
Macrophages at the Crossroads of Chronic Stress and Cancer

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Macrophages as Orchestrators of Tissue Homeostasis
3. Macrophages in Chronic Inflammation
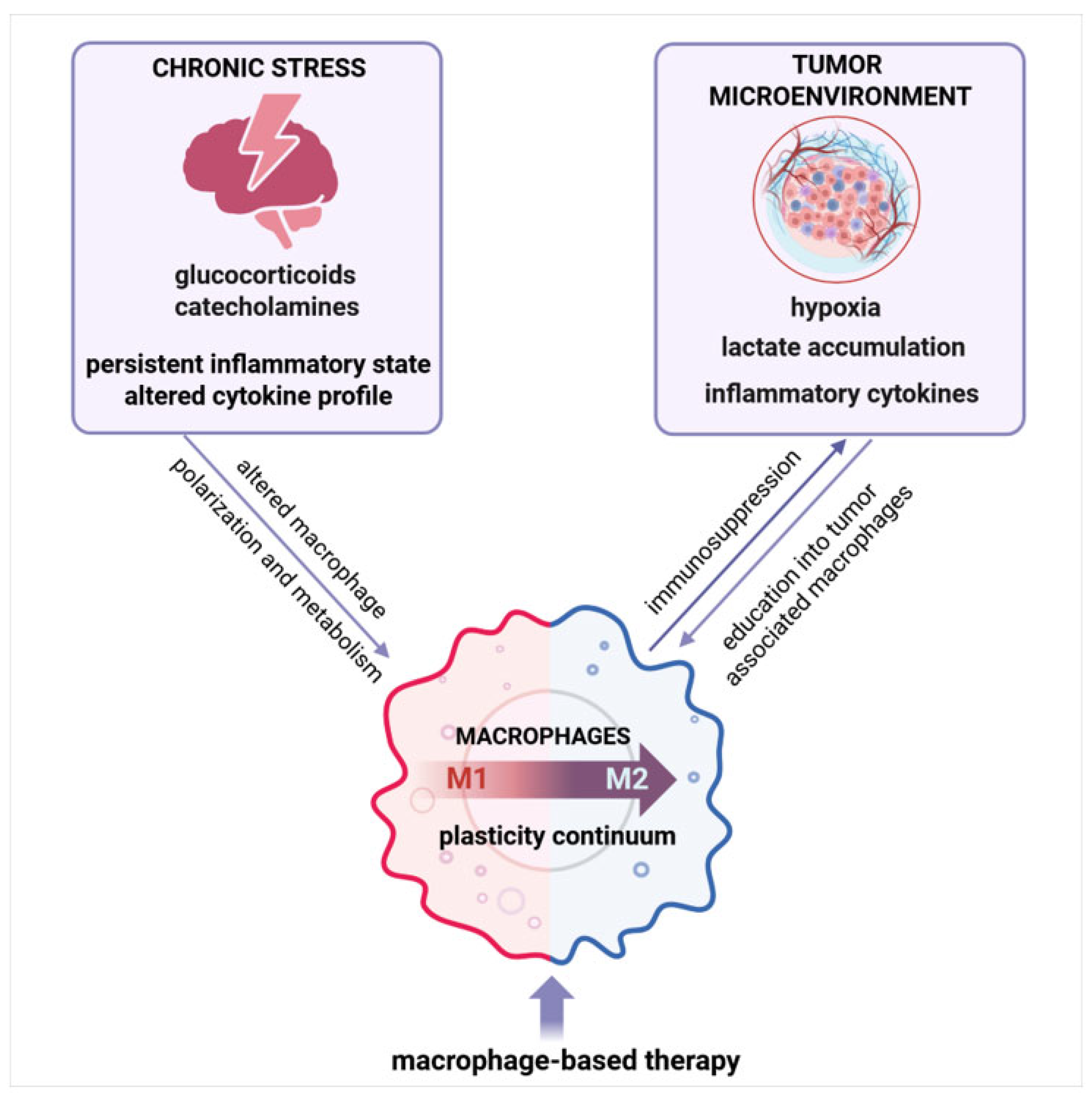
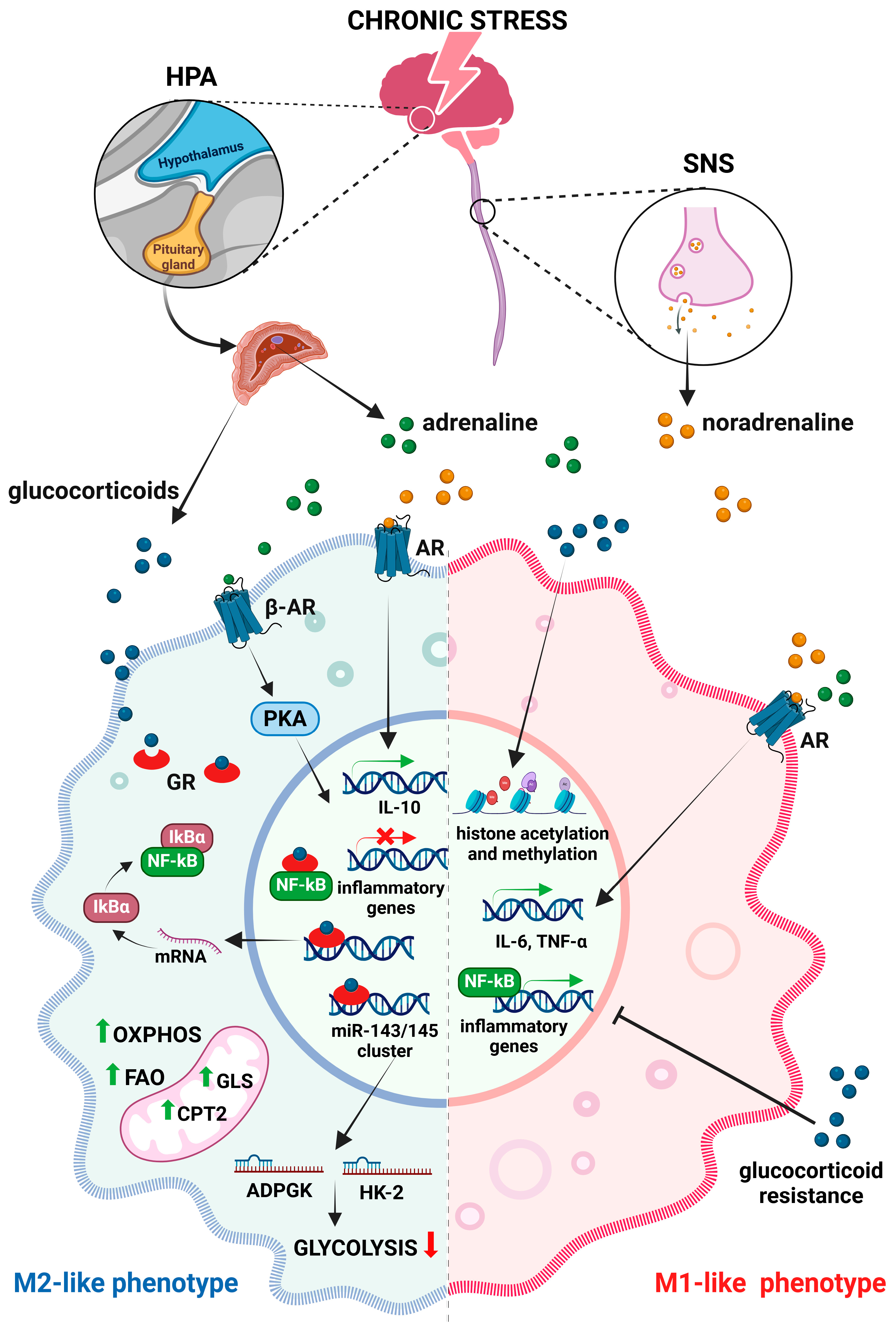
4. Chronic Stress and Macrophages
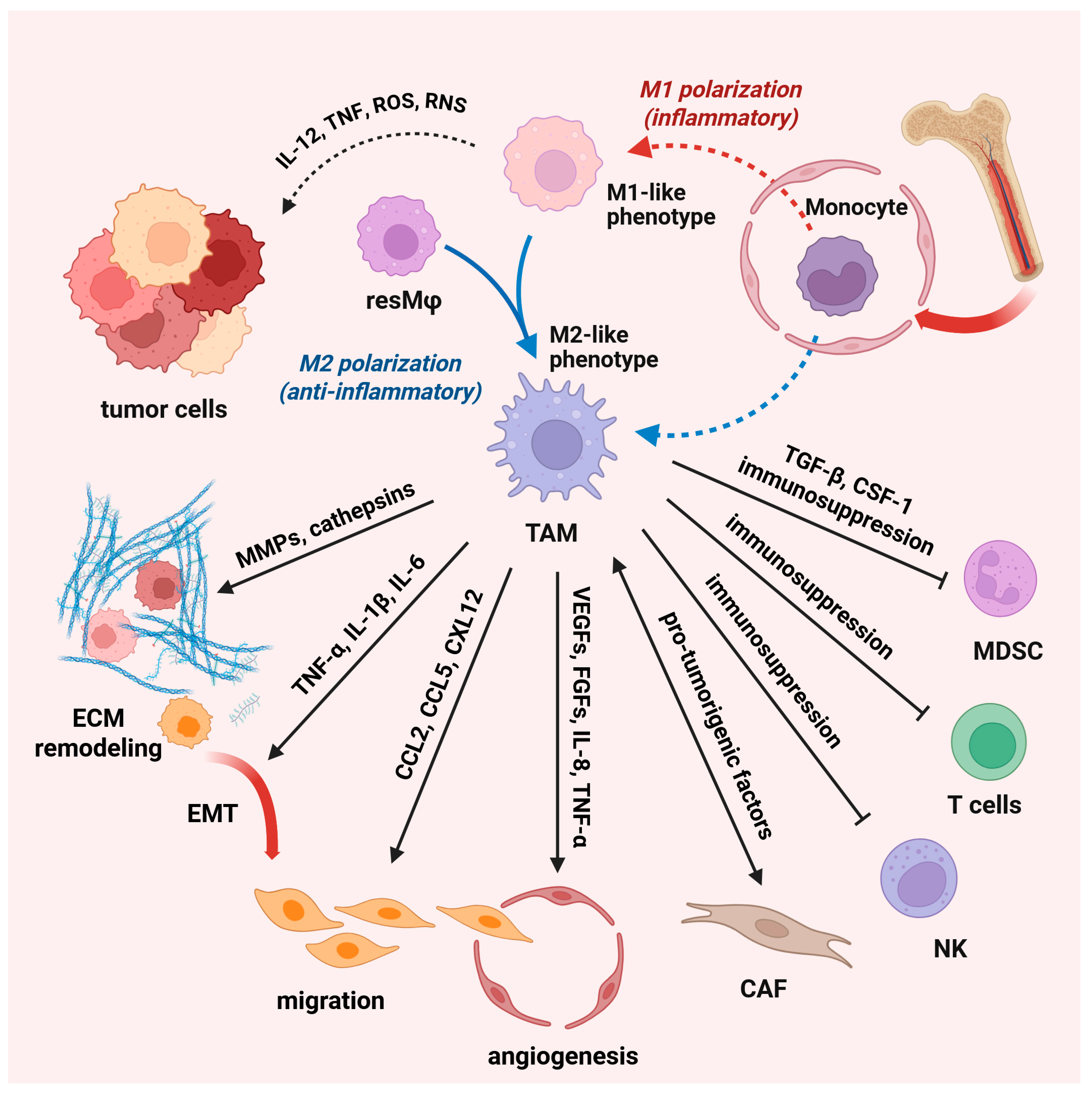
5. Macrophages Are Critical Players in Tumor Development and Progression
6. The Role of TAM in Cancer Immunotherapy
7. Targeting TAM-Associated Modulation of the Tumor Microenvironment: Focus on Their Functional Reprogramming
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage Biology in Development, Homeostasis and Disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Meizlish, M.L.; Franklin, R.A.; Zhou, X.; Medzhitov, R. Tissue Homeostasis and Inflammation. Annu. Rev. Immunol. 2021, 39, 557–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Labzin, L.I. Inflammatory Cell Death: How Macrophages Sense Neighbouring Cell Infection and Damage. Biochem. Soc. Trans. 2023, 51, 303–313. [Google Scholar] [CrossRef]
- Paterson, N.; Lämmermann, T. Macrophage Network Dynamics Depend on Haptokinesis for Optimal Local Surveillance. eLife 2022, 11, e75354. [Google Scholar] [CrossRef] [PubMed]
- Taghdiri, N.; Calcagno, D.M.; Fu, Z.; Huang, K.; Kohler, R.H.; Weissleder, R.; Coleman, T.P.; King, K.R. Macrophage Calcium Reporter Mice Reveal Immune Cell Communication In Vitro and In Vivo. Cell Rep. Methods 2021, 1, 100132. [Google Scholar] [CrossRef]
- Kulle, A.; Thanabalasuriar, A.; Cohen, T.S.; Szydlowska, M. Resident Macrophages of the Lung and Liver: The Guardians of Our Tissues. Front. Immunol. 2022, 13, 1029085. [Google Scholar] [CrossRef]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils Promote the Development of Reparative Macrophages Mediated by ROS to Orchestrate Liver Repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef]
- Franklin, R.A. Fibroblasts and Macrophages: Collaborators in Tissue Homeostasis. Immunol. Rev. 2021, 302, 86–103. [Google Scholar] [CrossRef]
- Gurevich, D.B.; Severn, C.E.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live Imaging of Wound Angiogenesis Reveals Macrophage Orchestrated Vessel Sprouting and Regression. EMBO J. 2018, 37, e97786. [Google Scholar] [CrossRef]
- Mass, E.; Nimmerjahn, F.; Kierdorf, K.; Schlitzer, A. Tissue-Specific Macrophages: How They Develop and Choreograph Tissue Biology. Nat. Rev. Immunol. 2023, 23, 563–579. [Google Scholar] [CrossRef]
- Park, M.D.; Silvin, A.; Ginhoux, F.; Merad, M. Macrophages in health and disease. Cell 2022, 185, 4259–4279. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Dick, S.A.; Wong, A.; Hamidzada, H.; Nejat, S.; Nechanitzky, R.; Vohra, S.; Mueller, B.; Zaman, R.; Kantores, C.; Aronoff, L.; et al. Three Tissue Resident Macrophage Subsets Coexist across Organs with Conserved Origins and Life Cycles. Sci. Immunol. 2022, 7, eabf7777. [Google Scholar] [CrossRef]
- Guilliams, M.; Svedberg, F.R. Does Tissue Imprinting Restrict Macrophage Plasticity? Nat. Immunol. 2021, 22, 118–127. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 Polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Li, P.; Ma, C.; Li, J.; You, S.; Dang, L.; Wu, J.; Hao, Z.; Li, J.; Zhi, Y.; Chen, L.; et al. Proteomic Characterization of Four Subtypes of M2 Macrophages Derived from Human THP-1 Cells. J. Zhejiang Univ.-Sci. B 2022, 23, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Zhang, Z.; Dayyani, F.; Zhang, Z.; Yaghmai, V.; Choi, A.; Valerin, J.; Imagawa, D.; Abi-Jaoudeh, N. Modulation of Tumor-Associated Macrophages to Overcome Immune Suppression in the Hepatocellular Carcinoma Microenvironment. Cancers 2024, 17, 66. [Google Scholar] [CrossRef]
