
Immune Checkpoint Inhibitors in Clear Cell Renal Cell Carcinoma (ccRCC)



Abstract
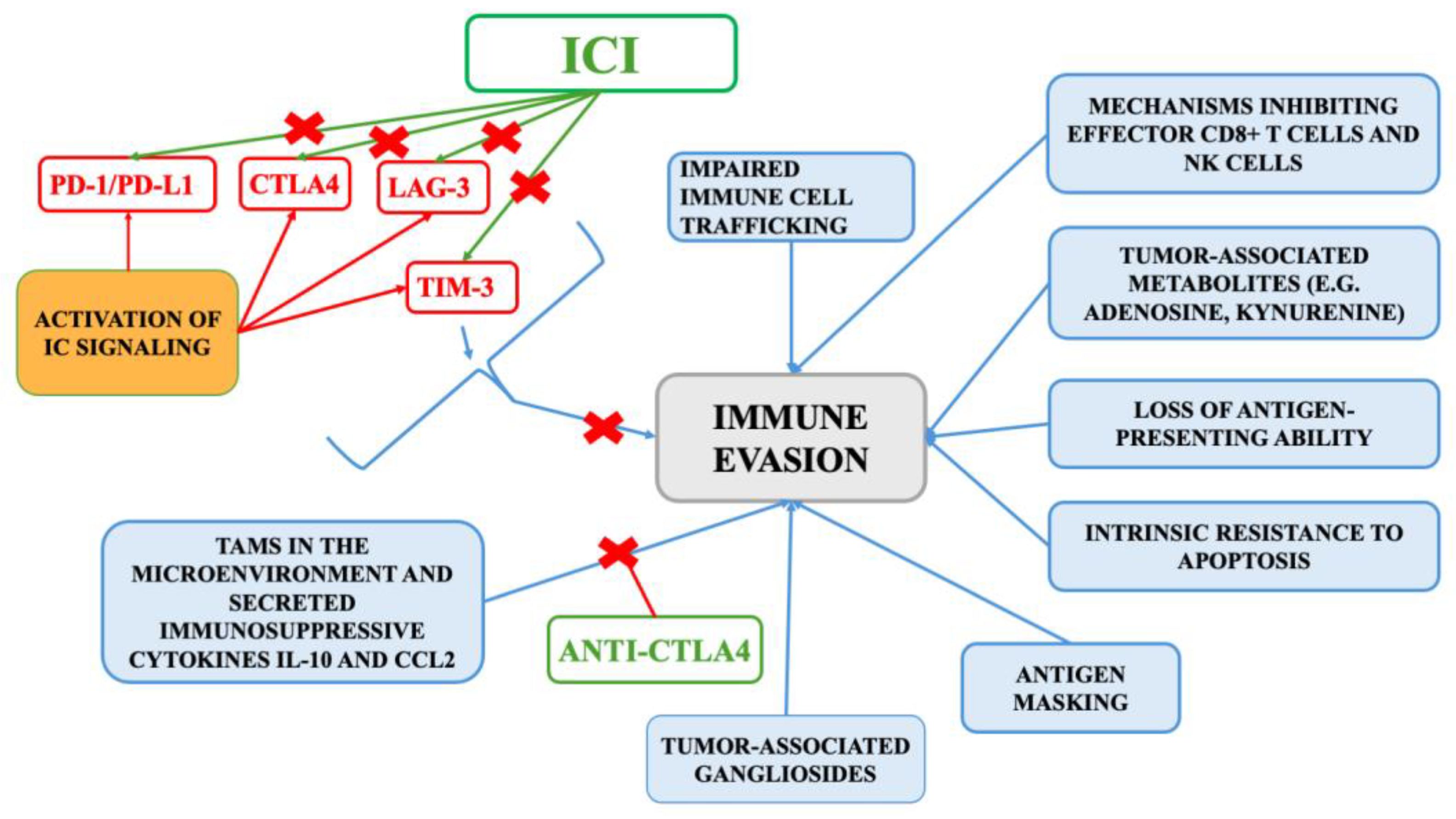
1. Introduction
2. Tumor Immunobiology of ccRCC
3. Overview of Immune Checkpoint Inhibitors
3.1. Monotherapy Clinical Trials
3.1.1. Nivolumab
3.1.2. Pembrolizumab
3.1.3. Avelumab
3.1.4. Atezolizumab
3.2. Combination Immunotherapy Trials
3.2.1. CHECKMATE 214: Nivolumab Plus and Ipilimumab Versus Sunitinib in Patients with Previously Untreated Advanced or Metastatic RCC
3.2.2. KEYNOTE-426: Pembrolizumab Plus Axitinib vs. Sunitinib for First-Line Treatment of Advanced Renal Cell Carcinoma
3.2.3. IMmotion151: A Phase III Trial of Atezolizumab Plus Bevacizumab Versus Sunitinib in Advanced Renal Cell Carcinoma
3.2.4. CheckMate 9ER: A Phase III Study of Nivolumab Plus Cabozantinib Versus Sunitinib in Treatment-Naïve Advanced Renal Cell Carcinoma
3.2.5. JAVELIN Renal 101: A Phase 3 Study of Avelumab Plus Axitinib Versus Sunitinib in First-Line Advanced Renal Cell Carcinoma
3.2.6. CLEAR Trial: Phase 3 Study Comparing Lenvatinib-Based Combinations Versus Sunitinib as First-Line Therapy for Advanced Renal Cell Carcinoma
3.2.7. COSMIC-313: Phase 3 Trial of Cabozantinib in Combination with Nivolumab and Ipilimumab for Advanced Renal Cell Carcinoma
3.2.8. RENOTORCH Phase III Trial: Toripalimab Plus Axitinib vs. Sunitinib in First-Line Treatment of Advanced RCC
3.2.9. CONTACT-03: Atezolizumab Combined with Cabozantinib Versus Cabozantinib Alone in Renal Cell Carcinoma Following Progression on Immune Checkpoint Inhibitors
4. ICI-Related Immune-Related Adverse Events
5. Current Role of ICIs in Clinical Practice
6. Conclusions
7. Future Perspectives
Funding
Conflicts of Interest
References
- Yang, D.C.; Chen, C.-H. Potential New Therapeutic Approaches for Renal Cell Carcinoma. Semin. Nephrol. 2020, 40, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Tronik-Le Roux, D.; Sautreuil, M.; Bentriou, M.; Vérine, J.; Palma, M.B.; Daouya, M.; Bouhidel, F.; Lemler, S.; LeMaoult, J.; Desgrandchamps, F.; et al. Comprehensive landscape of immune-checkpoints uncovered in clear cell renal cell carcinoma reveals new and emerging therapeutic targets. Cancer Immunol. Immunother. 2020, 69, 1237–1252. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Quinn, D.I.; Zhang, T.; Gurney, H.; Doshi, G.K.; Cobb, P.W.; Parnis, F.; Lee, J.-L.; Park, S.H.; Semenov, A.; et al. KEYNOTE-564: A phase 3, randomized, double blind, trial of pembrolizumab in the adjuvant treatment of renal cell carcinoma. J. Clin. Oncol. 2018, 36, TPS4599. [Google Scholar] [CrossRef]
- McDermott, D.F.; Atkins, M.B. PD-1 as a potential target in cancer therapy. Cancer Med. 2013, 2, 662–673. [Google Scholar] [CrossRef]
- George, D.J.; Lee, C.H.; Heng, D. New approaches to first-line treatment of advanced renal cell carcinoma. Ther. Adv. Med. Oncol. 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Shen, C.; Kaelin, W.G., Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol. 2013, 23, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J. 2013, 19, 324. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Le, V.H.; Oyama, T.; Ricketts, C.J.; Ho, T.H.; Cheng, E.H. Chromosome 3p loss–orchestrated VHL, HIF, and epigenetic deregulation in clear cell renal cell carcinoma. J. Clin. Oncol. 2018, 36, 3533. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F.; et al. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef]
