
Targeting the p53/xCT/GSH Axis with PRIMA-1Met Combined with Sulfasalazine Shows Therapeutic Potential in Chronic Lymphocytic Leukemia


 ,
,  , , , , , , ,
, , , , , , ,  ,
,  , , , , and add
Show full author list
, , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
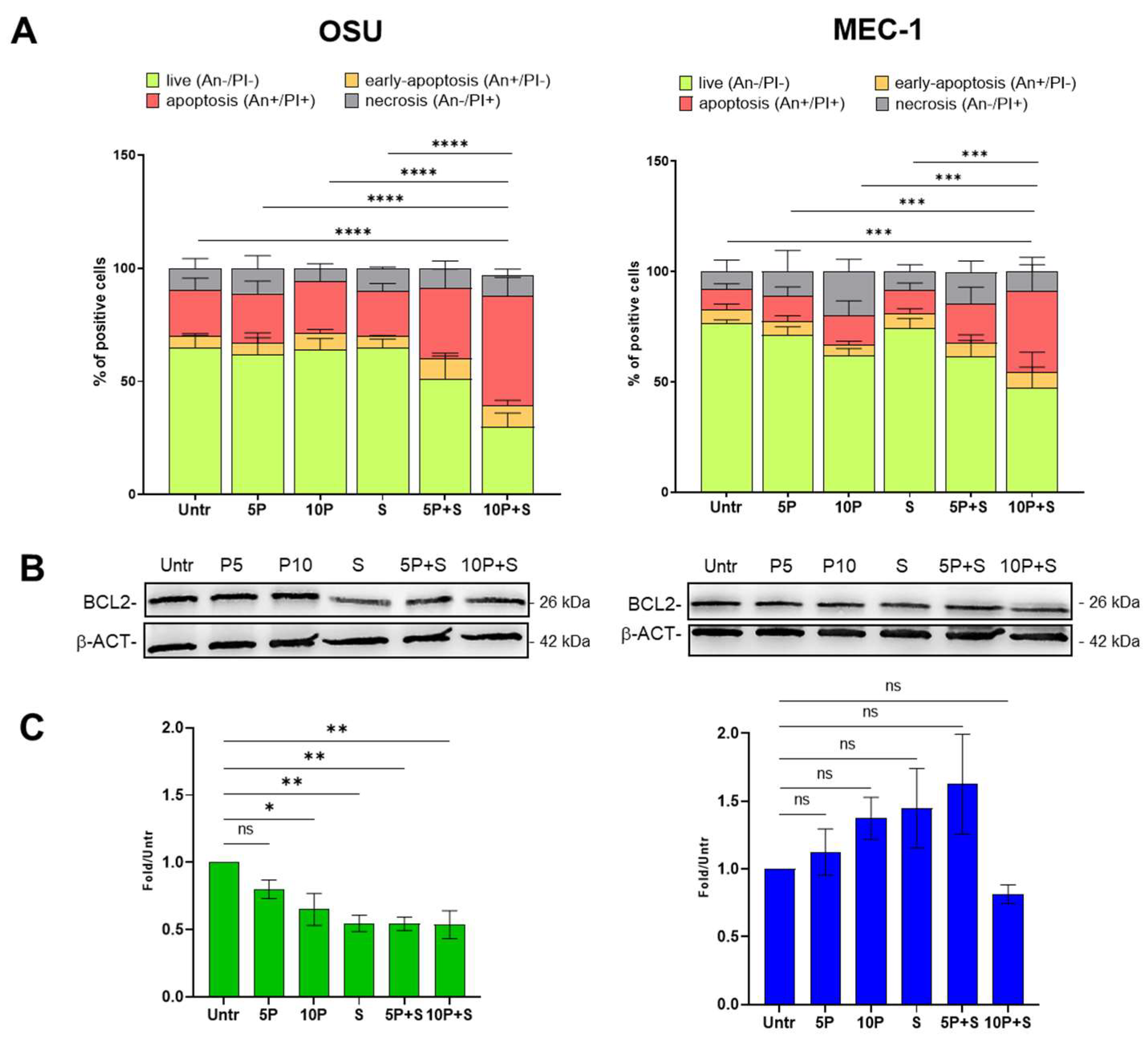
2.1. PRIMA1Met and SAS Combined Treatment Induces Apoptosis in CLL Cell Lines
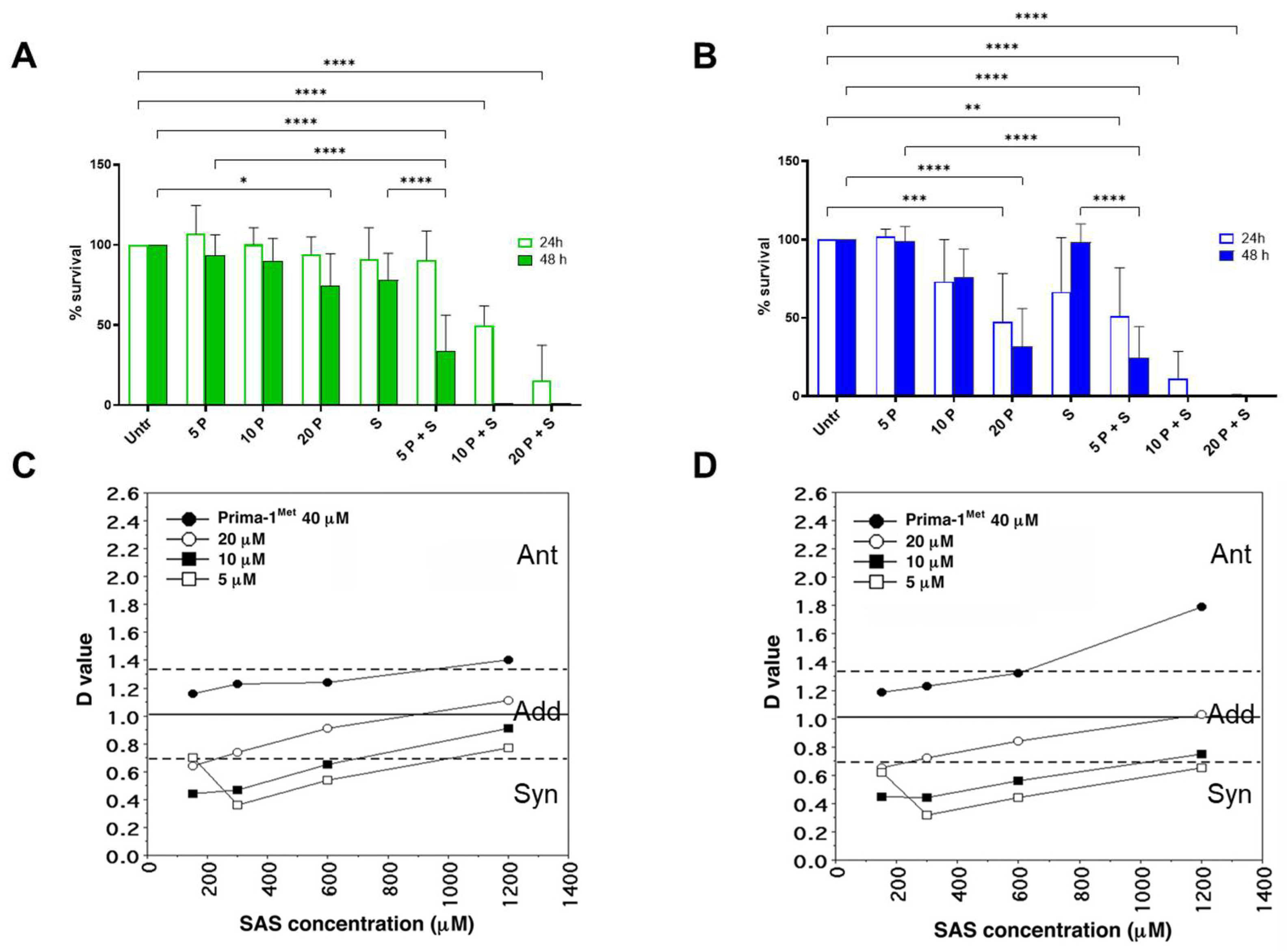
2.2. PRIMA1Met and SAS Synergistically Decrease Survival in CLL Cell Lines, Regardless of Their p53 Status
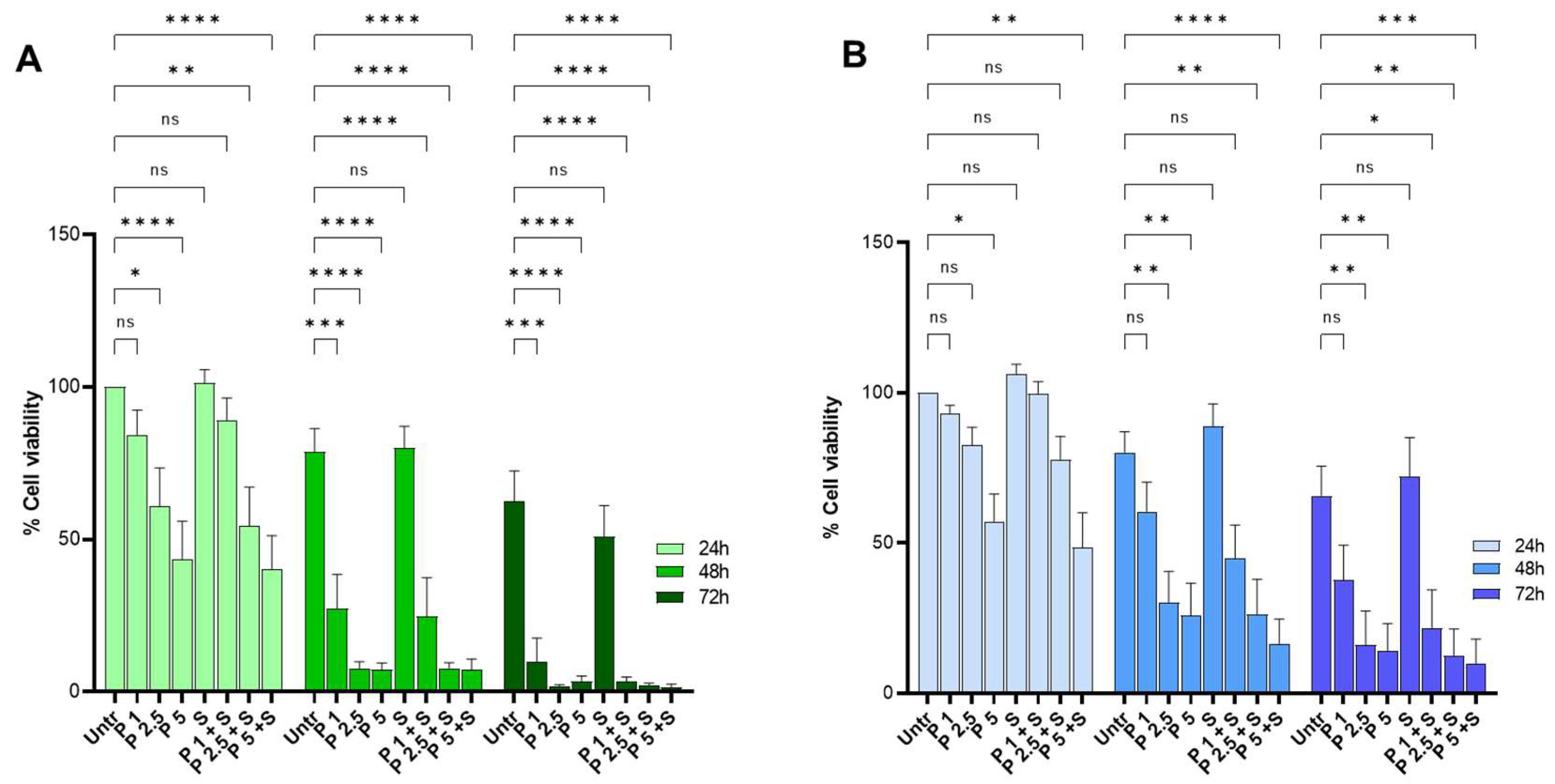
2.3. PRIMA-1Met and SAS Treatment Reduce the Survival of Primary CLL Cells Carrying Wild-Type and Mutant p53 Protein
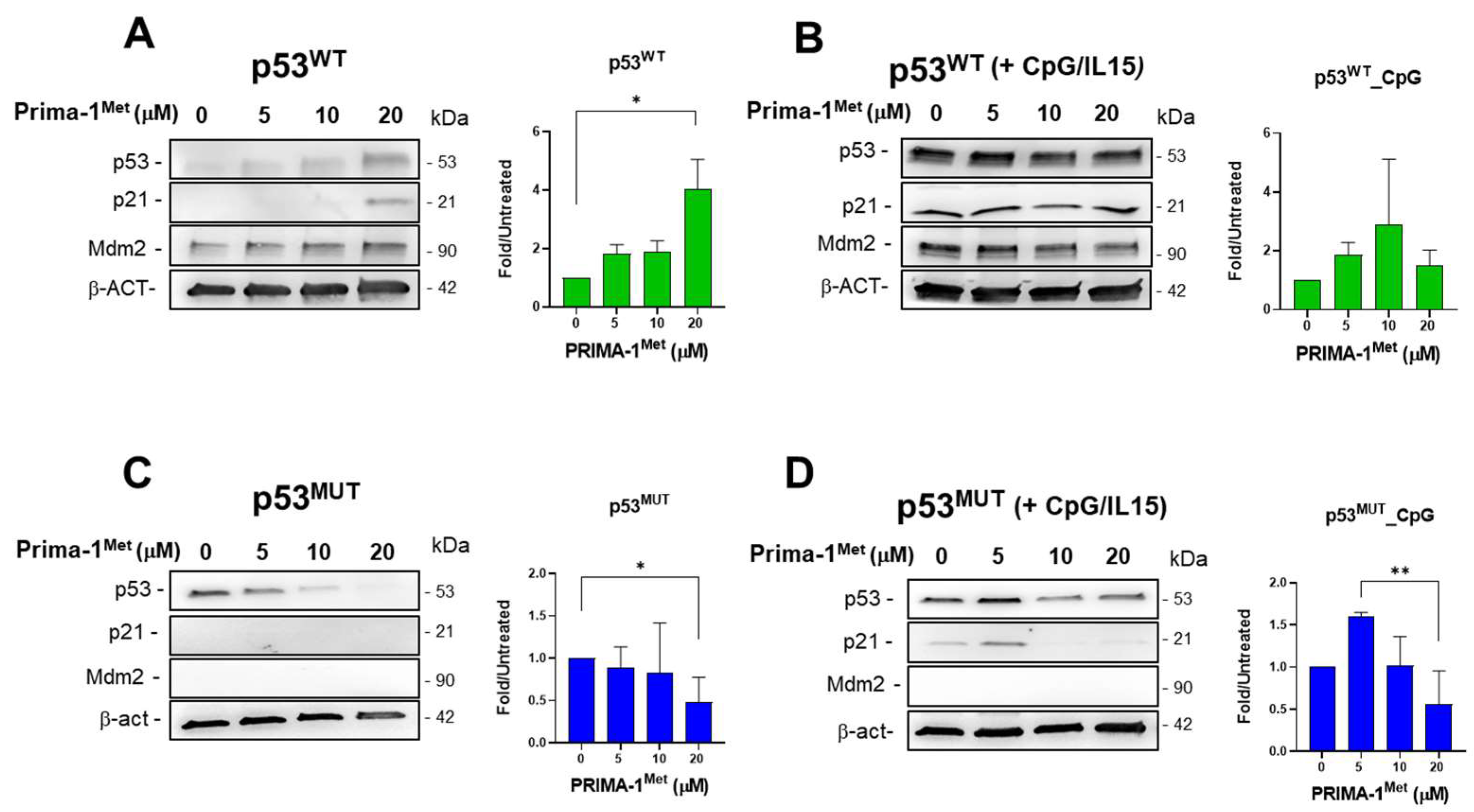
2.4. PRIMA-1Met Does Not Restore the Transcriptional Activity of Mutant p53 in CLL Cells
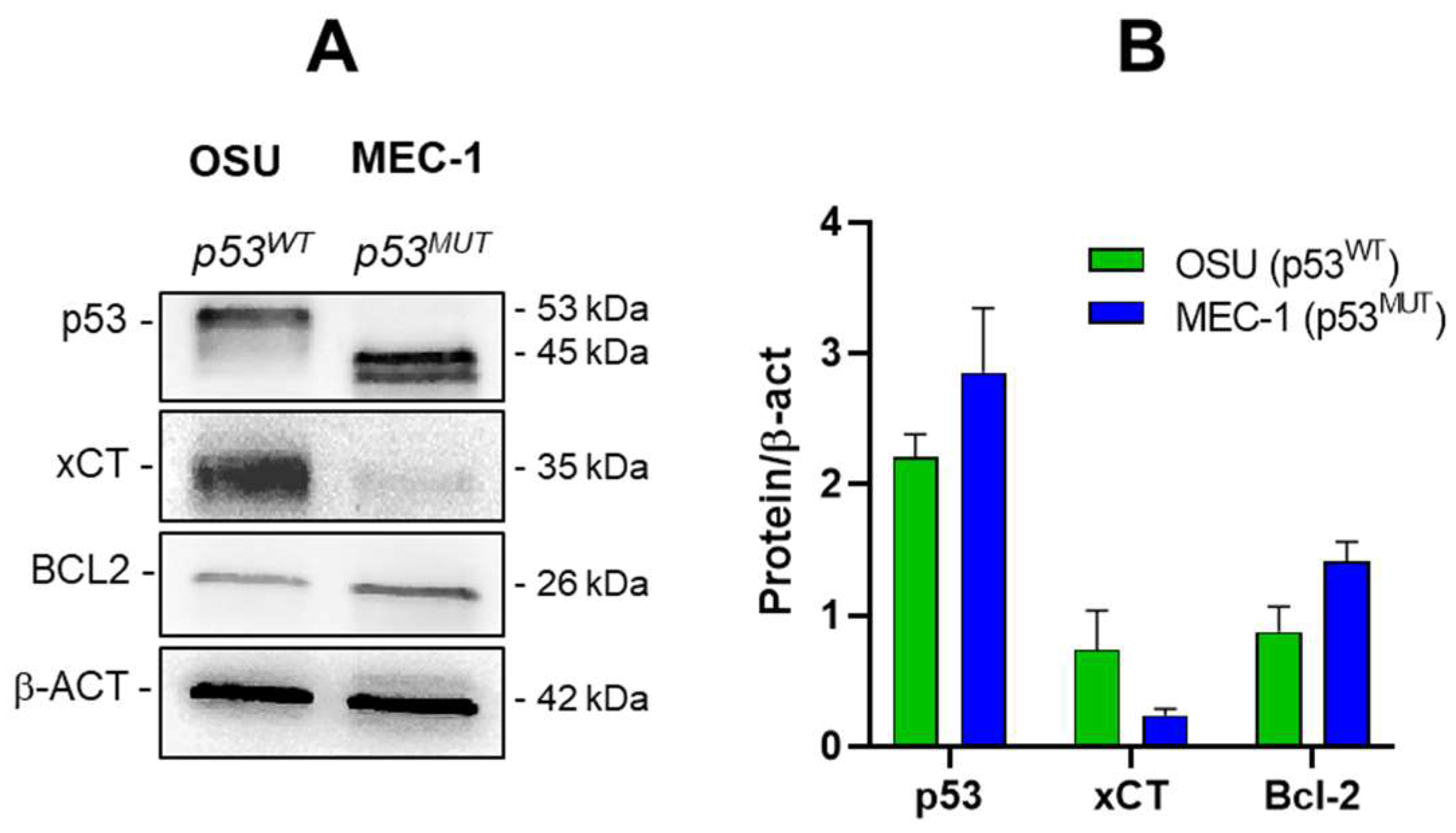
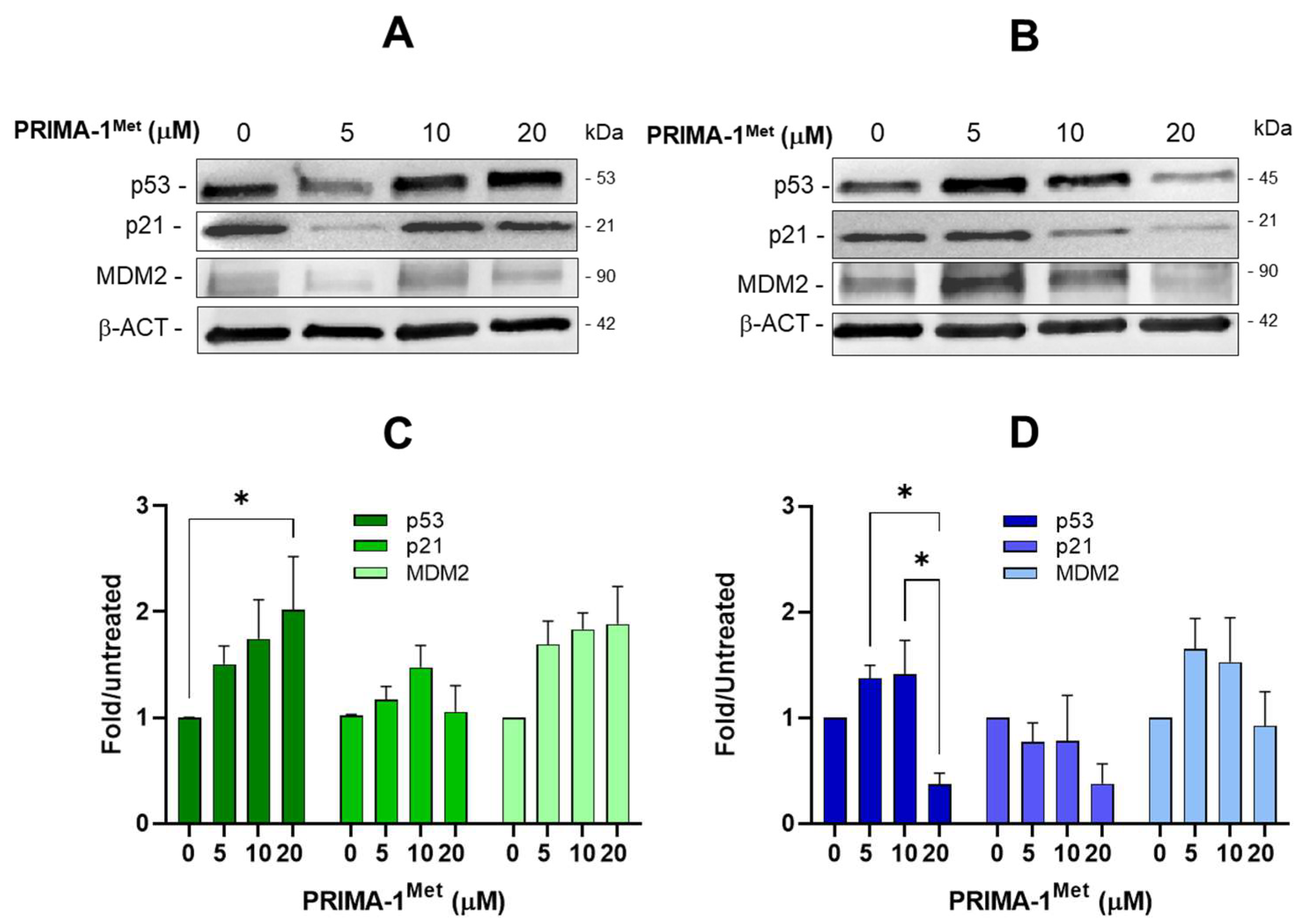
2.5. PRIMA-1Met and Sulfasalazine Significantly Induce SLC7A11/xCT in MEC-1 CLL Cell Line
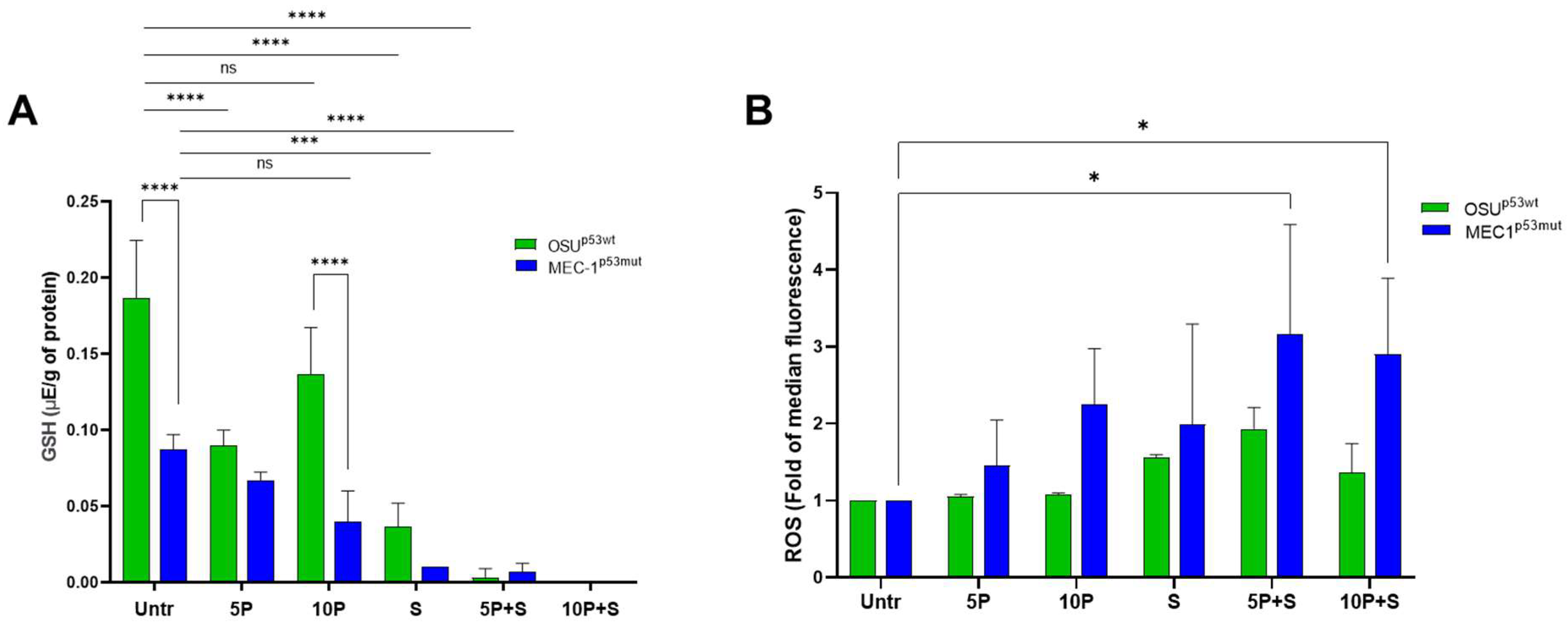
2.6. PRIMA1Met and SAS Deplete the Intracellular Reduced Glutathione (GSH) Content and Enhance ROS Levels Mainly in MEC-1 Cells
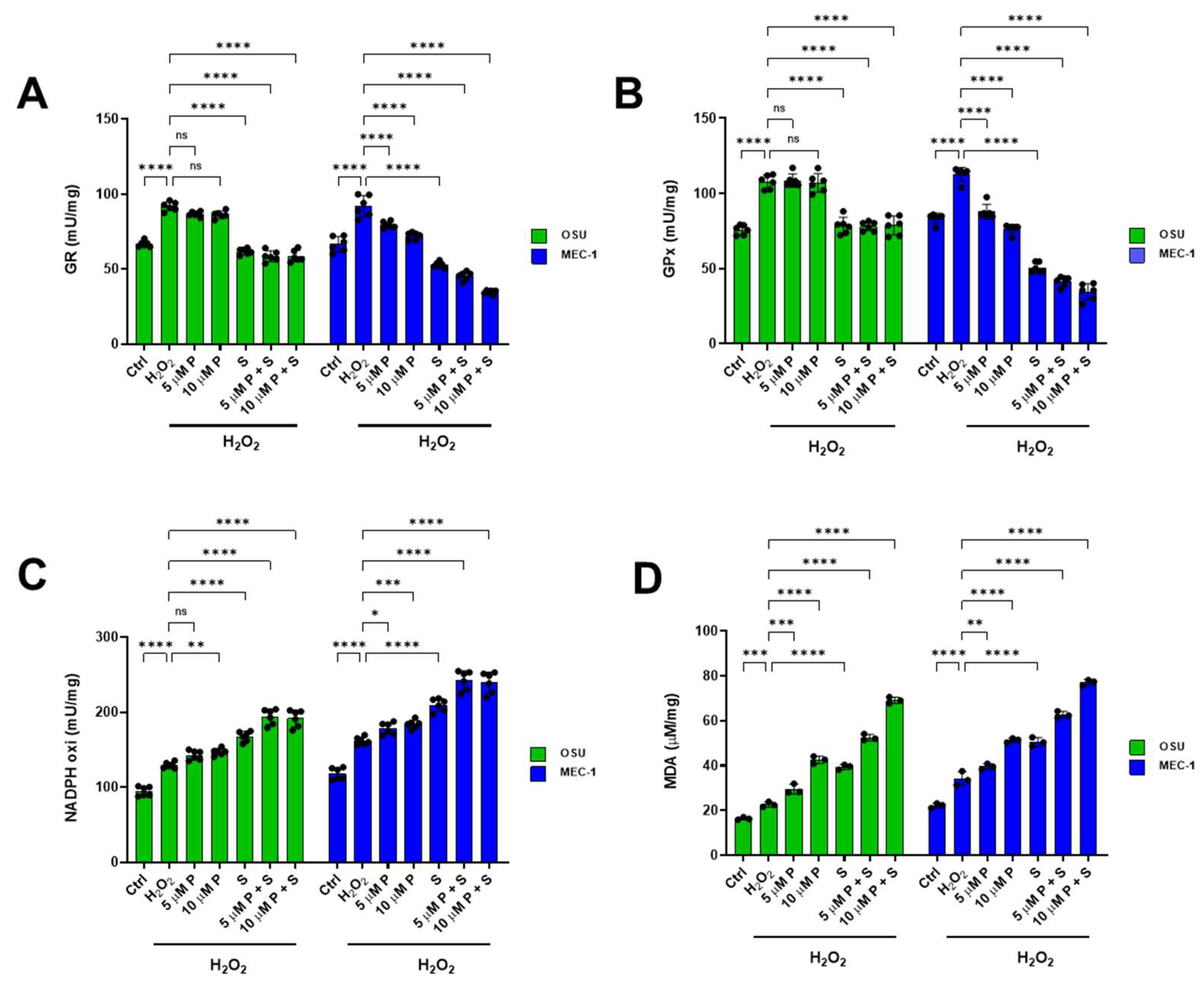
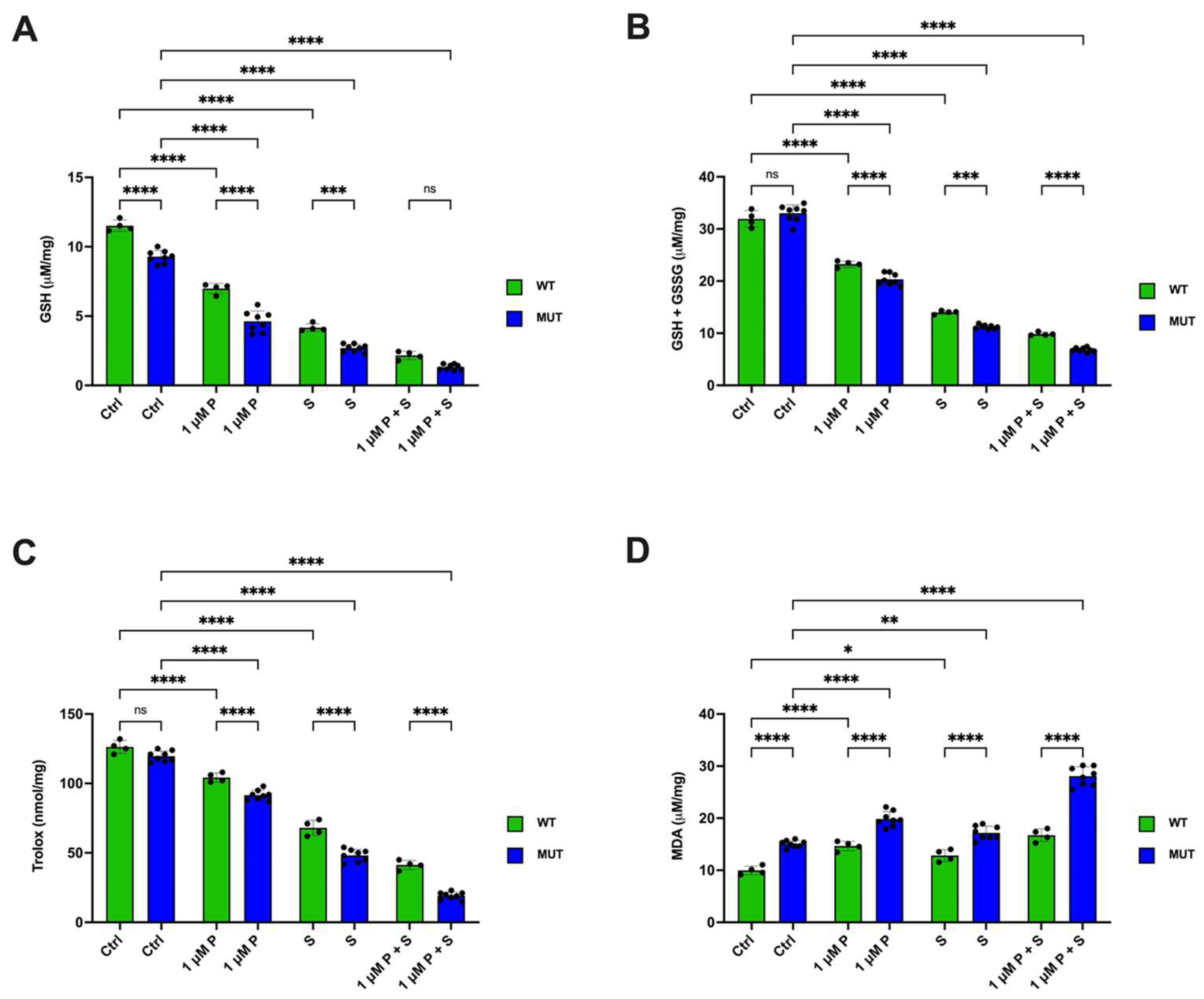
2.7. PRIMA 1Met and SAS Significantly Affect the Balance Between Oxidative Stress Production and Antioxidant Defenses in OSU and MEC-1 Cells
2.8. PRIMA-1Met and SAS Significantly Affect the Redox Status, Especially in TP53 Mutant CLL Primary Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Lines and Drug Treatments
5.2. CLL Patients’ Cell Preparations and Treatment
5.3. MTT Assay
5.4. Synergy Calculation
5.5. Western Blotting Analysis
5.6. Apoptosis Determination
5.7. ROS Determination by Flow Cytometry
5.8. Determination of the Intracellular Level of GSH by HPLC
5.9. Evaluation of Antioxidant Enzymatic Activity
