
Cell-Based Therapies for Solid Tumors: Challenges and Advances

Abstract
1. Introduction
2. Cell-Based Therapies in Solid Tumor Treatment
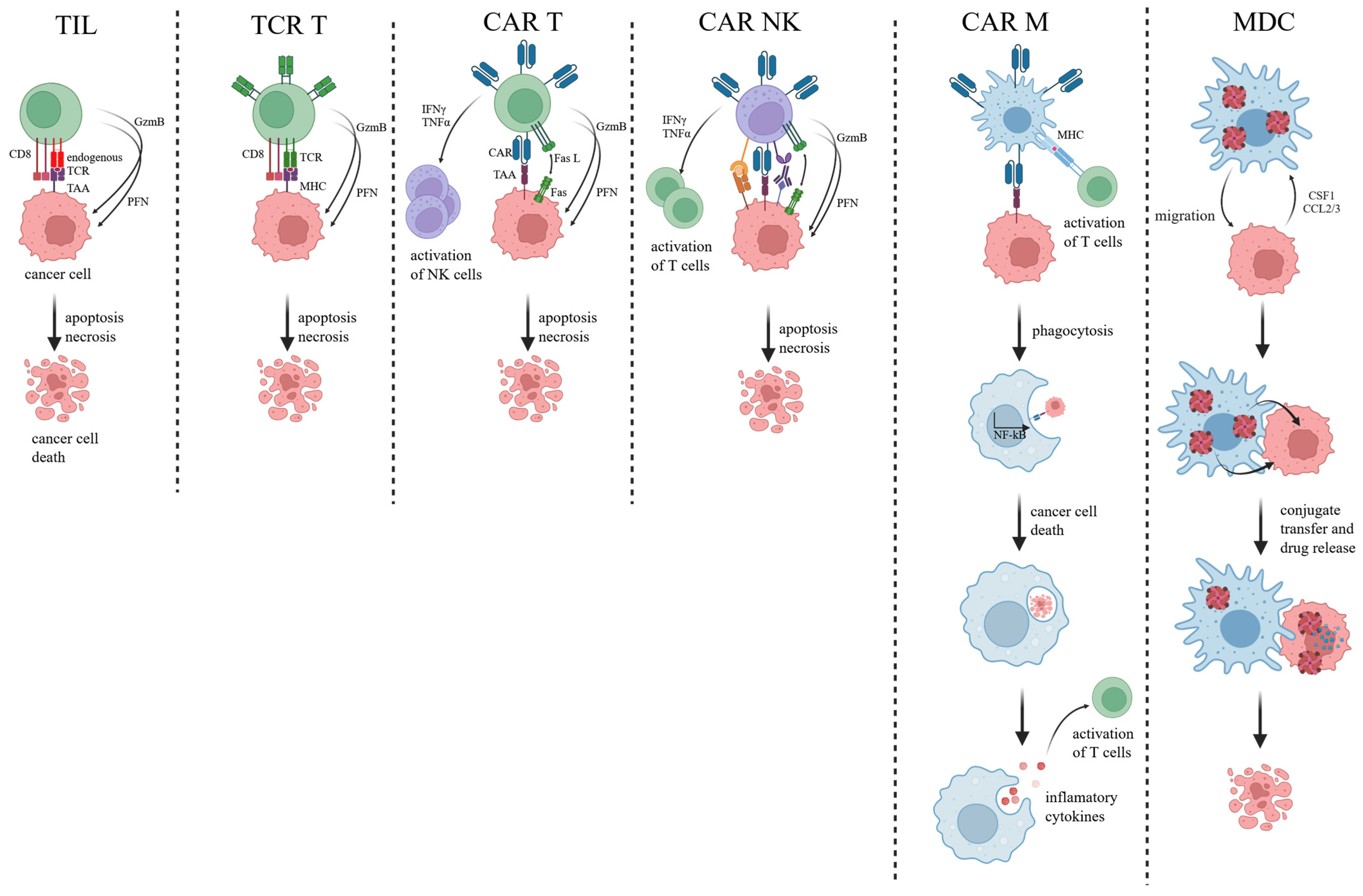
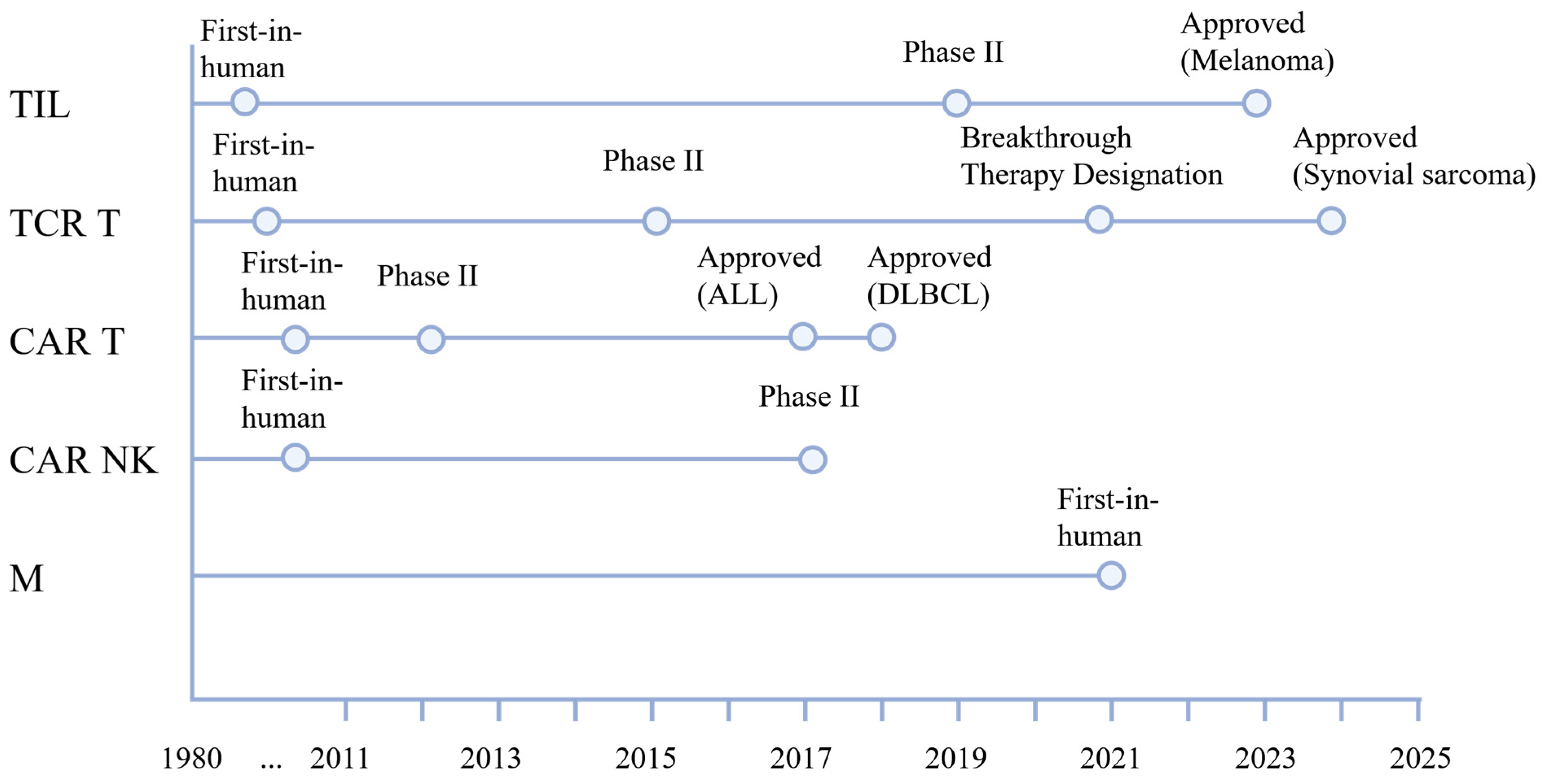
2.1. Tumor-Infiltrating Lymphocytes
2.1.1. Mechanism
2.1.2. Clinical Data
2.1.3. Limitations
2.2. T Cell Receptor-Engineered T Cells (TCR T)
2.2.1. Mechanism
2.2.2. Clinical Data
2.2.3. Limitations
2.3. Chimeric Antigen Receptor T Cells (CAR T)
2.3.1. Mechanism
2.3.2. Clinical Data
2.3.3. Limitations
2.4. CAR Natural Killer Cells
2.4.1. Mechanism
2.4.2. Clinical Data
2.4.3. Limitations
2.5. Macrophages
2.5.1. Mechanism
2.5.2. Clinical Data
3. Current Clinical Trials of Cell-Based Therapies Against Solid Tumors
4. Advances and Challenges in Cell-Based Therapy for Solid Tumors
5. The Implications of Autologous Versus Allogeneic Therapeutic Approaches in the Context of Cellular Immunotherapy for Solid Tumors
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | Adoptive Cell Therapy |
| ADC | Antibody-Dependent Cellular Cytotoxicity |
| ALL | Acute Lymphoblastic Leukemia |
| CAFs | Cancer-Associated Fibroblasts |
| CAR | Chimeric Antigen Receptor |
| CAR M | CAR Macrophages |
| CAR NK | CAR NK Cells |
| CAR T | CAR T Cells |
| CEA | Carcinoembryonic Antigen |
| CRS | Cytokine Release Syndrome |
| CTLs | Cytotoxic T Lymphocytes |
| CSC | Cancer Stem-Like Cells |
| DLBCL | Diffuse Large B Cell Lymphoma |
| ECM | Extracellular Matrix |
| EGFR | Epidermal Growth Factor Receptor |
| GD2 | Disialoganglioside |
| GvHD | Graft-Versus-Host Disease |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HLA | Human Leukocyte Antigen |
| HPV16 E7 | Human Papillomavirus 16 Protein E7 |
| iPSC | Induced Pluripotent Stem Cell |
| IL2 | Interleukin-2 |
| IL10 | Interleukin-10 |
| IL13Rα2 | Interleukin-13 Receptor Alpha 2 |
| IL15 | Interleukin-15 |
| MAGE-A4 | Melanoma Antigen Gene A4 |
| MAGE-C2 | Melanoma-Associated Gene C2 |
| MART-1 | Melanoma Antigen Recognized by T Cells 1 |
| mbIL-15 | Membrane-Bound Interleukin-15 |
| MDC | Macrophage–Drug Conjugate |
| MDSC | Myeloid-Derived Suppressor Cell |
| MHC | Major Histocompatibility Complex |
| MUC1 | Mucin 1 |
| NK | Natural Killer |
| NSCC | Non-Stem Cancer Cells |
| NY-ESO-1 | New York Esophageal Squamous Cell Carcinoma 1 |
| PBMC | Peripheral Blood Mononuclear Cell |
| PSMA | Prostate-Specific Membrane Antigen |
| ROR1 | Receptor Tyrosine Kinase Like Orphan Receptor 1 |
| scFv | Single-Chain Variable Fragment |
| TAA | Tumor-Associated Antigen |
| TCR | T Cell Receptor |
| TCR T | T Cell Receptor-Engineered T Cells |
| TGFβ | Transforming Growth Factor Beta |
| TILs | Tumor Infiltrating Lymphocytes |
| TME | Tumor Microenvironment |
| Treg | Regulatory T Cell |
| TROP2 | Trophoblast Cell Surface Antigen 2 |
| TRUCKs | T Cells Redirected For Universal Cytokine-Mediated Killing |
| WHO | World Health Organization |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Wu, Y.; Song, Y.; Wang, R.; Wang, T. Molecular Mechanisms of Tumor Resistance to Radiotherapy. Mol. Cancer 2023, 22, 96. [Google Scholar] [CrossRef]
- Rebucci, M.; Michiels, C. Molecular Aspects of Cancer Cell Resistance to Chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Mai, Y.; Su, J.; Yang, C.; Xia, C.; Fu, L. The Strategies to Cure Cancer Patients by Eradicating Cancer Stem-like Cells. Mol. Cancer 2023, 22, 171. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Xu, X.; Lin, S.; Zhang, Y.; Liu, H.; Zhang, C.; Mo, R. A Nanotherapeutic Strategy to Overcome Chemotherapeutic Resistance of Cancer Stem-like Cells. Nat. Nanotechnol. 2021, 16, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Lukong, K.E. Breast Cancer Stem-Like Cells in Drug Resistance: A Review of Mechanisms and Novel Therapeutic Strategies to Overcome Drug Resistance. Front. Oncol. 2022, 12, 856974. [Google Scholar] [CrossRef] [PubMed]