- Peng, Y.; Zhou, M.; Yang, H.; Qu, R.; Qiu, Y.; Hao, J.; Bi, H.; Guo, D. Regulatory Mechanism of M1/M2 Macrophage Polarization in the Development of Autoimmune Diseases. Mediators Inflamm. 2023, 2023, 8821610. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Wu, H.; Rong, X.; Guo, J. M2b Macrophage Polarization and Its Roles in Diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 Macrophages and Their Overlaps—Myth or Reality? Clin. Sci. 2023, 137, 1067–1093. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Ma, R.-Y.; Black, A.; Qian, B.-Z. Macrophage Diversity in Cancer Revisited in the Era of Single-Cell Omics. Trends Immunol. 2022, 43, 546–563. [Google Scholar] [CrossRef]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Luo, F.; Yan, T. Transcription Factor KLF4 Regulated STAT1 to Promote M1 Polarization of Macrophages in Rheumatoid Arthritis. Aging 2022, 14, 5669–5680. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Zhang, Z.; Cheng, C.; Tang, S.; Zhai, M.; Li, L.; Wei, F.; Ding, G. Small Extracellular Vesicles from Periodontal Ligament Stem Cells Primed by Lipopolysaccharide Regulate Macrophage M1 Polarization via miR-433-3p Targeting TLR2/TLR4/NF-κB. Inflammation 2023, 46, 1849–1858. [Google Scholar] [CrossRef]
- Lu, J.; Xie, L.; Liu, C.; Zhang, Q.; Sun, S. PTEN/PI3k/AKT Regulates Macrophage Polarization in Emphysematous Mice. Scand. J. Immunol. 2017, 85, 395–405. [Google Scholar] [CrossRef]
- Geng, K.; Ma, X.; Jiang, Z.; Gu, J.; Huang, W.; Wang, W.; Xu, Y.; Xu, Y. WDR74 Facilitates TGF-β/Smad Pathway Activation to Promote M2 Macrophage Polarization and Diabetic Foot Ulcer Wound Healing in Mice. Cell Biol. Toxicol. 2023, 39, 1577–1591. [Google Scholar] [CrossRef]
- Li, G.-S.; Cui, L.; Wang, G.-D. miR-155-5p Regulates Macrophage M1 Polarization and Apoptosis in the Synovial Fluid of Patients with Knee Osteoarthritis. Exp. Ther. Med. 2020, 21, 68. [Google Scholar] [CrossRef]
- Torres, M.; Wang, J.; Yannie, P.J.; Ghosh, S.; Segal, R.A.; Reynolds, A.M. Identifying Important Parameters in the Inflammatory Process with a Mathematical Model of Immune Cell Influx and Macrophage Polarization. PLOS Comput. Biol. 2019, 15, e1007172. [Google Scholar] [CrossRef]
- Hu, J.; Huang, S.; Liu, X.; Zhang, Y.; Wei, S.; Hu, X. miR-155: An Important Role in Inflammation Response. J. Immunol. Res. 2022, 2022, 7437281. [Google Scholar] [CrossRef] [PubMed]
- Gronau, L.; Duecker, R.P.; Jerkic, S.-P.; Eickmeier, O.; Trischler, J.; Chiocchetti, A.G.; Blumchen, K.; Zielen, S.; Schubert, R. Dual Role of microRNA-146a in Experimental Inflammation in Human Pulmonary Epithelial and Immune Cells and Expression in Inflammatory Lung Diseases. Int. J. Mol. Sci. 2024, 25, 7686. [Google Scholar] [CrossRef]
- Alotiby, A. Immunology of Stress: A Review Article. J. Clin. Med. 2024, 13, 6394. [Google Scholar] [CrossRef]
- Ehrchen, J.M.; Roth, J.; Barczyk-Kahlert, K. More Than Suppression: Glucocorticoid Action on Monocytes and Macrophages. Front. Immunol. 2019, 10, 2028. [Google Scholar] [CrossRef]
- Dohi, A.; Noguchi, T.; Yamashita, M.; Sasaguri, K.; Yamamoto, T.; Mori, Y. Acute Stress Transiently Activates Macrophages and Chemokines in Cervical Lymph Nodes. Immunol. Res. 2024, 72, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Kokkosis, A.G.; Madeira, M.M.; Hage, Z.; Valais, K.; Koliatsis, D.; Resutov, E.; Tsirka, S.E. Chronic Psychosocial Stress Triggers Microglial-/Macrophage-induced Inflammatory Responses Leading to Neuronal Dysfunction and Depressive-related Behavior. Glia 2024, 72, 111–132. [Google Scholar] [CrossRef]