- Wang, R.; Lan, C.; Benlagha, K.; Camara, N.O.S.; Miller, H.; Kubo, M.; Heegaard, S.; Lee, P.; Yang, L.; Forsman, H.; et al. The interaction of innate immune and adaptive immune system. MedComm 2024, 5, e714. [Google Scholar] [CrossRef]
- Monjaras-Avila, C.U.; Lorenzo-Leal, A.C.; Luque-Badillo, A.C.; D’Costa, N.; Chavez-Muñoz, C.; Bach, H. The Tumor Immune Microenvironment in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 7946. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, W.; Lin, K.; Ma, W.; Li, W.; Yao, X. Immunological perspective on the malignant progression of renal clear cell carcinoma. Ann. Transl. Med. 2021, 9, 1544. [Google Scholar] [CrossRef]
- Angulo, J.C.; Shapiro, O. The Changing Therapeutic Landscape of Metastatic Renal Cancer. Cancers 2019, 11, 1227. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Miller, C.P.; Warren, E.H.; Tykodi, S.S. Current status of antigen-specific T-cell immunotherapy for advanced renal-cell carcinoma. Hum. Vacc. Immunother. 2021, 17, 1882–1896. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Cervantes-Villagrana, R.D.; Albores-García, D.; Cervantes-Villagrana, A.R.; García-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef]
- McBride, M.A.; Patil, T.K.; Bohannon, J.K.; Hernandez, A.; Sherwood, E.R.; Patil, N.K. Immune Checkpoints: Novel Therapeutic Targets to Attenuate Sepsis-Induced Immunosuppression. Front. Immunol. 2021, 11, 624272. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Rezayi, M.; Hosseini, A. Structure of PD1 and its mechanism in the treatment of autoimmune diseases. Cell Biochem. Funct. 2023, 41, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2017, 8, 2171. [Google Scholar] [CrossRef]
- Escors, D.; Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; García-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.H.; Dong, H.; Lohse, C.M.; Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Kwon, E.D. PD-1 Is Expressed by Tumor-Infiltrating Immune Cells and Is Associated with Poor Outcome for Patients with Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 1757–1761. [Google Scholar] [CrossRef]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG 3 (CD 223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Mollica, V.; Di Nunno, V.; Gatto, L.; Santoni, M.; Cimadamore, A.; Cheng, L.; Lopez-Beltran, A.; Montironi, R.; Pisconti, S.; Battelli, N.; et al. Novel Therapeutic Approaches and Targets Currently Under Evaluation for Renal Cell Carcinoma: Waiting for the Revolution. Clin. Drug Investig. 2019, 39, 503–519. [Google Scholar] [CrossRef]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Chihara, N.; Madi, A.; Kondo, T.; Zhang, H.; Acharya, N.; Singer, M.; Nyman, J.; Marjanovic, N.D.; Kowalczyk, M.S.; Wang, C. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018, 558, 454–459. [Google Scholar] [CrossRef]
- DeLong, J.H.; Hall, A.O.H.; Rausch, M.; Moodley, D.; Perry, J.; Park, J.; Phan, A.T.; Beiting, D.P.; Kedl, R.M.; Hill, J.A. IL-27 and TCR stimulation promote T cell expression of multiple inhibitory receptors. Immunohorizons 2019, 3, 13–25. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e1000010. [Google Scholar] [CrossRef]
- Yuan, J.; Jiang, B.; Zhao, H.; Huang, Q. Prognostic implication of TIM-3 in clear cell renal cell carcinoma. Neoplasma 2014, 61, 35–40. [Google Scholar] [CrossRef]