- Comoli, P.; Chabannon, C.; Koehl, U.; Lanza, F.; Urbano-Ispizua, A.; Hudecek, M.; Ruggeri, A.; Secondino, S.; Bonini, C.; Pedrazzoli, P. Development of Adaptive Immune Effector Therapies in Solid Tumors. Ann. Oncol. 2019, 30, 1740–1750. [Google Scholar] [CrossRef]
- Bastien, J.P.; Minguy, A.; Dave, V.; Roy, D.C. Cellular Therapy Approaches Harnessing the Power of the Immune System for Personalized Cancer Treatment. Semin. Immunol. 2019, 42, 101306. [Google Scholar] [CrossRef]
- Grimes, J.M.; Carvajal, R.D.; Muranski, P. Cellular Therapy for the Treatment of Solid Tumors. Transfus. Apher. Sci. 2021, 60, 103056. [Google Scholar] [CrossRef]
- El-Kadiry, A.E.H.; Rafei, M.; Shammaa, R. Cell Therapy: Types, Regulation, and Clinical Benefits. Front. Med. 2021, 8, 756029. [Google Scholar] [CrossRef]
- Mundhara, N.; Sadhukhan, P. Cracking the Codes behind Cancer Cells’ Immune Evasion. Int. J. Mol. Sci. 2024, 25, 8899. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.E. Tumor-Induced Metabolic Immunosuppression: Mechanisms and Therapeutic Targets. Cell Rep. 2025, 44, 115206. [Google Scholar] [CrossRef]
- Xia, X.; Yang, Z.; Lu, Q.; Liu, Z.; Wang, L.; Du, J.; Li, Y.; Yang, D.H.; Wu, S. Reshaping the Tumor Immune Microenvironment to Improve CAR-T Cell-Based Cancer Immunotherapy. Mol. Cancer 2024, 23, 175. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wei, X.-W. Immunosuppressive Cells in Cancer: Mechanisms and Potential Therapeutic Targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Pogge von Strandmann, E.; Reinartz, S.; Wager, U.; Müller, R. Tumor–Host Cell Interactions in Ovarian Cancer: Pathways to Therapy Failure. Trends Cancer 2017, 3, 137–148. [Google Scholar] [CrossRef]
- Mandalà, M.; De Logu, F.; Merelli, B.; Nassini, R.; Massi, D. Immunomodulating Property of MAPK Inhibitors: From Translational Knowledge to Clinical Implementation. Lab. Investig. 2017, 97, 166–175. [Google Scholar] [CrossRef]
- Badoual, C.; Sandoval, F.; Pere, H.; Hans, S.; Gey, A.; Merillon, N.; Van Ryswick, C.; Quintin-Colonna, F.; Bruneval, P.; Brasnu, D.; et al. Better Understanding Tumor–Host Interaction in Head and Neck Cancer to Improve the Design and Development of Immunotherapeutic Strategies. Head Neck. 2010, 32, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Chen, D.S.; Powles, T.; Turley, S.J. The Cancer-Immunity Cycle: Indication, Genotype, and Immunotype. Immunity 2023, 56, 2188–2205. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Mount, N.M.; Ward, S.J.; Kefalas, P.; Hyllner, J. Cell-Based Therapy Technology Classifications and Translational Challenges. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20150017. [Google Scholar] [CrossRef] [PubMed]
- Dodson, B.P.; Levine, A.D. Challenges in the Translation and Commercialization of Cell Therapies. BMC Biotechnol. 2015, 15, 70. [Google Scholar] [CrossRef]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T Cells in Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef]
- Deo, A.S.; Shrijana; Sruthika, S.U.; Karun, S.; Bisaria, K.; Sarkar, K. Participation of T Cells in Generating Immune Protection against Cancers. Pathol. Res. Pract. 2024, 262, 155534. [Google Scholar] [CrossRef]
