
Homocysteinylation of Fibrinogen: A Post-Translational Link to Thrombosis
 ,
,  and
and 
Abstract
1. Introduction
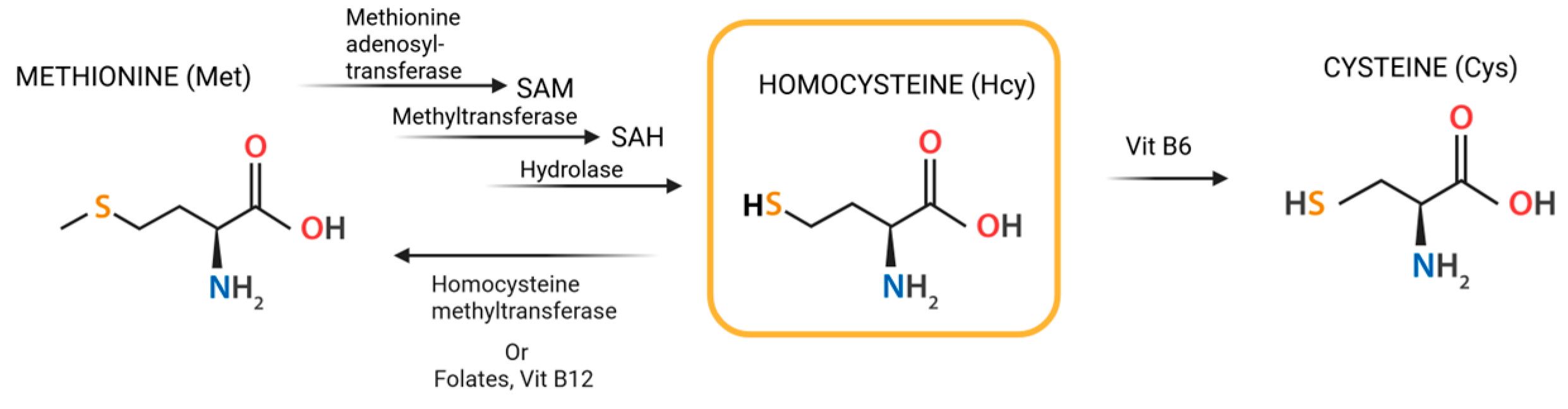
2. Homocysteine Metabolism
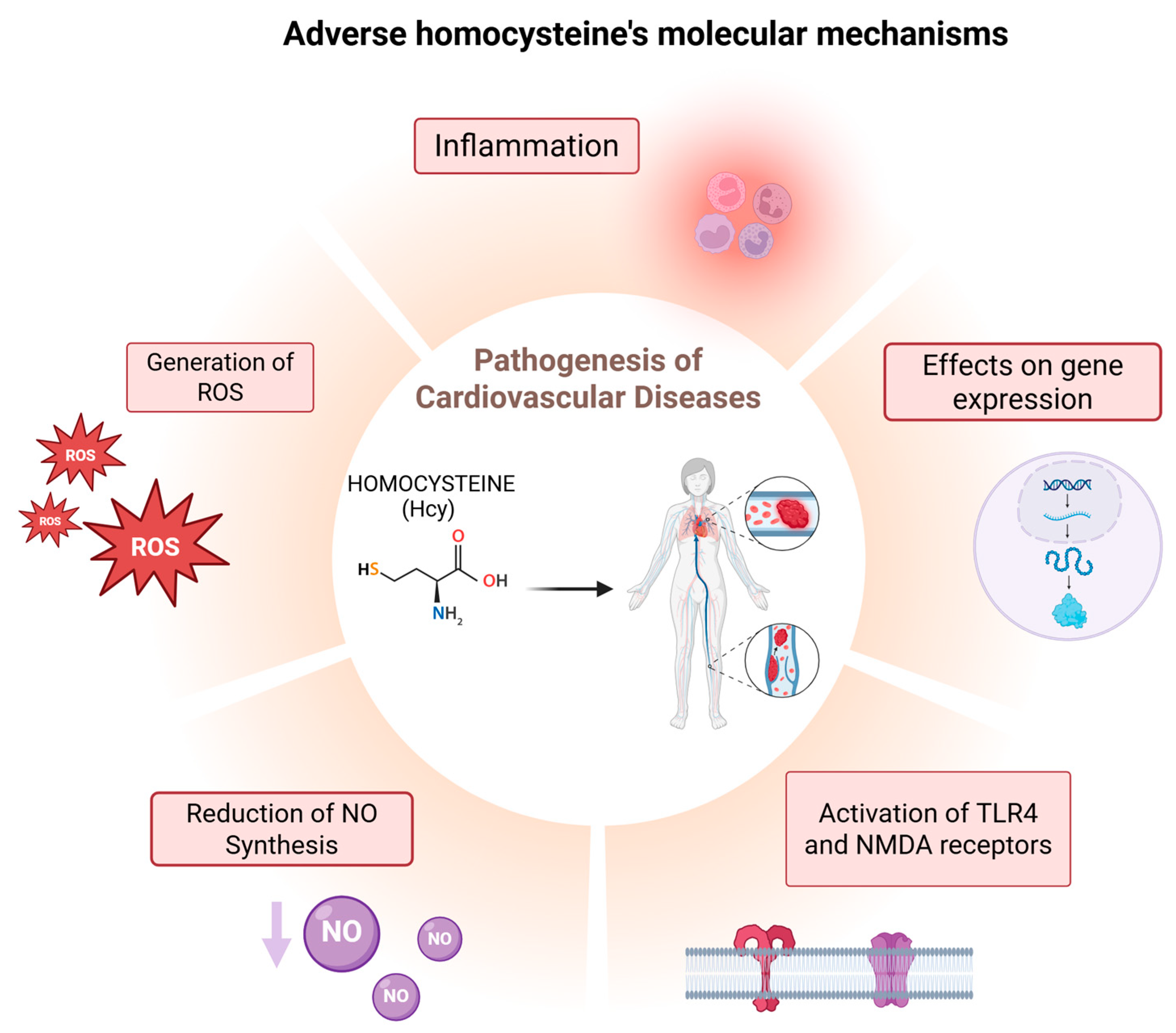
- Generation of Reactive Oxygen Species (ROS):
- Inhibition of Nitric Oxide (NO) Synthesis:
- Activation of Cellular Receptors:
- Increased Inflammation:
- Effects on gene expression:
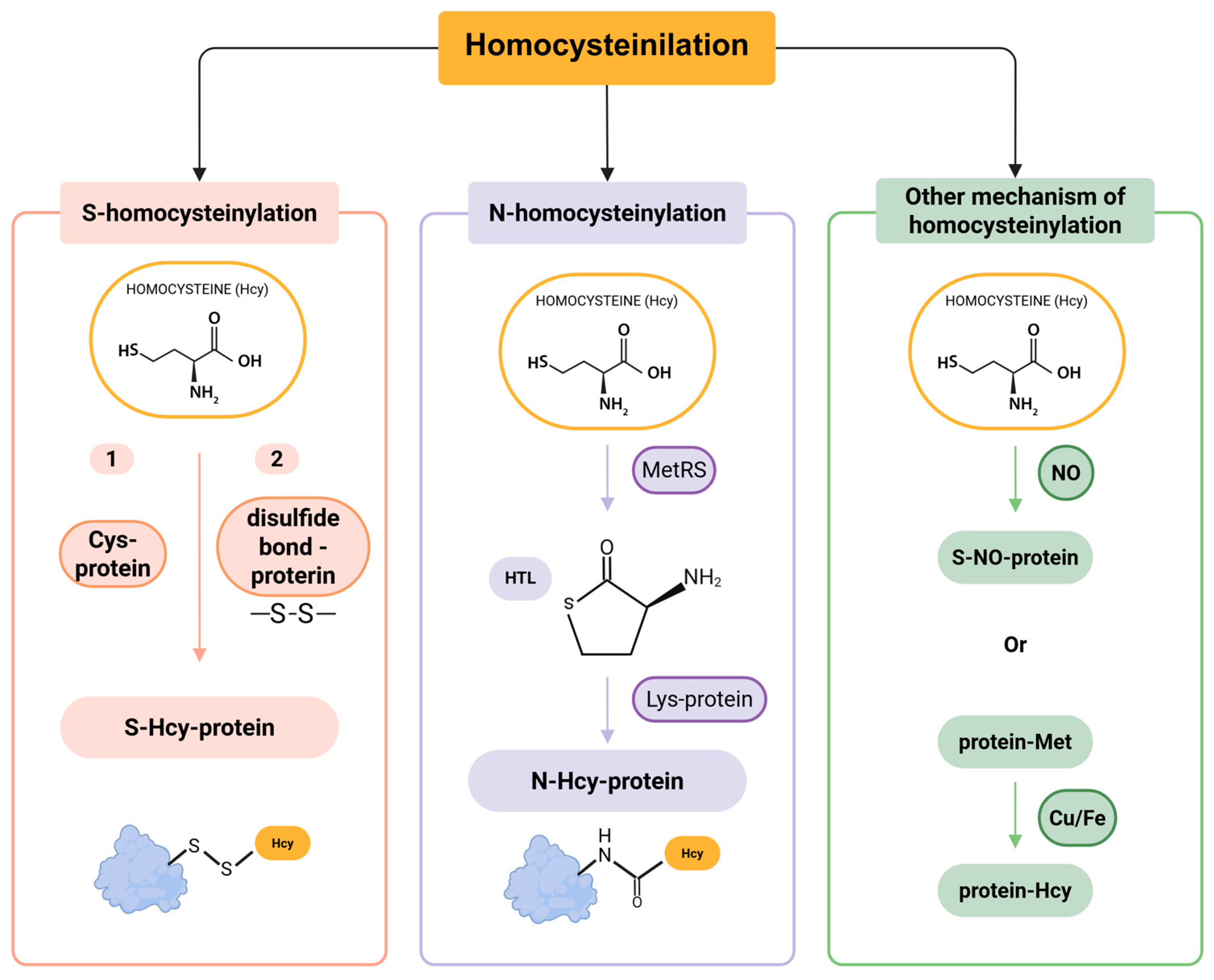
3. Homocysteinylation: Homocysteine Binding to Proteins
Mechanisms of Protein Homocysteinylation
4. Fibrinogen
Fibrinogen and PTMs
5. Fibrinogen Homocysteinylation and Clinical Consequences
6. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CVDs | Cardiovascular diseases |
| Cys | Cysteine |
| Hcy | Homocysteine |
| HHcy | Hyperhomocysteinemia |
| HTL | Homocysteine-thiolactone |
| ICAM-1 | Intracellular adhesion molecule-1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| Lys | Lysine |
| Met | Methionine |
| MI | Myocardial infarction |
| N-Hcy-proteins | N-homocysteinylated proteins |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric Oxide |
| PTMs | Post-translational modifications |
| ROS | Reactive Oxygen Species |
| SAM | S-adenosylmethionine |
| SAH | S-adenosylhomocysteine |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor-alpha |
References
- Hermann, A.; Sitdikova, G. Homocysteine: Biochemistry, Molecular Biology and Role in Disease. Biomolecules 2021, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78. [Google Scholar] [CrossRef]
- Wu, D.F.; Yin, R.X.; Deng, J.L. Homocysteine, hyperhomocysteinemia, and H-type hypertension. Eur. J. Prev. Cardiol. 2024, 31, 1092–1103. [Google Scholar] [CrossRef]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef] [PubMed]
