxamiRs: Regulators of cardiac hypoxia and energy metabolism. Trends Endocrinol. Metab. 2015, 26, 502–508. [Google Scholar] [CrossRef]
- Kuh, H.J.; Son, S.W.; Da Yun, B.; Song, M.G.; Lee, J.K.; Park, J.K.; Choi, S.Y. The Hypoxia–Long Noncoding RNA Interaction in Solid Cancers. Int. J. Mol. Sci. 2021, 22, 7261. [Google Scholar] [CrossRef]
- Ferracin, M.; Bayraktar, R.; Nemeth, K.; Calin, G.A. Non-coding RNAs in disease: From mechanisms to therapeutics. Nat. Rev. Genet. 2023, 25, 211–232. [Google Scholar] [CrossRef]
- Collawn, J.F.; Moszyńska, A.; Kochan-Jamrozy, K.; Bartoszewski, R.; Króliczewski, J. miRNA networks modulate human endothelial cell adaptation to cyclic hypoxia. Cell. Signal. 2019, 54, 150–160. [Google Scholar] [CrossRef]
- Kang, Y.; Hou, P.; Luo, J. Hypoxia-mediated miR-212-3p downregulation enhances progression of intrahepatic cholangiocarcinoma through upregulation of Rab1a. Cancer Biol. Ther. 2018, 19, 984–993. [Google Scholar] [CrossRef]
- Esposito, M.R.; Germano, G.; Micheli, S.; Fietta, A.; Zanella, L.; Borile, G.; Bova, L.; Fusco, P.; Bastianello, A.; Cimetta, E. miR-210-3p enriched extracellular vesicles from hypoxic neuroblastoma cells stimulate migration and invasion of target cells. Cell Biosci. 2023, 13, 1–17. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Z.; Li, X.; Cretoiu, D.; Cao, R.Y.; Zhang, Z.; Xiao, J.; Meng, X.; Zhu, Y.; Yin, M.; et al. Exercise-Induced miR-210 Promotes Cardiomyocyte Proliferation and Survival and Mediates Exercise-Induced Cardiac Protection against Ischemia/Reperfusion Injury. Research 2024, 7, 0327. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial Cell–Specific regulation of Hypoxia-Inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Collawn, J.F.; Bartoszewska, S.; Cabaj, A.; Dąbrowski, M.; Moszyńska, A.; Króliczewski, J.; Charzyńska, A.; Jaśkiewicz, M.; Gebert, M.; Bartoszewski, R. The transition from HIF-1 to HIF-2 during prolonged hypoxia results from reactivation of PHDs and HIF1A mRNA instability. Cell. Mol. Biol. Lett. 2022, 27, 1–19. [Google Scholar] [CrossRef]
- Aebersold, R.; Liu, Y.; Beyer, A. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Q.; Zhang, S.; Li, S.; Zhang, B.; Xu, J.; Bai, Y.-G.; Xie, M.-J.; Ma, J. Multiomics Analysis Reveals Role of ncRNA in Hypoxia of Mouse Brain Microvascular Endothelial Cells. Int. J. Mol. Sci. 2025, 26, 5629. https://doi.org/10.3390/ijms26125629
Shi Q, Zhang S, Li S, Zhang B, Xu J, Bai Y-G, Xie M-J, Ma J. Multiomics Analysis Reveals Role of ncRNA in Hypoxia of Mouse Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences. 2025; 26(12):5629. https://doi.org/10.3390/ijms26125629
Chicago/Turabian StyleShi, Qixin, Shuai Zhang, Shaohua Li, Bin Zhang, Jin Xu, Yun-Gang Bai, Man-Jiang Xie, and Jin Ma. 2025. "Multiomics Analysis Reveals Role of ncRNA in Hypoxia of Mouse Brain Microvascular Endothelial Cells" International Journal of Molecular Sciences 26, no. 12: 5629. https://doi.org/10.3390/ijms26125629
APA StyleShi, Q., Zhang, S., Li, S., Zhang, B., Xu, J., Bai, Y.-G., Xie, M.-J., & Ma, J. (2025). Multiomics Analysis Reveals Role of ncRNA in Hypoxia of Mouse Brain Microvascular Endothelial Cells. International Journal of Molecular Sciences, 26(12), 5629. https://doi.org/10.3390/ijms26125629