- Vignjević Petrinović, S.; Milošević, M.S.; Marković, D.; Momčilović, S. Interplay between Stress and Cancer—A Focus on Inflammation. Front. Physiol. 2023, 14, 1119095. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Vikramdeo, K.S.; Sudan, S.K.; Anand, S.; Deshmukh, S.K.; Singh, A.P.; Singh, S. Cortisol Affects Macrophage Polarization by Inducing miR-143/145 Cluster to Reprogram Glucose Metabolism and by Promoting TCA Cycle Anaplerosis. J. Biol. Chem. 2024, 300, 107753. [Google Scholar] [CrossRef]
- Sun, J.-X.; Xu, X.-H.; Jin, L. Effects of Metabolism on Macrophage Polarization Under Different Disease Backgrounds. Front. Immunol. 2022, 13, 880286. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in Biology and Targeted Therapy: New Insights and Translational Implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Niraula, A.; Wang, Y.; Godbout, J.P.; Sheridan, J.F. Corticosterone Production during Repeated Social Defeat Causes Monocyte Mobilization from the Bone Marrow, Glucocorticoid Resistance, and Neurovascular Adhesion Molecule Expression. J. Neurosci. 2018, 38, 2328–2340. [Google Scholar] [CrossRef] [PubMed]
- Deochand, D.K.; Dacic, M.; Bale, M.J.; Daman, A.W.; Chaudhary, V.; Josefowicz, S.Z.; Oliver, D.; Chinenov, Y.; Rogatsky, I. Mechanisms of Epigenomic and Functional Convergence between Glucocorticoid- and IL4-Driven Macrophage Programming. Nat. Commun. 2024, 15, 9000. [Google Scholar] [CrossRef] [PubMed]
- Freire, B.M.; De Melo, F.M.; Basso, A.S. Adrenergic Signaling Regulation of Macrophage Function: Do We Understand It Yet? Immunother. Adv. 2022, 2, ltac010. [Google Scholar] [CrossRef]
- Dragan, P.; Latek, D. The Two-Sided Impact of Beta-Adrenergic Receptor Ligands on Inflammation. Curr. Opin. Physiol. 2024, 41, 100779. [Google Scholar] [CrossRef]
- Ağaç, D.; Estrada, L.D.; Maples, R.; Hooper, L.V.; Farrar, J.D. The Β2-Adrenergic Receptor Controls Inflammation by Driving Rapid IL-10 Secretion. Brain. Behav. Immun. 2018, 74, 176–185. [Google Scholar] [CrossRef]
- Hu, D.; Wan, L.; Chen, M.; Caudle, Y.; LeSage, G.; Li, Q.; Yin, D. Essential Role of IL-10/STAT3 in Chronic Stress-Induced Immune Suppression. Brain. Behav. Immun. 2014, 36, 118–127. [Google Scholar] [CrossRef]
- McKim, D.B.; Yin, W.; Wang, Y.; Cole, S.W.; Godbout, J.P.; Sheridan, J.F. Social Stress Mobilizes Hematopoietic Stem Cells to Establish Persistent Splenic Myelopoiesis. Cell Rep. 2018, 25, 2552–2562.e3. [Google Scholar] [CrossRef]
- Momčilović, S.; Bogdanović, A.; Milošević, M.S.; Mojsilović, S.; Marković, D.C.; Kočović, D.M.; Vignjević Petrinović, S. Macrophages Provide Essential Support for Erythropoiesis, and Extracellular ATP Contributes to a Erythropoiesis-Supportive Microenvironment during Repeated Psychological Stress. Int. J. Mol. Sci. 2023, 24, 11373. [Google Scholar] [CrossRef] [PubMed]
- Vignjević Petrinović, S.; Budeč, M.; Marković, D.; Mitrović Ajtić, O.; Jovčić, G.; Milošević, M.; Momčilović, S.; Čokić, V. Nitric Oxide-Dependent Expansion of Erythroid Progenitors in a Murine Model of Chronic Psychological Stress. Histochem. Cell Biol. 2020, 153, 457–468. [Google Scholar] [CrossRef]
- Vignjević Petrinović, S.; Budeč, M.; Marković, D.; Gotić, M.; Mitrović Ajtić, O.; Mojsilović, S.; Stošić-Grujičić, S.; Ivanov, M.; Jovčić, G.; Čokić, V. Macrophage Migration Inhibitory Factor Is an Endogenous Regulator of Stress-Induced Extramedullary Erythropoiesis. Histochem. Cell Biol. 2016, 146, 311–324. [Google Scholar] [CrossRef]