- Daher, M.; Melo Garcia, L.; Li, Y.; Rezvani, K. CAR-NK Cells: The next Wave of Cellular Therapy for Cancer. Clin. Transl. Immunol. 2021, 10, e1274. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, J.; Liang, Z.; Dai, K.; Gan, J.; Wang, Q.; Xu, Y.; Chen, Y.H.; Wan, X. CAR-Macrophages and CAR-T Cells Synergistically Kill Tumor Cells In Vitro. Cells 2022, 11, 3692. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.; Toy, S.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.; et al. Use of Tumor-Infiltrating Lumphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalían, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients after Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-Infiltrating Lymphocytes in the Immunotherapy Era. Cell Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Saibil, S.D.; Sotov, V.; Le, M.X.; Khoja, L.; Ghazarian, D.; Bonilla, L.; Majeed, H.; Hogg, D.; Joshua, A.M.; et al. Phase II Clinical Trial of Adoptive Cell Therapy for Patients with Metastatic Melanoma with Autologous Tumor-Infiltrating Lymphocytes and Low-Dose Interleukin-2. Cancer Immunol. Immunother. 2019, 68, 773–785. [Google Scholar] [CrossRef]
- Han, E.; Choi, H.Y.; Kwon, H.J.; Chung, Y.R.; Shin, H.C.; Kim, E.K.; Suh, K.J.; Kim, S.H.; Kim, J.H.; Park, S.Y. Characterization of Tumor-Infiltrating Lymphocytes and Their Spatial Distribution in Triple-Negative Breast Cancer. Breast Cancer Res. 2024, 26, 180. [Google Scholar] [CrossRef]
- Liu, D.; Heij, L.R.; Czigany, Z.; Dahl, E.; Lang, S.A.; Ulmer, T.F.; Luedde, T.; Neumann, U.P.; Bednarsch, J. The Role of Tumor-Infiltrating Lymphocytes in Cholangiocarcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 127. [Google Scholar] [CrossRef]
- Zhao, X.; Pan, X.; Wang, Y.; Zhang, Y. Targeting Neoantigens for Cancer Immunotherapy. Biomark. Res. 2021, 9, 61. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Gerhardt, L.; Vareki, S.M. Immunosuppressive Effects of Myeloid-derived Suppressor Cells in Cancer and Immunotherapy. Cells 2021, 10, 1170. [Google Scholar] [CrossRef]
- Qin, D.; Zhang, Y.; Shu, P.; Lei, Y.; Li, X.; Wang, Y. Targeting Tumor-Infiltrating Tregs for Improved Antitumor Responses. Front. Immunol. 2024, 15, 1325946. [Google Scholar] [CrossRef]
- Hofer, F.; Sario, G.D.; Musiu, C.; Sartoris, S.; Sanctis, F.D.; Ugel, S. A Complex Metabolic Network Confers Immunosuppressive Functions to Myeloid-Derived Suppressor Cells (MDSCs) within the Tumour Microenvironment. Cells 2021, 10, 2700. [Google Scholar] [CrossRef]
- Amaria, R.; Knisely, A.; Vining, D.; Kopetz, S.; Overman, M.J.; Javle, M.; Antonoff, M.B.; Tzeng, C.W.D.; Wolff, R.A.; Pant, S.; et al. Efficacy and Safety of Autologous Tumor-Infiltrating Lymphocytes in Recurrent or Refractory Ovarian Cancer, Colorectal Cancer, and Pancreatic Ductal Adenocarcinoma. J. Immunother. Cancer 2024, 12, e006822. [Google Scholar] [CrossRef] [PubMed]
- Tarabay, J.; Ren, D.; Ifegu, I.; Zada, S.; Edwards, R.; Chan, J.; Kuan, E.C.; Wang, B. Prognostic Significance of Tumor-Infiltrating Lymphocytes and Anti-Programmed Death-Ligand 1 Therapy in Sinonasal Mucosal Melanoma: A 10-Year Experience at a Single Institution. J. Clin. Transl. Pathol. 2023, 3, 115–119. [Google Scholar] [CrossRef]