5.10. Evaluation of NADPH Oxidase Activity
5.11. Evaluation of Malondialdehyde Content and Antioxidant Capacity
5.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gentile, M.; Shanafelt, T.D.; Reda, G.; Mauro, F.R.; Zirlik, K.; Ciolli, S.; Laurenti, L.; Del Principe, M.I.; Rossi, D.; Di Renzo, N.; et al. Validation of a Biological Score to Predict Response in Chronic Lymphocytic Leukemia Patients Treated Front-Line with Bendamustine and Rituximab. Leukemia 2018, 32, 1869–1873. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, N.; Chen, S.-S.; Rai, K.R. Chronic Lymphocytic Leukemia. Cold Spring Harb. Perspect. Med. 2021, 11, a035220. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic Aberrations and Survival in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martín-García, D.; Beà, S.; et al. Clinical Impact of Clonal and Subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM Mutations in Chronic Lymphocytic Leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef]
- Buccheri, V.; Barreto, W.G.; Fogliatto, L.M.; Capra, M.; Marchiani, M.; Rocha, V. Prognostic and Therapeutic Stratification in CLL: Focus on 17p Deletion and P53 Mutation. Ann. Hematol. 2018, 97, 2269–2278. [Google Scholar] [CrossRef]
- Rossi, D.; Cerri, M.; Deambrogi, C.; Sozzi, E.; Cresta, S.; Rasi, S.; De Paoli, L.; Spina, V.; Gattei, V.; Capello, D.; et al. The Prognostic Value of TP53 Mutations in Chronic Lymphocytic Leukemia Is Independent of Del17p13: Implications for Overall Survival and Chemorefractoriness. Clin. Cancer Res. 2009, 15, 995–1004. [Google Scholar] [CrossRef]
- Zenz, T.; Vollmer, D.; Trbusek, M.; Smardova, J.; Benner, A.; Soussi, T.; Helfrich, H.; Heuberger, M.; Hoth, P.; Fuge, M.; et al. TP53 Mutation Profile in Chronic Lymphocytic Leukemia: Evidence for a Disease Specific Profile from a Comprehensive Analysis of 268 Mutations. Leukemia 2010, 24, 2072–2079. [Google Scholar] [CrossRef]