- Barrett, T.J.; Corr, E.M.; Van Solingen, C.; Schlamp, F.; Brown, E.J.; Koelwyn, G.J.; Lee, A.H.; Shanley, L.C.; Spruill, T.M.; Bozal, F.; et al. Chronic Stress Primes Innate Immune Responses in Mice and Humans. Cell Rep. 2021, 36, 109595. [Google Scholar] [CrossRef]
- Yang, J.; Wei, W.; Zhang, S.; Jiang, W. Chronic Stress Influences the Macrophage M1-M2 Polarization Balance through β-Adrenergic Signaling in Hepatoma Mice. Int. Immunopharmacol. 2024, 138, 112568. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- De Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Zhu, S.; Yi, M.; Wu, Y.; Dong, B.; Wu, K. Roles of Tumor-Associated Macrophages in Tumor Progression: Implications on Therapeutic Strategies. Exp. Hematol. Oncol. 2021, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-Associated Macrophages: An Effective Player of the Tumor Microenvironment. Front. Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef] [PubMed]
- Bied, M.; Ho, W.W.; Ginhoux, F.; Blériot, C. Roles of Macrophages in Tumor Development: A Spatiotemporal Perspective. Cell. Mol. Immunol. 2023, 20, 983–992. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Xia, H.; Chen, Y.H. Monocytes in Tumorigenesis and Tumor Immunotherapy. Cells 2023, 12, 1673. [Google Scholar] [CrossRef]
- Cui, B.; Luo, Y.; Tian, P.; Peng, F.; Lu, J.; Yang, Y.; Su, Q.; Liu, B.; Yu, J.; Luo, X.; et al. Stress-Induced Epinephrine Enhances Lactate Dehydrogenase A and Promotes Breast Cancer Stem-like Cells. J. Clin. Investig. 2019, 129, 1030–1046. [Google Scholar] [CrossRef]
- Hong, H.; Ji, M.; Lai, D. Chronic Stress Effects on Tumor: Pathway and Mechanism. Front. Oncol. 2021, 11, 738252. [Google Scholar] [CrossRef]
- Chi, J.; Gao, Q.; Liu, D. Tissue-Resident Macrophages in Cancer: Friend or Foe? Cancer Med. 2024, 13, e70387. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Tian, Y.; Lv, C. Decoding the Spatiotemporal Heterogeneity of Tumor-Associated Macrophages. Mol. Cancer 2024, 23, 150. [Google Scholar] [CrossRef]
- Lee, S.G.; Woo, S.M.; Seo, S.U.; Lee, C.-H.; Baek, M.-C.; Jang, S.H.; Park, Z.Y.; Yook, S.; Nam, J.-O.; Kwon, T.K. Cathepsin D Promotes Polarization of Tumor-Associated Macrophages and Metastasis Through TGFBI-CCL20 Signaling. Exp. Mol. Med. 2024, 56, 383–394. [Google Scholar] [CrossRef]
- Opzoomer, J.W.; Anstee, J.E.; Dean, I.; Hill, E.J.; Bouybayoune, I.; Caron, J.; Muliaditan, T.; Gordon, P.; Sosnowska, D.; Nuamah, R.; et al. Macrophages Orchestrate the Expansion of a Proangiogenic Perivascular Niche During Cancer Progression. Sci. Adv. 2021, 7, eabg9518. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-Education of Macrophages as a Therapeutic Strategy in Cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.P.; Tang, B.; Wang, Y.; Duran, C.L.; Karagiannis, G.S.; Xue, E.A.; Entenberg, D.; Borriello, L.; Coste, A.; Eddy, R.J.; et al. Live Tumor Imaging Shows Macrophage Induction and TMEM-Mediated Enrichment of Cancer Stem Cells during Metastatic Dissemination. Nat. Commun. 2021, 12, 7300. [Google Scholar] [CrossRef]
- Chen, W.; Chen, M.; Hong, L.; Xiahenazi, A.; Huang, M.; Tang, N.; Yang, X.; She, F.; Chen, Y. M2-like Tumor-Associated Macrophage-Secreted CCL2 Facilitates Gallbladder Cancer Stemness and Metastasis. Exp. Hematol. Oncol. 2024, 13, 83. [Google Scholar] [CrossRef]
- Friedman-DeLuca, M.; Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H.; Entenberg, D. Macrophages in Tumor Cell Migration and Metastasis. Front. Immunol. 2024, 15, 1494462. [Google Scholar] [CrossRef]