- Brummel, K.; Eerkens, A.L.; de Bruyn, M.; Nijman, H.W. Tumour-Infiltrating Lymphocytes: From Prognosis to Treatment Selection. Br. J. Cancer 2023, 128, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Haanen, J.; Los, C.; Phan, G.Q.; Betof Warner, A. Adoptive Cell Therapy for Solid Tumors: Current Status in Melanoma and Next-Generation Therapies. Am. Soc. Clin. Oncol. Educ. B 2024, 44, e431608. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.; Dudley, M.E.; Parkhurst, M.R.; Yang, J.C.; Rosenberg, S. A Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 9, 641–645. [Google Scholar] [CrossRef]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; De Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-Expression of CD39 and CD103 Identifies Tumor-Reactive CD8 T Cells in Human Solid Tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [CrossRef]
- Qiu, X.; Li, S.; Fan, T.; Zhang, Y.; Wang, B.; Zhang, B.; Zhang, M.; Zhang, L. Advances and Prospects in Tumor Infiltrating Lymphocyte Therapy. Discov. Oncol. 2024, 15, 630. [Google Scholar] [CrossRef]
- Matsueda, S.; Chen, L.; Li, H.; Yao, H.; Yu, F. Recent Clinical Researches and Technological Development in TIL Therapy. Cancer Immunol. Immunother. 2024, 73, 232. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. Perspective T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Li, D.; Liu, S. Research Progress on Tumor- in Fi Ltrating Lymphocyte Therapy for Cervical Cancer. Front. Immunol. 2025, 16, 1524842. [Google Scholar] [CrossRef]
- Tas, L.; Jedema, I.; Haanen, J.B.A.G. Novel Strategies to Improve Efficacy of Treatment with Tumor-Infiltrating Lymphocytes (TILs) for Patients with Solid Cancers. Curr. Opin. Oncol. 2023, 35, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.F.; Hoyos, V.; Savoldo, B.; Quintarelli, C.; Giordano Attianese, G.M.P.; Leen, A.M.; Liu, H.; Foster, A.E.; Heslop, H.E.; Rooney, C.M.; et al. Genetic Manipulation of Tumor-Specific Cytotoxic T Lymphocytes to Restore Responsiveness to IL-7. Mol. Ther. 2009, 17, 880–888. [Google Scholar] [CrossRef]
- Lee, K.L.; Schlom, J.; Hamilton, D.H. Combination Therapies Utilizing Neoepitope-Targeted Vaccines. Cancer Immunol. Immunother. 2021, 70, 875–885. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a Systems Understanding of MHC Class I and MHC Class II Antigen Presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor Regression in Patients with Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive with NY-ESO-1. J. Clin. Neurosci. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Miao, R.; Yu, J.; Kim, R.D. Targeting the KRAS Oncogene for Patients with Metastatic Colorectal Cancer. Cancers 2025, 17, 1336–1347. [Google Scholar] [CrossRef]
- Bollard, C.M.; Aguilar, L.; Straathof, K.C.; Gahn, B.; Huls, M.H.; Rousseau, A.; Sixbey, J.; Gresik, M.V.; Carrum, G.; Hudson, M.; et al. Cytotoxic T Lymphocyte Therapy for Epstein-Barr Virus+ Hodgkin’s Disease. J. Exp. Med. 2004, 200, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy with TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 C259 T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K. Immune Escape: A Critical Hallmark in Solid Tumors. Life Sci. 2020, 258, 118110. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Wang, T.; Wang, X.F.; Zhang, Y.Q.; Song, S.X.; Ma, C. qing Improving the Ability of CAR-T Cells to Hit Solid Tumors: Challenges and Strategies. Pharmacol. Res. 2022, 175, 106036. [Google Scholar] [CrossRef]