- Zaric, B.L.; Obradovic, M.; Bajic, V.; Haidara, M.A.; Jovanovic, M.; Isenovic, E.R. Homocysteine and Hyperhomocysteinaemia. Curr. Med. Chem. 2019, 26, 2948–2961. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Mason, A.M.; Carter, P.; Burgess, S.; Larsson, S.C. Homocysteine, B vitamins, and cardiovascular disease: A Mendelian randomization study. BMC Med. 2021, 19, 97. [Google Scholar] [CrossRef]
- Rehman, T.; Shabbir, M.A.; Inam-Ur-Raheem, M.; Manzoor, M.F.; Ahmad, N.; Liu, Z.W.; Ahmad, M.H.; Siddeeg, A.; Abid, M.; Aadil, R.M. Cysteine and homocysteine as biomarker of various diseases. Food Sci. Nutr. 2020, 8, 4696–4707. [Google Scholar] [CrossRef]
- Guieu, R.; Ruf, J.; Mottola, G. Hyperhomocysteinemia and cardiovascular diseases. Ann. Biol. Clin. 2022, 80, 7–14. [Google Scholar] [CrossRef]
- Cordaro, M.; Siracusa, R.; Fusco, R.; Cuzzocrea, S.; Di Paola, R.; Impellizzeri, D. Involvements of Hyperhomocysteinemia in Neurological Disorders. Metabolites 2021, 11, 37. [Google Scholar] [CrossRef]
- Karmin, O.; Siow, Y.L. Metabolic Imbalance of Homocysteine and Hydrogen Sulfide in Kidney Disease. Curr. Med. Chem. 2018, 25, 367–377. [Google Scholar] [CrossRef]
- Keller, A.C.; Klawitter, J.; Hildreth, K.L.; Christians, U.; Putnam, K.; Kohrt, W.M.; Reusch, J.E.B.; Moreau, K.L. Elevated plasma homocysteine and cysteine are associated with endothelial dysfunction across menopausal stages in healthy women. J. Appl. Physiol. (1985) 2019, 126, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.S. Homocysteine and cardiovascular disease: A review of the evidence. Diabetes Vasc. Dis. Res. 2007, 4, 143–150. [Google Scholar] [CrossRef]
- Djuric, D.; Jakovljevic, V.; Zivkovic, V.; Srejovic, I. Homocysteine and homocysteine-related compounds: An overview of the roles in the pathology of the cardiovascular and nervous systems. Can. J. Physiol. Pharmacol. 2018, 96, 991–1003. [Google Scholar] [CrossRef]