- Sailer, V.; Sailer, U.; Bawden, E.G.; Zarbl, R.; Wiek, C.; Vogt, T.J.; Dietrich, J.; Loick, S.; Grünwald, I.; Toma, M.; et al. DNA methylation of indoleamine 2,3-dioxygenase 1 (IDO1) in head and neck squamous cell carcinomas correlates with IDO1 expression, HPV status, patients’ survival, immune cell infiltrates, mutational load, and interferon γ signature. eBioMedicine 2019, 48, 341–352. [Google Scholar] [CrossRef]
- Adams, S.; Braidy, N.; Bessesde, A.; Brew, B.J.; Grant, R.; Teo, C.; Guillemin, G.J. The kynurenine pathway in brain tumor pathogenesis. Cancer Res. 2012, 72, 5649–5657. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2, 3-dioxygenase and its therapeutic inhibition in cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar]
- Maliniemi, P.; Laukkanen, K.; Väkevä, L.; Dettmer, K.; Lipsanen, T.; Jeskanen, L.; Bessede, A.; Oefner, P.J.; Kadin, M.E.; Ranki, A. Biological and clinical significance of tryptophan-catabolizing enzymes in cutaneous T-cell lymphomas. Oncoimmunology 2017, 6, e1273310. [Google Scholar] [CrossRef]
- Yentz, S.; Smith, D. Indoleamine 2, 3-dioxygenase (IDO) inhibition as a strategy to augment cancer immunotherapy. BioDrugs 2018, 32, 311–317. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR pathway for cancer immunotherapy–challenges and opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Zhai, L.; Spranger, S.; Binder, D.C.; Gritsina, G.; Lauing, K.L.; Giles, F.J.; Wainwright, D.A. Molecular pathways: Targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 5427–5433. [Google Scholar] [CrossRef] [PubMed]
- Godin-Ethier, J.; Hanafi, L.-A.; Piccirillo, C.A.; Lapointe, R. Indoleamine 2, 3-dioxygenase expression in human cancers: Clinical and immunologic perspectives. Clin. Cancer Res. 2011, 17, 6985–6991. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Górska, M.; Polityńska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Duan, J.; Cui, L.; Zhao, X.; Bai, H.; Cai, S.; Wang, G.; Zhao, Z.; Zhao, J.; Chen, S.; Song, J.; et al. Use of Immunotherapy with Programmed Cell Death 1 vs Programmed Cell Death Ligand 1 Inhibitors in Patients with Cancer: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 6, 375–384. [Google Scholar] [CrossRef]
- Gaynor, N.; Crown, J.; Collins, D.M. Immune checkpoint inhibitors: Key trials and an emerging role in breast cancer. Semin. Cancer Biol. 2022, 79, 44–57. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Yang, S.; Yang, X.; Hou, Z.; Zhu, L.; Yao, Z.; Zhang, Y.; Chen, Y.; Teng, J.; Fang, C.; Chen, S.; et al. Rationale for immune checkpoint inhibitors plus targeted therapy for advanced renal cell carcinoma. Heliyon 2024, 10, e29215. [Google Scholar] [CrossRef]
- Motzer, R.J.; Agarwal, N.; Beard, C.; Bolger, G.B.; Boston, B.; Carducci, M.A.; Choueiri, T.K.; Figlin, R.A.; Fishman, M.; Hancock, S.L.; et al. NCCN clinical practice guidelines in oncology: Kidney cancer. J. Natl. Com. Canc Netw. 2009, 7, 618–630. [Google Scholar] [CrossRef]
- Rini, B.I.; Battle, D.; Figlin, R.A.; George, D.J.; Hammers, H.; Hutson, T.; Jonasch, E.; Joseph, R.W.; McDermott, D.F.; Motzer, R.J.; et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of advanced renal cell carcinoma (RCC). J. Immunother. Cancer 2019, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- Ornstein, M.C.; Wood, L.S.; Hobbs, B.P.; Allman, K.D.; Martin, A.; Bevan, M.; Gilligan, T.D.; Garcia, J.A.; Rini, B.I. A phase II trial of intermittent nivolumab in patients with metastatic renal cell carcinoma (mRCC) who have received prior anti-angiogenic therapy. J. Immunother. Cancer 2019, 7, 127. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tykodi, S.S.; Escudier, B.; Oudard, S.; Hammers, H.J.; McDermott, D.F.; George, S.; Castellano, D.; Choueiri, T.K.; Alva, A.S.; et al. Final analysis of the CheckMate 025 trial comparing nivolumab (NIVO) versus everolimus (EVE) with >5 years of follow-up in patients with advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 2020, 38, 617. [Google Scholar] [CrossRef]