- Bomben, R.; Rossi, F.M.; Vit, F.; Bittolo, T.; D’Agaro, T.; Zucchetto, A.; Tissino, E.; Pozzo, F.; Vendramini, E.; Degan, M.; et al. TP53 Mutations with Low Variant Allele Frequency Predict Short Survival in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2021, 27, 5566–5575. [Google Scholar] [CrossRef]
- Morabito, F.; Del Poeta, G.; Mauro, F.R.; Reda, G.; Sportoletti, P.; Laurenti, L.; Coscia, M.; Herishanu, Y.; Bossio, S.; Varettoni, M.; et al. TP53 Disruption as a Risk Factor in the Era of Targeted Therapies: A Multicenter Retrospective Study of 525 Chronic Lymphocytic Leukemia Cases. Am. J. Hematol. 2021, 96, E306–E310. [Google Scholar] [CrossRef]
- Malcikova, J.; Tausch, E.; Rossi, D.; Sutton, L.A.; Soussi, T.; Zenz, T.; Kater, A.P.; Niemann, C.U.; Gonzalez, D.; Davi, F.; et al. ERIC Recommendations for TP53 Mutation Analysis in Chronic Lymphocytic Leukemia-Update on Methodological Approaches and Results Interpretation. Leukemia 2018, 32, 1070–1080. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hao, Q.; Lu, H. Mutant P53 in Cancer Therapy—The Barrier or the Path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic Functions of the Tumor Suppressor P53: Implications in Normal Physiology, Metabolic Disorders, and Cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W. The Complexity of P53-Mediated Metabolic Regulation in Tumor Suppression. Semin. Cancer Biol. 2022, 85, 4–32. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Ravera, S.; Speciale, A.; Velkova, I.; Foggetti, G.; Degan, P.; Fronza, G.; Menichini, P. Mutant P53K120R Expression Enables a Partial Capacity to Modulate Metabolism. Front. Genet. 2022, 13, 974662. [Google Scholar] [CrossRef]