- Sitdikova, G.; Hermann, A. Homocysteine: Biochemistry, Molecular Biology, and Role in Disease 2021. Biomolecules 2023, 13, 1111. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Fang, C.; Wang, J.; Tian, Y.; Zou, T. Association between homocysteine levels and mortality in CVD: A cohort study based on NHANES database. BMC Cardiovasc. Disord. 2024, 24, 652. [Google Scholar] [CrossRef]
- Kim, M.; Shin, S.; Yoo, E.; Kang, J.H.; Sung, E.; Kim, C.H.; Shin, H.; Lee, M.Y. Serum Homocysteine Levels and All-Cause and Cause-Specific Mortality in Korean Adult Men: A Cohort Study. Nutrients 2024, 16, 2759. [Google Scholar] [CrossRef]
- Catena, C.; Colussi, G.; Nait, F.; Capobianco, F.; Sechi, L.A. Elevated Homocysteine Levels Are Associated With the Metabolic Syndrome and Cardiovascular Events in Hypertensive Patients. Am. J. Hypertens. 2015, 28, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Riba, R.; Nicolaou, A.; Troxler, M.; Homer-Vaniasinkam, S.; Naseem, K.M. Altered platelet reactivity in peripheral vascular disease complicated with elevated plasma homocysteine levels. Atherosclerosis 2004, 175, 69–75. [Google Scholar] [CrossRef]
- Bosevski, M.; Zlatanovikj, N.; Petkoska, D.; Gjorgievski, A.; Lazarova, E.; Stojanovska, L. Plasma Homocysteine in Patients with Coronary and Carotid Artery Disease: A Case Control Study. Prilozi 2020, 41, 15–22. [Google Scholar] [CrossRef]
- Guéant, J.L.; Guéant-Rodriguez, R.M.; Oussalah, A.; Zuily, S.; Rosenberg, I. Hyperhomocysteinemia in Cardiovascular Diseases: Revisiting Observational Studies and Clinical Trials. Thromb. Haemost. 2023, 123, 270–282. [Google Scholar] [CrossRef]