- Xu, J.X.; Maher, V.E.; Zhang, L.; Tang, S.; Sridhara, R.; Ibrahim, A.; Kim, G.; Pazdur, R. FDA approval summary: Nivolumab in advanced renal cell carcinoma after anti-angiogenic therapy and exploratory predictive biomarker analysis. Oncologist 2017, 22, 311–317. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Chang, Y.-H.; Hajek, J.; Symeonides, S.N.; Lee, J.L.; Sarwar, N.; et al. Adjuvant Pembrolizumab after Nephrectomy in Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 385, 683–694. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Lee, J.-L.; Bjarnason, G.A.; Larkin, J.M.G.; Gafanov, R.A.; Kochenderfer, M.D.; Jensen, N.V.; Donskov, F.; Malik, J.; Poprach, A.; et al. Open-Label, Single-Arm Phase II Study of Pembrolizumab Monotherapy as First-Line Therapy in Patients with Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2021, 39, 1020–1028. [Google Scholar] [CrossRef]
- Vaishampayan, U.; Schöffski, P.; Ravaud, A.; Borel, C.; Peguero, J.; Chaves, J.; Morris, J.C.; Kotecki, N.; Smakal, M.; Zhou, D.; et al. Avelumab monotherapy as first-line or second-line treatment in patients with metastatic renal cell carcinoma: Phase Ib results from the JAVELIN Solid Tumor trial. J. Immunother. Cancer 2019, 7, 275. [Google Scholar] [CrossRef]
- McDermott, D.F.; Sosman, J.A.; Sznol, M.; Massard, C.; Gordon, M.S.; Hamid, O.; Powderly, J.D.; Infante, J.R.; Fassò, M.; Wang, Y.V.; et al. Atezolizumab, an Anti-Programmed Death-Ligand 1 Antibody, in Metastatic Renal Cell Carcinoma: Long-Term Safety, Clinical Activity, and Immune Correlates from a Phase Ia Study. J. Clin. Oncol. 2016, 34, 833–842. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Tannir, N.M.; Albigès, L.; McDermott, D.F.; Burotto, M.; Choueiri, T.K.; Hammers, H.J.; Barthélémy, P.; Plimack, E.R.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 8-year follow-up results of efficacy and safety from the phase III CheckMate 214 trial. Ann. Oncol. 2024, 35, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Plimack, E.R.; Powles, T.; Stus, V.; Gafanov, R.; Nosov, D.; Waddell, T.; Alekseev, B.; Pouliot, F.; Melichar, B.; Soulières, D.; et al. Pembrolizumab Plus Axitinib Versus Sunitinib as First-line Treatment of Advanced Renal Cell Carcinoma: 43-month Follow-up of the Phase 3 KEYNOTE-426 Study. Eur. Urol. 2023, 84, 449–454. [Google Scholar] [CrossRef]
- Bedke, J.; Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Waddell, T.; Nosov, D.; Pouliot, F.; Soulières, D.; Melichar, B.; et al. Health-related Quality of Life Analysis from KEYNOTE-426: Pembrolizumab plus Axitinib Versus Sunitinib for Advanced Renal Cell Carcinoma. Eur. Urol. 2022, 82, 427–439. [Google Scholar] [CrossRef] [PubMed]
- A Study of Atezolizumab in Combination with Bevacizumab Versus Sunitinib in Participants with Untreated Advanced Renal Cell Carcinoma (RCC) (IMmotion151). Available online: https://ctv.veeva.com/study/a-study-of-atezolizumab-in-combination-with-bevacizumab-versus-sunitinib-in-participants-with-untrea (accessed on 9 April 2025).