- Morabito, F.; Gentile, M.; Monti, P.; Recchia, A.G.; Menichini, P.; Skafi, M.; Atrash, M.; De Luca, G.; Bossio, S.; Al-Janazreh, H.; et al. TP53 Dysfunction in Chronic Lymphocytic Leukemia: Clinical Relevance in the Era of B-Cell Receptors and BCL-2 Inhibitors. Expert. Opin. Investig. Drugs 2020, 29, 869–880. [Google Scholar] [CrossRef]
- Hallek, M.; Al-Sawaf, O. Chronic Lymphocytic Leukemia: 2022 Update on Diagnostic and Therapeutic Procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting Mutant P53 for Efficient Cancer Therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Menichini, P.; Monti, P.; Speciale, A.; Cutrona, G.; Matis, S.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; et al. Antitumor Effects of PRIMA-1 and PRIMA-1Met (APR246) in Hematological Malignancies: Still a Mutant P53-Dependent Affair? Cells 2021, 10, 98. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, M.-Q.-Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET Inhibits Thioredoxin Reductase 1 and Converts the Enzyme to a Dedicated NADPH Oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met Induces Myeloma Cell Death Independent of P53 by Impairing the GSH/ROS Balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Valenti, G.E.; Roveri, A.; Venerando, R.; Menichini, P.; Monti, P.; Tasso, B.; Traverso, N.; Domenicotti, C.; Marengo, B. PTC596-Induced BMI-1 Inhibition Fights Neuroblastoma Multidrug Resistance by Inducing Ferroptosis. Antioxidants 2023, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the System XC-/Glutathione Axis Selectively Targets Cancers with Mutant-P53 Accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, K.M.; Corrales Benitez, M.; Cabalag, C.S.; Zhang, B.Z.; Ko, H.S.; Liu, D.S.; Simpson, K.J.; Haupt, Y.; Lipton, L.; Haupt, S.; et al. SLC7A11 Is a Superior Determinant of APR-246 (Eprenetapopt) Response than TP53 Mutation Status. Mol. Cancer Ther. 2021, 20, 1858–1867. [Google Scholar] [CrossRef]