- Zhong, Q.; Fang, Y.; Lai, Q.; Wang, S.; He, C.; Li, A.; Liu, S.; Yan, Q. CPEB3 Inhibits Epithelial-Mesenchymal Transition by Disrupting the Crosstalk between Colorectal Cancer Cells and Tumor-Associated Macrophages via IL-6R/STAT3 Signaling. J. Exp. Clin. Cancer Res. 2020, 39, 132. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, W.; Wang, J.; Si, T.; Xing, W. Tumor-associated macrophage-derived transforming growth factor-β promotes colorectal cancer progression through HIF1-TRIB3 signaling. Cancer Sci. 2021, 112, 4198–4207. [Google Scholar] [CrossRef]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leucoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef]
- Cao, P.; Sun, Z.; Zhang, F.; Zhang, J.; Zheng, X.; Yu, B.; Zhao, Y.; Wang, W. TGF-β Enhances Immunosuppression of Myeloid-Derived Suppressor Cells to Induce Transplant Immune Tolerance Through Affecting Arg-1 Expression. Front. Immunol. 2022, 13, 919674. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol. Cell. Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef]
- Andersen, M.H. Novel Immunotherapeutic Combinations Moving Forward: The Modulation of the Immunosuppressive Microenvironment. Semin. Immunopathol. 2023, 45, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Kzhyshkowska, J.; Shen, J.; Larionova, I. Targeting of TAMs: Can We Be More Clever than Cancer Cells? Cell. Mol. Immunol. 2024, 21, 1376–1409. [Google Scholar] [CrossRef]
- Xu, S.; Wang, C.; Yang, L.; Wu, J.; Li, M.; Xiao, P.; Xu, Z.; Xu, Y.; Wang, K. Targeting Immune Checkpoints on Tumor-Associated Macrophages in Tumor Immunotherapy. Front. Immunol. 2023, 14, 1199631. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, M.; Wang, J.; Fang, S. CAR-Macrophage versus CAR-T for Solid Tumors: The Race between a Rising Star and a Superstar. Biomol. Biomed. 2024, 24, 465–476. [Google Scholar] [CrossRef]
- Kirthiga Devi, S.S.; Singh, S.; Joga, R.; Patil, S.Y.; Meghana Devi, V.; Chetan Dushantrao, S.; Dwivedi, F.; Kumar, G.; Kumar Jindal, D.; Singh, C.; et al. Enhancing Cancer Immunotherapy: Exploring Strategies to Target the PD-1/PD-L1 Axis and Analyzing the Associated Patent, Regulatory, and Clinical Trial Landscape. Eur. J. Pharm. Biopharm. 2024, 200, 114323. [Google Scholar] [CrossRef]
- Taefehshokr, N.; Baradaran, B.; Baghbanzadeh, A.; Taefehshokr, S. Promising Approaches in Cancer Immunotherapy. Immunobiology 2020, 225, 151875. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune Checkpoint Blockade Therapy for Cancer: An Overview of FDA-Approved Immune Checkpoint Inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Jalil, A.R.; Andrechak, J.C.; Discher, D.E. Macrophage Checkpoint Blockade: Results from Initial Clinical Trials, Binding Analyses, and CD47-SIRPα Structure–Function. Antib. Ther. 2020, 3, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Kono, Y.; Saito, H.; Miyauchi, W.; Shimizu, S.; Murakami, Y.; Shishido, Y.; Miyatani, K.; Matsunaga, T.; Fukumoto, Y.; Nakayama, Y.; et al. Increased PD-1-Positive Macrophages in the Tissue of Gastric Cancer Are Closely Associated with Poor Prognosis in Gastric Cancer Patients. BMC Cancer 2020, 20, 175. [Google Scholar] [CrossRef] [PubMed]
- Diskin, B.; Adam, S.; Cassini, M.F.; Sanchez, G.; Liria, M.; Aykut, B.; Buttar, C.; Li, E.; Sundberg, B.; Salas, R.D.; et al. PD-L1 Engagement on T Cells Promotes Self-Tolerance and Suppression of Neighboring Macrophages and Effector T Cells in Cancer. Nat. Immunol. 2020, 21, 442–454. [Google Scholar] [CrossRef]
- Park, D.-J.; Sung, P.-S.; Lee, G.-W.; Cho, S.-W.; Kim, S.-M.; Kang, B.-Y.; Hur, W.-H.; Yang, H.; Lee, S.-K.; Lee, S.-H.; et al. Preferential Expression of Programmed Death Ligand 1 Protein in Tumor-Associated Macrophages and Its Potential Role in Immunotherapy for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 4710. [Google Scholar] [CrossRef]