- Kankeu Fonkoua, L.A.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T Cell Therapy and the Tumor Microenvironment: Current Challenges and Opportunities. Mol. Ther. Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gkoss, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the γ or ζ Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Ayala Ceja, M.; Khericha, M.; Harris, C.M.; Puig-Saus, C.; Chen, Y.Y. CAR-T Cell Manufacturing: Major Process Parameters and next-Generation Strategies. J. Exp. Med. 2024, 221, e20230903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T Cells. Biomark. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Dotti, G. Chimeric Antigen Receptor (CAR)-Engineered Lymphocytes for Cancer Therapy. Expert Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Mazinani, M.; Rahbarizadeh, F. CAR-T Cell Potency: From Structural Elements to Vector Backbone Components. Biomark. Res. 2022, 10, 70. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T Design: Elements and Their Synergistic Function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 Costimulation Improves Expansion and Persistence of Chimeric Antigen Receptor–Modified T Cells in Lymphoma Patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI 3 Kinase/AKT/Bcl-X L Activation and CD8 T Cell-Mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Khan, S.H.; Choi, Y.; Veena, M.; Lee, J.K.; Shin, D.S. Advances in CAR T Cell Therapy: Antigen Selection, Modifications, and Current Trials for Solid Tumors. Front. Immunol. 2024, 15, 1489827. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Klysz, D.; Sotillo, E.; Fisher, C.J.; Vilches-Moure, J.G.; Pacheco, K.Z.B.; Malipatlolla, M.; Xu, P.; Hui, J.H.; et al. Enhanced Safety and Efficacy of Protease-Regulated CAR-T Cell Receptors. Cell 2022, 185, 1745–1763.e22. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T Cell Therapy for H3K27M-Mutated Diffuse Midline Gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-Specific CAR T Cells in Gastrointestinal Cancers: Phase 1 Trial Interim Results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Haas, A.R.; Tanyi, J.L.; Hara, M.H.O.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; Mcgarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of t Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from Car T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef]
- Priceman, S.; Aguilar, B.; Starr, R.; Tilakawardane, D.; Park, A.; Gerdts, E.; Chang, W.; Wright, S.; Forman, S.J.; Brown, C.E. Development of HER2-Specific Chimeric Antigen Receptor T Cells for the Treatment of Breast-to-Brain Metastasis. J. Immunother. Cancer 2015, 3, P121. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-Specific T Cells Engineered to Coexpress Tumor-Specific Receptors: Persistence and Antitumor Activity in Individuals with Neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Jiang, H.; Shi, Z.; Wang, P.; Wang, C.; Yang, L.; Du, G.; Zhang, H.; Shi, B.; Jia, J.; Li, Q.; et al. Claudin18. 2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J. Natl. Cancer Inst. 2019, 111, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor Heterogeneity: Evolution through Space and Time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational Targeting Offsets Antigen Escape and Enhances Effector Functions of Adoptively Transferred T Cells in Glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef]
- Huang, B.; Zheng, S.; Sudarshan, K.; Mukkamala, R.; Srinivasarao, M.; Sardesai, T.; Yang, X.; Chu, H.; Low, P.S. Use of a Universal Targeting CAR T Cell to Simultaneously Kill Cancer Cells and Cancer-Associated Fibroblasts. Front. Immunol. 2025, 16, 1539265. [Google Scholar] [CrossRef]