- Clemons, N.J.; Liu, D.S.; Duong, C.P.; Phillips, W.A. Inhibiting System xC− and Glutathione Biosynthesis—A Potential Achilles’ Heel in Mutant-P53 Cancers. Mol. Cell. Oncol. 2017, 4, e1344757. [Google Scholar] [CrossRef]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A Potent and Specific Inhibitor of Nuclear Factor Kappa B. J. Clin. Investig. 1998, 101, 1163–1174. [Google Scholar] [CrossRef]
- Mansouri, L.; Papakonstantinou, N.; Ntoufa, S.; Stamatopoulos, K.; Rosenquist, R. NF-ΚB Activation in Chronic Lymphocytic Leukemia: A Point of Convergence of External Triggers and Intrinsic Lesions. Semin. Cancer Biol. 2016, 39, 40–48. [Google Scholar] [CrossRef]
- Jaskova, Z.; Pavlova, S.; Malcikova, J.; Brychtova, Y.; Trbusek, M. PRIMA-1MET Cytotoxic Effect Correlates with P53 Protein Reduction in TP53-Mutated Chronic Lymphocytic Leukemia Cells. Leuk. Res. 2020, 89, 106288. [Google Scholar] [CrossRef]
- Stacchini, A.; Aragno, M.; Vallario, A.; Alfarano, A.; Circosta, P.; Gottardi, D.; Faldella, A.; Rege-Cambrin, G.; Thunberg, U.; Nilsson, K.; et al. MEC1 and MEC2: Two New Cell Lines Derived from B-Chronic Lymphocytic Leukaemia in Prolymphocytoid Transformation. Leuk. Res. 1999, 23, 127–136. [Google Scholar] [CrossRef]
- Gupta, R.; Yan, X.J.; Barrientos, J.; Kolitz, J.E.; Allen, S.L.; Rai, K.; Chiorazzi, N.; Mongini, P.K.A. Mechanistic Insights into CpG DNA and IL-15 Synergy in Promoting B Cell Chronic Lymphocytic Leukemia Clonal Expansion. J. Immunol. 2018, 201, 1570–1585. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the Tumor Suppressor Function to Mutant P53 by a Low-Molecular-Weight Compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.; Mohammad, D.K.; Mujahed, H.; Jonson-Videsäter, K.; Nore, B.; Paul, C.; Lehmann, S. Anti-Leukaemic Effects Induced by APR-246 Are Dependent on Induction of Oxidative Stress and the NFE2L2/HMOX1 Axis That Can Be Targeted by PI3K and MTOR Inhibitors in Acute Myeloid Leukaemia Cells. Br. J. Haematol. 