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Motzer, R.J.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Alekseev, B.Y.; Lee, J.L.; Suarez, C.; Stroyakovskiy, D.; De Giorgi, U.; et al. Final Overall Survival and Molecular Analysis in IMmotion151, a Phase 3 Trial Comparing Atezolizumab Plus Bevacizumab vs Sunitinib in Patients with Previously Untreated Metastatic Renal Cell Carcinoma. JAMA Oncol. 2022, 8, 275–280. [Google Scholar] [CrossRef]
- Atkins, M.B.; Rini, B.I.; Motzer, R.J.; Powles, T.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Gurney, H.; et al. Patient-Reported Outcomes from the Phase III Randomized IMmotion151 Trial: Atezolizumab + Bevacizumab versus Sunitinib in Treatment-Naïve Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2506–2514. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Shah, A.Y.; Suárez, C.; Hamzaj, A.; Porta, C.; Hocking, C.M.; et al. Nivolumab plus cabozantinib versus sunitinib in first-line treatment for advanced renal cell carcinoma (CheckMate 9ER): Long-term follow-up results from an open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 888–898. [Google Scholar] [CrossRef]
- Hamuro, L.; Hu, Z.; Passarell, J.; Barcomb, H.; Zhang, J.; Goldstein, S.; Bello, A.; Roy, A.; Zhu, L. Exposure-Response Analysis to Support Nivolumab Once Every 4 Weeks Dosing in Combination with Cabozantinib in Renal Cell Carcinoma. Clin. Cancer Res. 2022, 28, 1603–1613. [Google Scholar] [CrossRef]
- Apolo, A.B.; Powles, T.; Escudier, B.; Burotto, M.; Zhang, J.; Simsek, B.; Scheffold, C.; Motzer, R.J.; Choueiri, T.K. Nivolumab plus ipilimumab plus cabozantinib triplet combination for patients with previously untreated advanced renal cell carcinoma: Results from a discontinued arm of the phase III CheckMate 9ER trial. Eur. J. Cancer 2022, 177, 63–71. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Motzer, R.J.; Rini, B.I.; Haanen, J.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Gravis-Mescam, G.; Uemura, M.; Lee, J.L.; et al. Updated efficacy results from the JAVELIN Renal 101 trial: First-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann. Oncol. 2020, 31, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Larkin, J.; Pal, S.; Motzer, R.J.; Rini, B.I.; Venugopal, B.; Alekseev, B.; Miyake, H.; Gravis, G.; Bilen, M.A.; et al. Efficacy and correlative analyses of avelumab plus axitinib versus sunitinib in sarcomatoid renal cell carcinoma: Post hoc analysis of a randomized clinical trial. ESMO Open 2021, 6, 100101. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Motzer, R.; Porta, C.; Alekseev, B.; Rha, S.Y.; Choueiri, T.K.; Mendez-Vidal, M.J.; Hong, S.H.; Kapoor, A.; Goh, J.C.; Eto, M.; et al. Health-related quality-of-life outcomes in patients with advanced renal cell carcinoma treated with lenvatinib plus pembrolizumab or everolimus versus sunitinib (CLEAR): A randomised, phase 3 study. Lancet Oncol. 2022, 23, 768–780. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Albiges, L.; Burotto, M.; Szczylik, C.; Zurawski, B.; Yanez Ruiz, E.; Maruzzo, M.; Suarez Zaizar, A.; Fein, L.E.; et al. Cabozantinib plus Nivolumab and Ipilimumab in Renal-Cell Carcinoma. N. Engl. J. Med. 2023, 388, 1767–1778. [Google Scholar] [CrossRef]
- Yan, X.Q.; Ye, M.J.; Zou, Q.; Chen, P.; He, Z.S.; Wu, B.; He, D.L.; He, C.H.; Xue, X.Y.; Ji, Z.G.; et al. Toripalimab plus axitinib versus sunitinib as first-line treatment for advanced renal cell carcinoma: RENOTORCH, a randomized, open-label, phase III study. Ann. Oncol. 2024, 35, 190–199. [Google Scholar] [CrossRef]
- Pal, S.K.; Albiges, L.; Tomczak, P.; Suárez, C.; Voss, M.H.; de Velasco, G.; Chahoud, J.; Mochalova, A.; Procopio, G.; Mahammedi, H.; et al. Atezolizumab plus cabozantinib versus cabozantinib monotherapy for patients with renal cell carcinoma after progression with previous immune checkpoint inhibitor treatment (CONTACT-03): A multicentre, randomised, open-label, phase 3 trial. Lancet 2023, 402, 185–195. [Google Scholar] [CrossRef]