- Yang, X.; Wang, G.X.; Zhou, J.F. CAR T Cell Therapy for Hematological Malignancies. Curr. Med. Sci. 2019, 39, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Pasqui, D.M.; Latorraca, C.d.O.C.; Pacheco, R.L.; Riera, R. CAR-T Cell Therapy for Patients with Hematological Malignancies. A Systematic Review. Eur. J. Haematol. 2022, 109, 601–618. [Google Scholar] [CrossRef]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK Cells Mediate Reduction of GVHD by Inhibiting Activated, Alloreactive T Cells While Retaining GVT Effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Tam, Y.K.; Miyagawa, B.; Ho, V.C.; Klingemann, H.G. Immunotherapy of Malignant Melanoma in a SCID Mouse Model Using the Highly Cytotoxic Natural Killer Cell Line NK-92. J. Hematother. 1999, 8, 281–290. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of Natural Killer Cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.; Bentele, M.; Kutle, I.; Zimmermann, K.; Lühmann, J.L.; Steinemann, D.; Kloess, S.; Koehl, U.; Roßberg, W.; Ahmed, A.; et al. CAR-NK Cells Targeting HER1 (EGFR) Show Efficient Anti-Tumor Activity against Head and Neck Squamous Cell Carcinoma (HNSCC). Cancers 2023, 15, 3169. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Chen, J.; Hou, W.; Chen, J.; Xiong, Y.; Li, H.; Qi, X.; Xu, H.; Xie, Z.; Li, M.; et al. Engineering a HER2-CAR-NK Cell Secreting Soluble Programmed Cell Death Protein with Superior Antitumor Efficacy. Int. J. Mol. Sci. 2023, 24, 6843. [Google Scholar] [CrossRef] [PubMed]
- Kutle, I.; Polten, R.; Stalp, J.L.; Hachenberg, J.; Todzey, F.; Hass, R.; Zimmermann, K.; von der Ohe, J.; von Kaisenberg, C.; Neubert, L.; et al. Anti-Mesothelin CAR-NK Cells as a Novel Targeted Therapy against Cervical Cancer. Front. Immunol. 2024, 15, 1485461. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, M.; Shen, X.; Xia, C.; Hu, F.; Huang, D.; Weng, Q.; Zhang, Q.; Liu, L.; Zhu, Y.; et al. Mesothelin CAR-Engineered NK Cells Derived from Human Embryonic Stem Cells Suppress the Progression of Human Ovarian Cancer in Animals. Cell Prolif. 2024, 57, e13727. [Google Scholar] [CrossRef]
- Ren, Y.; Xue, M.; Hui, X.; Liu, X.; Farooq, M.A.; Chen, Y.; Ji, Y.; Duan, Y.; Ajmal, I.; Yao, J.; et al. Chimeric Cytokine Receptor TGF-β RII/IL-21R Improves CAR-NK Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Gastric Cancer. Pharmacol. Res. 2025, 212, 107637. [Google Scholar] [CrossRef]
- Lin, X.; Sun, Y.; Dong, X.; Liu, Z.; Sugimura, R.; Xie, G. IPSC-Derived CAR-NK Cells for Cancer Immunotherapy. Biomed. Pharmacother. 2023, 165, 115123. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Rojas-Quintero, J.; Díaz, M.P.; Palmar, J.; Galan-Freyle, N.J.; Morillo, V.; Escalona, D.; González-Torres, H.J.; Torres, W.; Navarro-Quiroz, E.; Rivera-Porras, D.; et al. Car T Cells in Solid Tumors: Overcoming Obstacles. Int. J. Mol. Sci. 2024, 25, 4170. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical Lessons Learned from the First Leg of the CAR T Cell Journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ma, Y.; Li, Q.; Xu, Y.; Xue, Y.; Xu, S. CAR Macrophages: A Promising Novel Immunotherapy for Solid Tumors and Beyond. Biomark. Res. 2024, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and Regulators of the Immune System. Cell. Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Pierini, S.; Gabbasov, R.; Oliveira-Nunes, M.C.; Qureshi, R.; Worth, A.; Huang, S.; Nagar, K.; Griffin, C.; Lian, L.; Yashiro-Ohtani, Y.; et al. Chimeric Antigen Receptor Macrophages (CAR-M) Sensitize HER2+ Solid Tumors to PD1 Blockade in Pre-Clinical Models. Nat. Commun. 2025, 16, 706. [Google Scholar] [CrossRef]