2016, 174, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Synnott, N.C.; Madden, S.F.; Bykov, V.J.N.; Crown, J.; Wiman, K.G.; Duffy, M.J. The Mutant P53-Targeting Compound APR-246 Induces ROS-Modulating Genes in Breast Cancer Cells. Transl. Oncol. 2018, 11, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type P53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]
- Tuval, A.; Strandgren, C.; Heldin, A.; Palomar-Siles, M.; Wiman, K.G. Pharmacological Reactivation of P53 in the Era of Precision Anticancer Medicine. Nat. Rev. Clin. Oncol. 2024, 21, 106–120. [Google Scholar] [CrossRef]
- Peuget, S.; Zhou, X.; Selivanova, G. Translating P53-Based Therapies for Cancer into the Clinic. Nat. Rev. Cancer 2024, 24, 192–215. [Google Scholar] [CrossRef]
- Pozzo, F.; Dal Bo, M.; Peragine, N.; Bomben, R.; Zucchetto, A.; Rossi, F.; Degan, M.; Rossi, D.; Chiarenza, A.; Grossi, A.; et al. Detection of TP53 Dysfunction in Chronic Lymphocytic Leukemia by an in Vitro Functional Assay Based on TP53 Activation by the Non-Genotoxic Drug Nutlin-3: A Proposal for Clinical Application. J. Hematol. Oncol. 2013, 6, 83. [Google Scholar] [CrossRef]
- Russo, D.; Ottaggio, L.; Penna, I.; Foggetti, G.; Fronza, G.; Inga, A.; Menichini, P. PRIMA-1 Cytotoxicity Correlates with Nucleolar Localization and Degradation of Mutant P53 in Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2010, 402, 345–350. [Google Scholar] [CrossRef]
- Hertlein, E.; Beckwith, K.A.; Lozanski, G.; Chen, T.L.; Towns, W.H.; Johnson, A.J.; Lehman, A.; Ruppert, A.S.; Bolon, B.; Andritsos, L.; et al. Characterization of a New Chronic Lymphocytic Leukemia Cell Line for Mechanistic in Vitro and in Vivo Studies Relevant to Disease. PLoS ONE 2013, 8, e76607. [Google Scholar] [CrossRef]