- Candido, W.; Eggen, A.C.; Jalving, M.; Bosma, I.; Horinga, R.D.; van Heuvelen, K.C.; Hiltermann, T.J.N.; Oosting, S.; Racz, E.; van der Klauw, M.M.; et al. Quality of life, neurocognitive functioning, psychological issues, sexuality and comorbidity more than 2 years after commencing immune checkpoint inhibitor treatment. J. Immunother. Cancer 2025, 13, e011168. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Nemoto, Y.; Tachibana, H.; Fukuda, H.; Yoshida, K.; Kobayashi, H.; Iizuka, J.; Hashimoto, Y.; Takagi, T.; Ishida, H.; et al. Outcomes of nivolumab monotherapy for previously treated metastatic renal cell carcinoma: A real-world multi-institution data with a minimum of 2 years of follow-up. Jpn. J. Clin. Oncol. 2022, 52, 785–790. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Liu, H.; Dai, J.; Zhao, J.; Zhu, S.; Zhang, X.; Liang, J.; Hu, X.; Zhao, J.; et al. The incidence of immune-related adverse events (irAEs) and their association with clinical outcomes in advanced renal cell carcinoma and urothelial carcinoma patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. Cancer Treat. Rev. 2024, 129, 102787. [Google Scholar] [CrossRef]
- Gupta, A.K.; Macleod, M.A.; Abramovits, W. OPDIVO (Nivolumab). Skinmed 2015, 13, 471–474. [Google Scholar]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Grover, S.; Rahma, O.E.; Hashemi, N.; Lim, R.M. Gastrointestinal and hepatic toxicities of checkpoint inhibitors: Algorithms for management. In Proceedings of the American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 1–5 June 2018; pp. 13–19. [Google Scholar]
- McGregor, B.; Mortazavi, A.; Cordes, L.; Salabao, C.; Vandlik, S.; Apolo, A.B. Management of adverse events associated with cabozantinib plus nivolumab in renal cell carcinoma: A review. Cancer Treat. Rev. 2022, 103, 102333. [Google Scholar] [CrossRef]
- Osorio, J.; Ni, A.; Chaft, J.; Pollina, R.; Kasler, M.; Stephens, D.; Rodriguez, C.; Cambridge, L.; Rizvi, H.; Wolchok, J. Antibody-mediated thyroid dysfunction during T-cell checkpoint blockade in patients with non-small-cell lung cancer. Ann. Oncol. 2017, 28, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L. Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Naidoo, J.; Wang, X.; Woo, K.M.; Iyriboz, T.; Halpenny, D.; Cunningham, J.; Chaft, J.E.; Segal, N.H.; Callahan, M.K.; Lesokhin, A.M. Pneumonitis in patients treated with anti–programmed death-1/programmed death ligand 1 therapy. J. Clin. Oncol. 2017, 35, 709–717. [Google Scholar] [CrossRef]
- Mamlouk, O.; Selamet, U.; Machado, S.; Abdelrahim, M.; Glass, W.F.; Tchakarov, A.; Gaber, L.; Lahoti, A.; Workeneh, B.; Chen, S. Nephrotoxicity of immune checkpoint inhibitors beyond tubulointerstitial nephritis: Single-center experience. J. Immunother. Cancer 2019, 7, 2. [Google Scholar] [CrossRef]
- Mirabile, A.; Brioschi, E.; Ducceschi, M.; Piva, S.; Lazzari, C.; Bulotta, A.; Viganò, M.G.; Petrella, G.; Gianni, L.; Gregorc, V. PD-1 inhibitors-related neurological toxicities in patients with non-small-cell lung cancer: A literature review. Cancers 2019, 11, 296. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, H.; Zhou, L.; Li, W.; Yang, L.; Li, W.; Li, K.; Liu, X. Immunotherapy-associated pancreatic adverse events: Current understanding of their mechanism, diagnosis, and management. Front. Oncol. 2021, 11, 627612. [Google Scholar] [CrossRef] [PubMed]
- Belum, V.; Benhuri, B.; Postow, M.; Hellmann, M.; Lesokhin, A.; Segal, N.; Motzer, R.; Wu, S.; Busam, K.; Wolchok, J. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur. J. Cancer 2016, 60, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lacouture, M.E. Pruritus associated with targeted anticancer therapies and their management. Dermatol. Clin. 2018, 36, 315–324. [Google Scholar] [CrossRef]
- Phillips, G.S.; Wu, J.; Hellmann, M.D.; Postow, M.A.; Rizvi, N.A.; Freites-Martinez, A.; Chan, D.; Dusza, S.; Motzer, R.J.; Rosenberg, J.E. Treatment outcomes of immune-related cutaneous adverse events. J. Clin. Oncol. 2019, 37, 2746–2758. [Google Scholar] [CrossRef]
- Herrmann, S.M.; Perazella, M.A. Immune checkpoint inhibitors and immune-related adverse renal events. Kidney Int. Rep. 2020, 5, 1139–1148. [Google Scholar] [CrossRef]
- Tucci, M.; Passarelli, A.; Todisco, A.; Mannavola, F.; Stucci, L.S.; D’Oronzo, S.; Rossini, M.; Taurisano, M.; Gesualdo, L.; Silvestris, F. The mechanisms of acute interstitial nephritis in the era of immune checkpoint inhibitors in melanoma. Ther. Adv. Med. Oncol. 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Nephrotic Syndrome. Available online: https://www.mayoclinic.org/diseases-conditions/nephrotic-syndrome/symptoms-causes/syc-20375608 (accessed on 17 May 2025).