- Jelinic, P.; Ricca, J.; Van Oudenhove, E.; Olvera, N.; Merghoub, T.; Levine, D.A.; Zamarin, D. Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. JNCI J. Natl. Cancer Inst. 2018, 110, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Guo, X.; Wang, G.; Bi, Y.; Han, L.; Zhu, Q.; Qiu, C.; Tanaka, M.; Zhao, Y. Breast Cancer Cells Promote CD169+ Macrophage-Associated Immunosuppression through JAK2-Mediated PD-L1 Upregulation on Macrophages. Int. Immunopharmacol. 2020, 78, 106012. [Google Scholar] [CrossRef]
- Liu, Y.; Zugazagoitia, J.; Ahmed, F.S.; Henick, B.S.; Gettinger, S.N.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Immune Cell PD-L1 Colocalizes with Macrophages and Is Associated with Outcome in PD-1 Pathway Blockade Therapy. Clin. Cancer Res. 2020, 26, 970–977. [Google Scholar] [CrossRef]
- Petty, A.J.; Dai, R.; Lapalombella, R.; Baiocchi, R.A.; Benson, D.M.; Li, Z.; Huang, X.; Yang, Y. Hedgehog-Induced PD-L1 on Tumor-Associated Macrophages Is Critical for Suppression of Tumor-Infiltrating CD8+ T Cell Function. JCI Insight 2021, 6, e146707. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, H.; Chen, B.; Liu, X.; Zhang, S.; Zong, Z.; Gao, M. PD-L1-Mediated Immunosuppression in Glioblastoma Is Associated With the Infiltration and M2-Polarization of Tumor-Associated Macrophages. Front. Immunol. 2020, 11, 588552. [Google Scholar] [CrossRef]
- Wang, L.; Guo, W.; Guo, Z.; Yu, J.; Tan, J.; Simons, D.L.; Hu, K.; Liu, X.; Zhou, Q.; Zheng, Y.; et al. PD-L1-Expressing Tumor-Associated Macrophages Are Immunostimulatory and Associate with Good Clinical Outcome in Human Breast Cancer. Cell Rep. Med. 2024, 5, 101420. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage–Mediated Resistance Pathway in Anti–PD-1 Therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, L.; Li, Z.; Zhang, W.; Luo, F.; Chu, Y.; Chen, G. Glycocalyx-Mimicking Nanoparticles Improve Anti-PD-L1 Cancer Immunotherapy through Reversion of Tumor-Associated Macrophages. Biomacromolecules 2018, 19, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Luo, X.; Zhou, Q.; Gong, H.; Gao, H.; Liu, T.; Chen, J.; Liang, L.; Kurihara, H.; Li, Y.-F.; et al. The Disbalance of LRP1 and SIRPα by Psychological Stress Dampens the Clearance of Tumor Cells by Macrophages. Acta Pharm. Sin. B 2022, 12, 197–209. [Google Scholar] [CrossRef]
- Qu, T.; Li, B.; Wang, Y. Targeting CD47/SIRPα as a Therapeutic Strategy, Where We Are and Where We Are Headed. Biomark. Res. 2022, 10, 20. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.H.; Tsai, R.K.; Dooling, L.J.; Kadu, S.; Lee, J.Y.; Pantano, D.; Rodriguez, P.L.; Subramanian, S.; Shin, J.-W.; Discher, D.E. Macrophages Show Higher Levels of Engulfment After Disruption of Cis Interactions Between CD47 and the Checkpoint Receptor SIRPα. J. Cell Sci. 2020, 133, jcs237800. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, L.; Xu, Y.; Liang, L.; Liu, L.; Chen, X.; Li, H.; Liu, H. The Progress and Prospects of Targeting the Adenosine Pathway in Cancer Immunotherapy. Biomark. Res. 2025, 13, 75. [Google Scholar] [CrossRef]
- Chen, S.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Matei, D.E.; Zhang, Y.; Zhang, B. Cd73: An Emerging Checkpoint for Cancer Immunotherapy. Immunotherapy 2019, 11, 983–997. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 Signalling Through Macrophage Siglec-10 Is a Target for Cancer Immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically Engineered T Cells for Cancer Immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef]
- Huang, T.; Bei, C.; Hu, Z.; Li, Y. CAR-Macrophage: Breaking New Ground in Cellular Immunotherapy. Front. Cell Dev. Biol. 2024, 12, 1464218. [Google Scholar] [CrossRef] [PubMed]