- Magrini, R.; Russo, D.; Ottaggio, L.; Fronza, G.; Inga, A.; Menichini, P. PRIMA-1 Synergizes with Adriamycin to Induce Cell Death in Non-Small Cell Lung Cancer Cells. J. Cell. Biochem. 2008, 104, 2363–2373. [Google Scholar] [CrossRef]
- Berenbaum, M.C. What Is Synergy? Pharmacol. Rev. 1989, 41, 93–141. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, M.C. Criteria for Analyzing Interactions between Biologically Active Agents. Adv. Cancer Res. 1981, 35, 269–335. [Google Scholar] [CrossRef] [PubMed]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy Induced by SAHA Affects Mutant P53 Degradation and Cancer Cell Survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef] [PubMed]
- Cossarizza, A.; Ferraresi, R.; Troiano, L.; Roat, E.; Gibellini, L.; Bertoncelli, L.; Nasi, M.; Pinti, M. Simultaneous Analysis of Reactive Oxygen Species and Reduced Glutathione Content in Living Cells by Polychromatic Flow Cytometry. Nat. Protoc. 2009, 4, 1790–1797. [Google Scholar] [CrossRef]
- Fariss, M.W.; Reed, D.J. High-Performance Liquid Chromatography of Thiols and Disulfides: Dinitrophenol Derivatives. Methods Enzymol. 1987, 143, 101–109. [Google Scholar] [CrossRef]
- Ravera, S.; Bertola, N.; Puddu, A.; Bruno, S.; Maggi, D.; Panfoli, I. Crosstalk between the Rod Outer Segments and Retinal Pigmented Epithelium in the Generation of Oxidative Stress in an In Vitro Model. Cells 2023, 12, 2173. [Google Scholar] [CrossRef]
- Brullo, C.; Russo, E.; Garibaldi, S.; Altieri, P.; Ameri, P.; Ravera, S.; Signorello, M.G. Inside the Mechanism of Action of Three Pyrazole Derivatives in Human Platelets and Endothelial Cells. Antioxidants 2023, 12, 216. [Google Scholar] [CrossRef]