- Kim, D. Posterior reversible encephalopathy syndrome induced by nivolumab immunotherapy for non–small-cell lung cancer. Clin. Case Rep. 2019, 7, 935. [Google Scholar] [CrossRef]
- Patwari, A.; Bhatlapenumarthi, V.; Pascual, S.K. Atypical posterior reversible encephalopathy syndrome due to oral tyrosine kinase inhibitor cabozantinib: First case report. Case Rep. Oncol. 2020, 13, 1013–1019. [Google Scholar] [CrossRef]
- Stuby, J.; Herren, T.; Naumburger, G.S.; Papet, C.; Rudiger, A. Immune checkpoint inhibitor therapy-associated encephalitis: A case series and review of the literature. Swiss Med. Wkly. 2020, 150, w20377. [Google Scholar] [CrossRef]
- Cozma, A.; Sporis, N.D.; Lazar, A.L.; Buruiana, A.; Ganea, A.M.; Malinescu, T.V.; Berechet, B.M.; Fodor, A.; Sitar-Taut, A.V.; Vlad, V.C.; et al. Cardiac Toxicity Associated with Immune Checkpoint Inhibitors: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 10948. [Google Scholar] [CrossRef]
- Moriyama, S.; Fukata, M.; Tatsumoto, R.; Kono, M. Refractory constrictive pericarditis caused by an immune checkpoint inhibitor properly managed with infliximab: A case report. Eur. Heart J. Case Rep. 2021, 5, ytab002. [Google Scholar] [CrossRef]
- Oristrell, G.; Bañeras, J.; Ros, J.; Muñoz, E. Cardiac tamponade and adrenal insufficiency due to pembrolizumab: A case report. Eur. Heart J. Case Rep. 2018, 2, yty038. [Google Scholar] [CrossRef]
- Dasanu, C.A.; Jen, T.; Skulski, R. Late-onset pericardial tamponade, bilateral pleural effusions and recurrent immune monoarthritis induced by ipilimumab use for metastatic melanoma. J. Oncol. Pharm. Pract. 2017, 23, 231–234. [Google Scholar] [CrossRef]
- Öztürk, C.; Luetkens, J.A. Cardiac MRI in immune checkpoint inhibitor associated pericarditis. Clin. Case Rep. 2021, 9, e04926. [Google Scholar] [CrossRef]
- Reddy, N.; Moudgil, R.; Lopez-Mattei, J.C.; Karimzad, K.; Mouhayar, E.N.; Somaiah, N.; Conley, A.P.; Patel, S.; Giza, D.E.; Iliescu, C. Progressive and reversible conduction disease with checkpoint inhibitors. Can. J. Cardiol. 2017, 33, 1335. [Google Scholar] [CrossRef]
- Giancaterino, S.; Abushamat, F.; Duran, J.; Lupercio, F.; DeMaria, A.; Hsu, J.C. Complete heart block and subsequent sudden cardiac death from immune checkpoint inhibitor–associated myocarditis. HearthRhythm Case Rep. 2020, 6, 761–764. [Google Scholar] [CrossRef]
- Patrinely, J.R.; Johnson, R.; Lawless, A.R.; Bhave, P.; Sawyers, A.; Dimitrova, M.; Yeoh, H.L.; Palmeri, M.; Ye, F.; Fan, R. Chronic immune-related adverse events following adjuvant anti–PD-1 therapy for high-risk resected melanoma. JAMA Oncol. 2021, 7, 744–748. [Google Scholar] [CrossRef]
- Pacholczak-Madej, R.; Drobniak, A.; Stokłosa, Ł.; Bidas, A.; Dobrzańska, J.; Grela-Wojewoda, A.; Roman, A.; Tusień-Małecka, D.; Walocha, J.; Blecharz, P.; et al. Adverse events after nivolumab and ipilimumab combined immunotherapy in advanced renal cell carcinoma: A multicentre experience in Poland. BMC Cancer 2024, 24, 1411. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Zhu, T.C.; Nie, R.C.; Lu, L.H.; Xiang, Z.C.; Xie, D.; Luo, R.Z.; Cai, M.Y. Association between Early Immune-Related Adverse Events and Survival in Patients Treated with PD-1/PD-L1 Inhibitors. J. Clin. Med. 2023, 12, 736. [Google Scholar] [CrossRef]
- Lin, L.; Liu, Y.; Chen, C.; Wei, A.; Li, W. Association between immune-related adverse events and immunotherapy efficacy in non-small-cell lung cancer: A meta-analysis. Front. Pharmacol. 2023, 14, 1190001. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Naqash, A.R.; Naidoo, J.; Sehgal, K.; Miller, A.; Kehl, K.; Venkatraman, D.; Sands, J.; Lamberti, G.; Recondo, G.; et al. Association Between Immune-Related Adverse Events and Clinical Outcomes to Programmed Cell Death Protein 1/Programmed Death-Ligand 1 Blockade in SCLC. JTO Clin. Res. Rep. 2020, 1, 100074. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, S.C.; Pisetsky, D.S. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology 2019, 58 (Suppl. S7), vii59–vii67. [Google Scholar] [CrossRef] [PubMed]
- Blum, S.M.; Rouhani, S.J.; Sullivan, R.J. Effects of immune-related adverse events (irAEs) and their treatment on antitumor immune responses. Immunol. Rev. 2023, 318, 167–178. [Google Scholar] [CrossRef]
- Fukushima, T.; Kobayashi, S.; Ueno, M. The correlation between immune-related adverse events and efficacy of immune checkpoint inhibitors. Jpn. J. Clin. Oncol. 2024, 54, 949–958. [Google Scholar] [CrossRef]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Alva, A.; Bagshaw, H. NCCN Guidelines Version 3.2025 Kidney Cancer. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1440 (accessed on 15 April 2025).