- Bialasek, M.; Sun, M.; Taciak, B.; Gorczak, M.; Marszalek, I.; Osadchuk, O.; Kurpiel, D.; Lipinski, W.; Sas, Z.; Weller, M.; et al. Immunotherapy: Macrophage-Drug Conjugate As a Cell-Based Therapy for Glioblastoma. Cytotherapy 2023, 25, S215. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-Inducible Factors: Master Regulators of Hypoxic Tumor Immune Escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, X.; Qin, J.; Sun, Y.; Xiong, H.; Lin, B.; Liu, K.; Tan, B.; Zhang, C.; Huang, C.; et al. LIGHT/TNFSF14 Promotes CAR-T Cell Trafficking and Cytotoxicity through Reversing Immunosuppressive Tumor Microenvironment. Mol. Ther. 2023, 31, 2575–2590. [Google Scholar] [CrossRef]
- Ribatti, D. Tertiary Lymphoid Structures, a Historical Reappraisal. Tissue Cell 2024, 86, 102288. [Google Scholar] [CrossRef]
- He, B.; Johansson-Percival, A.; Backhouse, J.; Li, J.; Lee, G.Y.F.; Hamzah, J.; Ganss, R. Remodeling of Metastatic Vasculature Reduces Lung Colonization and Sensitizes Overt Metastases to Immunotherapy. Cell Rep. 2020, 30, 714–724.e5. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Jabouille, A.; Steri, V.; Johansson-Percival, A.; Michael, I.P.; Kotamraju, V.R.; Junckerstorff, R.; Nowak, A.K.; Hamzah, J.; Lee, G.; et al. Vascular Targeting of LIGHT Normalizes Blood Vessels in Primary Brain Cancer and Induces Intratumoural High Endothelial Venules. J. Pathol. 2018, 245, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.N.; Rathmell, J.C. Metabolic Programming and Immune Suppression in the Tumor Microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Bou-Puerto, A.; Carda, M.; Falomir, E. Styryl Carbamate Backbones for the Discovery of TME-Disrupting Agents. Results Chem. 2024, 7, 101372. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFV by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat. Biotechnol. 2018, 36, 847–858. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A Novel Chimeric Antigen Receptor Containing a JAK-STAT Signaling Domain Mediates Superior Antitumor Effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, Y.; Ji, Y.; Gao, Y.; Zhang, M.; Liu, Y.; Zhu, W.; Lu, P. Cytokine Release Syndrome After Modified CAR-NK Therapy in an Advanced Non-Small Cell Lung Cancer Patient: A Case Report. Cell Transplant. 2022, 31, 9636897221094244. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, Q.; Wang, F.; Zhang, C.; Xu, C.; Gu, A.; Zhong, W.H.; Hu, Y.; Zhong, X. Co-Expression IL-15 Receptor Alpha with IL-15 Reduces Toxicity via Limiting IL-15 Systemic Exposure during CAR-T Immunotherapy. J. Transl. Med. 2022, 20, 432. [Google Scholar] [CrossRef]
- Borgers, J.S.W.; Lenkala, D.; Kohler, V.; Jackson, E.K.; Linssen, M.D.; Hymson, S.; McCarthy, B.; O’Reilly Cosgrove, E.; Balogh, K.N.; Esaulova, E.; et al. Personalized, Autologous Neoantigen-Specific T Cell Therapy in Metastatic Melanoma: A Phase 1 Trial. Nat. Med. 2025, 31, 881–893. [Google Scholar] [CrossRef]
- DiNofia, A.M.; Grupp, S.A. Will Allogeneic CAR T Cells for CD19+ Malignancies Take Autologous CAR T Cells ‘off the Shelf’? Nat. Rev. Clin. Oncol. 2021, 18, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic Modification of Primary Natural Killer Cells Overcomes Inhibitory Signals and Induces Specific Killing of Leukemic Cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef]
- Kweon, S.; Phan, M.T.T.; Chun, S.; Yu, H.B.; Kim, J.; Kim, S.; Lee, J.; Ali, A.K.; Lee, S.H.; Kim, S.K.; et al. Expansion of Human NK Cells Using K562 Cells Expressing OX40 Ligand and Short Exposure to IL-21. Front. Immunol. 2019, 10, 879. [Google Scholar] [CrossRef]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Goldenson, B.H.; Hor, P.; Kaufman, D.S. IPSC-Derived Natural Killer Cell Therapies—Expansion and Targeting. Front. Immunol. 2022, 13, 841107. [Google Scholar] [CrossRef]