- Colla, R.; Izzotti, A.; De Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Ricciarelli, R.; Furfaro, A.L.; Pulliero, A.; Passalacqua, M.; et al. Glutathione-Mediated Antioxidant Response and Aerobic Metabolism: Two Crucial Factors Involved in Determining the Multi-Drug Resistance of High-Risk Neuroblastoma. Oncotarget 2016, 7, 70715–70737. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasino, M.; Speciale, A.; Ravera, S.; Cutrona, G.; Massara, R.; Bertola, N.; Viale, M.; Velkova, I.; Mazzarello, A.N.; Fais, F.; et al. Targeting the p53/xCT/GSH Axis with PRIMA-1Met Combined with Sulfasalazine Shows Therapeutic Potential in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2025, 26, 5559. https://doi.org/10.3390/ijms26125559
Pasino M, Speciale A, Ravera S, Cutrona G, Massara R, Bertola N, Viale M, Velkova I, Mazzarello AN, Fais F, et al. Targeting the p53/xCT/GSH Axis with PRIMA-1Met Combined with Sulfasalazine Shows Therapeutic Potential in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences. 2025; 26(12):5559. https://doi.org/10.3390/ijms26125559
Chicago/Turabian StylePasino, Martina, Andrea Speciale, Silvia Ravera, Giovanna Cutrona, Rosanna Massara, Nadia Bertola, Maurizio Viale, Irena Velkova, Andrea Nicola Mazzarello, Franco Fais, and et al. 2025. "Targeting the p53/xCT/GSH Axis with PRIMA-1Met Combined with Sulfasalazine Shows Therapeutic Potential in Chronic Lymphocytic Leukemia" International Journal of Molecular Sciences 26, no. 12: 5559. https://doi.org/10.3390/ijms26125559
APA StylePasino, M., Speciale, A., Ravera, S., Cutrona, G., Massara, R., Bertola, N., Viale, M., Velkova, I., Mazzarello, A. N., Fais, F., Loiacono, F., Matis, S., Valenti, G. E., Traverso, N., Domenicotti, C., Marengo, B., Tasso, B., Ibatici, A., Angelucci, E., ... Menichini, P. (2025). Targeting the p53/xCT/GSH Axis with PRIMA-1Met Combined with Sulfasalazine Shows Therapeutic Potential in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences, 26(12), 5559. https://doi.org/10.3390/ijms26125559