- Abdin, S.M.; Paasch, D.; Lachmann, N. CAR Macrophages on a Fast Track to Solid Tumor Therapy. Nat. Immunol. 2024, 25, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.J.A.; Monnikhof, M.; Meeldijk, J.; Koorman, T.; Bovenschen, N. Chimeric Antigen Receptor-Macrophages: Emerging next-Generation Cell Therapy for Brain Cancer. Neuro-Oncol. Adv. 2025, 7, vdaf059. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, S.-R.; Yu, Z.-L. Targeting the Lipid Metabolic Reprogramming of Tumor-Associated Macrophages: A Novel Insight into Cancer Immunotherapy. Cell. Oncol. 2024, 47, 415–428. [Google Scholar] [CrossRef]
- Reiss, K.A.; Angelos, M.G.; Dees, E.C.; Yuan, Y.; Ueno, N.T.; Pohlmann, P.R.; Johnson, M.L.; Chao, J.; Shestova, O.; Serody, J.S.; et al. CAR-Macrophage Therapy for HER2-Overexpressing Advanced Solid Tumors: A Phase 1 Trial. Nat. Med. 2025, 31, 1171–1182. [Google Scholar] [CrossRef]
- Lim, C.Y.; Chang, J.H.; Lee, W.S.; Kim, J.; Park, I.Y. CD40 Agonists Alter the Pancreatic Cancer Microenvironment by Shifting the Macrophage Phenotype toward M1 and Suppress Human Pancreatic Cancer in Organotypic Slice Cultures. Gut Liver 2022, 16, 645–659. [Google Scholar] [CrossRef]
- Chakraborty, S.; Ye, J.; Wang, H.; Sun, M.; Zhang, Y.; Sang, X.; Zhuang, Z. Application of Toll-like Receptors (TLRs) and Their Agonists in Cancer Vaccines and Immunotherapy. Front. Immunol. 2023, 14, 1227833. [Google Scholar] [CrossRef]
- Zeng, W.; Li, F.; Jin, S.; Ho, P.-C.; Liu, P.-S.; Xie, X. Functional Polarization of Tumor-Associated Macrophages Dictated by Metabolic Reprogramming. J. Exp. Clin. Cancer Res. 2023, 42, 245. [Google Scholar] [CrossRef]
- Qian, Y.; Yin, Y.; Zheng, X.; Liu, Z.; Wang, X. Metabolic Regulation of Tumor-Associated Macrophage Heterogeneity: Insights into the Tumor Microenvironment and Immunotherapeutic Opportunities. Biomark. Res. 2024, 12, 1. [Google Scholar] [CrossRef]
- Wang, S.; Liu, G.; Li, Y.; Pan, Y. Metabolic Reprogramming Induces Macrophage Polarization in the Tumor Microenvironment. Front. Immunol. 2022, 13, 840029. [Google Scholar] [CrossRef]
- Praharaj, M.; Shen, F.; Lee, A.J.; Zhao, L.; Nirschl, T.R.; Theodros, D.; Singh, A.K.; Wang, X.; Adusei, K.M.; Lombardo, K.A.; et al. Metabolic Reprogramming of Tumor-Associated Macrophages Using Glutamine Antagonist JHU083 Drives Tumor Immunity in Myeloid-Rich Prostate and Bladder Cancers. Cancer Immunol. Res. 2024, 12, 854–875. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Khan, M.U.; Azhar Ud Din, M.; Khan, I.M.; Khan, M.I.; Bungau, S.; Hassan, S.S.U. Reprogramming Tumor-Associated Macrophages as a Unique Approach to Target Tumor Immunotherapy. Front. Immunol. 2023, 14, 1166487. [Google Scholar] [CrossRef]
- Xie, Q.; Zeng, Y.; Zhang, X.; Yu, F. The Significance of Lipid Metabolism Reprogramming of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Cancer Immunol. Immunother. 2024, 73, 171. [Google Scholar] [CrossRef]
- Li, N.; Geng, S.; Dong, Z.; Jin, Y.; Ying, H.; Li, H.-W.; Shi, L. A New Era of Cancer Immunotherapy: Combining Revolutionary Technologies for Enhanced CAR-M Therapy. Mol. Cancer 2024, 23, 117. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, S.; Xue, D.; Neculai, D.; Zhang, J. Metabolic Reprogramming of Macrophages in Cancer Therapy. Trends Endocrinol. Metab. 2024, S1043276024002443. [Google Scholar] [CrossRef]