- Lei, T.; Wang, Y.; Zhang, Y.; Yang, Y.; Cao, J.; Huang, J.; Chen, J.; Chen, H.; Zhang, J.; Wang, L.; et al. Leveraging CRISPR Gene Editing Technology to Optimize the Efficacy, Safety and Accessibility of CAR T-Cell Therapy. Leukemia 2024, 38, 2517–2543. [Google Scholar] [CrossRef]
- Feng, X.; Li, Z.; Liu, Y.; Chen, D.; Zhou, Z. CRISPR/Cas9 Technology for Advancements in Cancer Immunotherapy: From Uncovering Regulatory Mechanisms to Therapeutic Applications. Exp. Hematol. Oncol. 2024, 13, 102. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cell Type | Trial Name/ID | ID | Cancer Type(s) | Phase |
|---|---|---|---|---|
| TIL | NCT05727904 | - | Advanced Melanoma | Phase 3 |
| NCT05361174 | - | Melanoma, NSCLC | Phase 1/2 | |
| NCT06481592 | - | Endometrial Cancer | Phase 2 | |
| NCT06060613 | - | Melanoma, NSCLC, Lung Cancer | Phase 1/2 | |
| NCT05470283 | - | Melanoma | Phase 1 | |
| TCR T | NCT04044768 | MAGE-A4 | Synovial Sarcoma | Phase 2 |
| NCT04526509 | NY-ESO-1/LAGE-1a | Various Solid Tumors | Phase 1 | |
| NCT04729543 | MAGE-C2 | Melanoma, HNSCC | Phase 1/2 | |
| NCT03912831 | HPV16 E7 | HPV-Associated Cancers | Phase 1 | |
| NCT02650986 | NY-ESO-1 | Melanoma, Synovial Sarcoma, Ovarian Carcinoma, Peritoneal Carcinoma | Phase 1/2 | |
| NCT00670748 | NY-ESO-1 | Various Solid Tumors | Phase 2 | |
| CAR T | NCT00910650 | MART-1 | Melanoma | Phase 2 |
| NCT04581473 | Claudin18.2 | Gastric, Pancreatic Cancers | Phase 1/2 | |
| NCT02208362 | IL13Rα2 | Glioblastoma | Phase 1 | |
| NCT04196413 | GD2 | Diffuse Midline Gliomas | Phase 1 | |
| NCT02706392 | ROR1 | Triple-Negative Breast Cancer, NSCLC | Phase 1 | |
| NCT01869166 | EGFR | Advanced EGFR-positive Solid Tumors | Phase 1/2 | |
| NCT02349724 | CEA | Colorectal, Lung, Gastric, Breast, Pancreatic Cancers | Phase 1 | |
| NCT02159716 | Mesothelin | Pancreatic, Ovarian, Mesothelioma | Phase 1 | |
| NCT05239143 | MUC1 | Breast, Ovarian, Pancreatic, Colorectal, Gastric Cancers, NSCLC, HNSCC | Phase 1 | |
| NCT02541370 | CD133 | Liver, Pancreatic, Brain, Breast, Ovarian, Colorectal Cancers, Hepatocellular Carcinoma | Phase 1/2 | |
| NCT04897321 | B7-H3 | Pediatric Solid Tumors | Phase 1 | |
| CAR NK | NCT06066424 | TROP2 | Advanced Solid Tumors | Phase 1 |
| NCT05410717 | Claudin6, GPC3, Mesothelin, AXL | Ovarian, Endometrial, Urologic Cancers | Phase 1 | |
| NCT06572956 | Various (CAR-T/CAR-NK) | Pancreatic, Prostate, Breast, Glioma | Early Phase 1 | |
| CAR M | NCT04660929 | HER2 | HER2-overexpressing Solid Tumors | Phase 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smolarska, A.; Kokoszka, Z.; Naliwajko, M.; Strupczewska, J.; Tondera, J.; Wiater, M.; Orzechowska, R. Cell-Based Therapies for Solid Tumors: Challenges and Advances. Int. J. Mol. Sci. 2025, 26, 5524. https://doi.org/10.3390/ijms26125524
Smolarska A, Kokoszka Z, Naliwajko M, Strupczewska J, Tondera J, Wiater M, Orzechowska R. Cell-Based Therapies for Solid Tumors: Challenges and Advances. International Journal of Molecular Sciences. 2025; 26(12):5524. https://doi.org/10.3390/ijms26125524
Chicago/Turabian StyleSmolarska, Anna, Zuzanna Kokoszka, Marcelina Naliwajko, Julia Strupczewska, Jędrzej Tondera, Maja Wiater, and Roksana Orzechowska. 2025. "Cell-Based Therapies for Solid Tumors: Challenges and Advances" International Journal of Molecular Sciences 26, no. 12: 5524. https://doi.org/10.3390/ijms26125524
APA StyleSmolarska, A., Kokoszka, Z., Naliwajko, M., Strupczewska, J., Tondera, J., Wiater, M., & Orzechowska, R. (2025). Cell-Based Therapies for Solid Tumors: Challenges and Advances. International Journal of Molecular Sciences, 26(12), 5524. https://doi.org/10.3390/ijms26125524