- Powles, T.; Albiges, L.; Bex, A.; Comperat, E.; Grünwald, V.; Kanesvaran, R.; Kitamura, H.; McKay, R.; Porta, C.; Procopio, G.; et al. Renal cell carcinoma: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2024, 35, 692–706. [Google Scholar] [CrossRef]
- Rathmell, W.K.; Rumble, R.B.; Veldhuizen, P.J.V.; Al-Ahmadie, H.; Emamekhoo, H.; Hauke, R.J.; Louie, A.V.; Milowsky, M.I.; Molina, A.M.; Rose, T.L.; et al. Management of Metastatic Clear Cell Renal Cell Carcinoma: ASCO Guideline. J. Clin. Oncol. 2022, 40, 2957–2995. [Google Scholar] [CrossRef]

{kind=link}
| Guideline | Risk Stratification (IMDC) | First-Line Treatment Recommendations | Second-Line Recommendations |
|---|---|---|---|
| NCCN [129] | Preferred Regimens | Favorable * Axitinib + pembrolizumab (category 1) Cabozantinib + nivolumab (category 1) Lenvatinib + pembrolizumab (category 1) Ipilimumab + nivolumab | IO Therapy Naïve None |
| Poor/Intermediate * Axitinib + pembrolizumab (category 1) Cabozantinib + nivolumab (category 1) Ipilimumab + nivolumab (category 1) Lenvatinib + pembrolizumab (category 1) | Prior IO Therapy None | ||
| Other Recommended Regimens | Favorable * Axitinib + avelumab | IO Therapy Naïve Axitinib + pembrolizumab Cabozantinib + nivolumab Ipilimumab + nivolumab Lenvatinib + pembrolizumab Nivolumab | |
| Poor/Intermediate * Axitinib + avelumab | Prior IO Therapy None | ||
| ESMO [130] | Local and locoregional ccRCC | Intermediate–high- or high-risk operable ccRCC Adjuvant pembrolizumab [I, A; ESMO-MCBS v1.1 score: A]. | |
| Advanced or metastatic ccRCC | Favorable * Lenvatinib–pembrolizumab [I, A; MCBS 4] Axitinib–pembrolizumab [I, A; MCBS 4] Cabozantinib–nivolumab [I, A; MCBS 1] Ipilimumab–nivolumab [I, C; MCBS 4] | Further ICI therapy after first-line PD-1-targeted combination therapy is not recommended | |
| Intermediate- and poor * Lenvatinib–pembrolizumab [I, A; MCBS 4] Axitinib–pembrolizumab [I, A; MCBS 4] Cabozantinib–nivolumab [I, A; MCBS 1] Ipilimumab–nivolumab [I, A; MCBS 4] Axitinib–toripalimab [I,C] | |||
| ASCO [131] | metastatic ccRCC | Intermediate- or poor-risk disease Combination treatment with two ICI, e.g., ipilimumab and nivolumab) or an ICI in combination with a VEGFR TKI; [Type: Evidence based, benefits outweigh harms; Evidence quality: High; Strength of recommendation: Strong]. | Nivolumab or cabozantinib should be offered to patients who progressed on a VEGFR TKI alone [Type: Evidence based, benefits outweigh harms; Evidence quality: High; Strength of recommendation: Strong]. |
| Favorable risk * ICI in combination with a VEGFR TKI [Type: Evidence based, benefits outweigh harms; Evidence quality: High; Strength of recommendation: Strong] | |||
| Select patients ICI monotherapy, e.g., pembrolizumab [Type: Evidence based, benefits outweigh harms; Evidence quality: Moderate; Strength of recommendation: Strong] | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rysz, J.; Ławiński, J.; Franczyk, B.; Gluba-Sagr, A. Immune Checkpoint Inhibitors in Clear Cell Renal Cell Carcinoma (ccRCC). Int. J. Mol. Sci. 2025, 26, 5577. https://doi.org/10.3390/ijms26125577
Rysz J, Ławiński J, Franczyk B, Gluba-Sagr A. Immune Checkpoint Inhibitors in Clear Cell Renal Cell Carcinoma (ccRCC). International Journal of Molecular Sciences. 2025; 26(12):5577. https://doi.org/10.3390/ijms26125577
Chicago/Turabian StyleRysz, Jacek, Janusz Ławiński, Beata Franczyk, and Anna Gluba-Sagr. 2025. "Immune Checkpoint Inhibitors in Clear Cell Renal Cell Carcinoma (ccRCC)" International Journal of Molecular Sciences 26, no. 12: 5577. https://doi.org/10.3390/ijms26125577
APA StyleRysz, J., Ławiński, J., Franczyk, B., & Gluba-Sagr, A. (2025). Immune Checkpoint Inhibitors in Clear Cell Renal Cell Carcinoma (ccRCC). International Journal of Molecular Sciences, 26(12), 5577. https://doi.org/10.3390/ijms26125577