- Cao, M.; Wang, Z.; Lan, W.; Xiang, B.; Liao, W.; Zhou, J.; Liu, X.; Wang, Y.; Zhang, S.; Lu, S.; et al. The Roles of Tissue Resident Macrophages in Health and Cancer. Exp. Hematol. Oncol. 2024, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Terock, J.; Hannemann, A.; Van Der Auwera, S.; Janowitz, D.; Spitzer, C.; Bonk, S.; Völzke, H.; Grabe, H.J. Posttraumatic Stress Disorder Is Associated with Reduced Vitamin D Levels and Functional Polymorphisms of the Vitamin D Binding-Protein in a Population-Based Sample. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 96, 109760. [Google Scholar] [CrossRef]
- Trovato, B.; Godos, J.; Varrasi, S.; Roggio, F.; Castellano, S.; Musumeci, G. Physical Activity, Sun Exposure, Vitamin D Intake and Perceived Stress in Italian Adults. Nutrients 2023, 15, 2301. [Google Scholar] [CrossRef]
- Fernandez, G.J.; Ramírez-Mejía, J.M.; Urcuqui-Inchima, S. Vitamin D Boosts Immune Response of Macrophages through a Regulatory Network of microRNAs and mRNAs. J. Nutr. Biochem. 2022, 109, 109105. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, J.; Xin, S.; Lin, Y.; Chen, Y.; Zhou, X.; Chen, H.; Li, X. CYP24A1 Affected Macrophage Polarization through Degradation of Vitamin D as a Candidate Biomarker for Ovarian Cancer Prognosis. Int. Immunopharmacol. 2024, 138, 112575. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, M.; Guo, Y.; Song, Z.; Liu, B. 1,25-Dihydroxyvitamin D3 Promotes High Glucose-Induced M1 Macrophage Switching to M2 via the VDR-PPAR γ Signaling Pathway. BioMed Res. Int. 2015, 2015, 157834. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Cai, J.; Li, Y.; Yang, R. 1,25-Dihydroxy-Vitamin D3 Induces Macrophage Polarization to M2 by Upregulating T-cell Ig-mucin-3 Expression. Mol. Med. Rep. 2019, 19, 3707–3713. [Google Scholar] [CrossRef] [PubMed]
- Stachowicz-Suhs, M.; Łabędź, N.; Anisiewicz, A.; Banach, J.; Kłopotowska, D.; Milczarek, M.; Piotrowska, A.; Dzięgiel, P.; Maciejczyk, A.; Matkowski, R.; et al. Calcitriol Promotes M2 Polarization of Tumor-Associated Macrophages in 4T1 Mouse Mammary Gland Cancer via the Induction of Proinflammatory Cytokines. Sci. Rep. 2024, 14, 3778. [Google Scholar] [CrossRef] [PubMed]
- Stachowicz-Suhs, M.; Łabędź, N.; Milczarek, M.; Kłopotowska, D.; Filip-Psurska, B.; Maciejczyk, A.; Matkowski, R.; Wietrzyk, J. Vitamin D3 Reduces the Expression of M1 and M2 Macrophage Markers in Breast Cancer Patients. Sci. Rep. 2024, 14, 22126. [Google Scholar] [CrossRef]
- Karkeni, E.; Marcotorchino, J.; Tourniaire, F.; Astier, J.; Peiretti, F.; Darmon, P.; Landrier, J.-F. Vitamin D Limits Chemokine Expression in Adipocytes and Macrophage Migration In Vitro and in Male Mice. Endocrinology 2015, 156, 1782–1793. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Momčilović, S.; Milošević, M.; Kočović, D.M.; Marković, D.; Zdravković, D.; Vignjević Petrinović, S. Macrophages at the Crossroads of Chronic Stress and Cancer. Int. J. Mol. Sci. 2025, 26, 6838. https://doi.org/10.3390/ijms26146838
Momčilović S, Milošević M, Kočović DM, Marković D, Zdravković D, Vignjević Petrinović S. Macrophages at the Crossroads of Chronic Stress and Cancer. International Journal of Molecular Sciences. 2025; 26(14):6838. https://doi.org/10.3390/ijms26146838
Chicago/Turabian StyleMomčilović, Sanja, Maja Milošević, Dušica M. Kočović, Dragana Marković, Darko Zdravković, and Sanja Vignjević Petrinović. 2025. "Macrophages at the Crossroads of Chronic Stress and Cancer" International Journal of Molecular Sciences 26, no. 14: 6838. https://doi.org/10.3390/ijms26146838
APA StyleMomčilović, S., Milošević, M., Kočović, D. M., Marković, D., Zdravković, D., & Vignjević Petrinović, S. (2025). Macrophages at the Crossroads of Chronic Stress and Cancer. International Journal of Molecular Sciences, 26(14), 6838. https://doi.org/10.3390/ijms26146838