- Habib, S.S.; Al-Khlaiwi, T.; Almushawah, A.; Alsomali, A.; Habib, S.A. Homocysteine as a predictor and prognostic marker of atherosclerotic cardiovascular disease: A systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 8598–8608. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H. Homocysteine—From disease biomarker to disease prevention. J. Intern. Med. 2021, 290, 826–854. [Google Scholar] [CrossRef]
- Stabler, S.P. Alterations in Sulfur Amino Acids as Biomarkers of Disease. J. Nutr. 2020, 150, 2532S–2537S. [Google Scholar] [CrossRef]
- Gospodarczyk, A.; Marczewski, K.; Gospodarczyk, N.; Widuch, M.; Tkocz, M.; Zalejska-Fiolka, J. Homocysteine and Cardiovascular Disease—A Current Review. Wiad. Lek. 2022, 75, 2862–2866. [Google Scholar] [CrossRef] [PubMed]
- Raghubeer, S.; Matsha, T.E. Methylenetetrahydrofolate (MTHFR), the One-Carbon Cycle, and Cardiovascular Risks. Nutrients 2021, 13, 4562. [Google Scholar] [CrossRef]
- Hoţoleanu, C.; Porojan-Iuga, M.; Rusu, M.L.; Andercou, A. Hyperhomocysteinemia: Clinical and therapeutical involvement in venous thrombosis. Rom. J. Intern. Med. 2007, 45, 159–164. [Google Scholar] [PubMed]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef]
- Suematsu, N.; Ojaimi, C.; Kinugawa, S.; Wang, Z.; Xu, X.; Koller, A.; Recchia, F.A.; Hintze, T.H. Hyperhomocysteinemia alters cardiac substrate metabolism by impairing nitric oxide bioavailability through oxidative stress. Circulation 2007, 115, 255–262. [Google Scholar] [CrossRef]
- Kolling, J.; Scherer, E.B.; da Cunha, A.A.; da Cunha, M.J.; Wyse, A.T. Homocysteine induces oxidative-nitrative stress in heart of rats: Prevention by folic acid. Cardiovasc. Toxicol. 2011, 11, 67–73. [Google Scholar] [CrossRef]
- Scherer, E.B.; da Cunha, A.A.; Kolling, J.; da Cunha, M.J.; Schmitz, F.; Sitta, A.; Lima, D.D.; Delwing, D.; Vargas, C.R.; Wyse, A.T. Development of an animal model for chronic mild hyperhomocysteinemia and its response to oxidative damage. Int. J. Dev. Neurosci. 2011, 29, 693–699. [Google Scholar] [CrossRef]
- Petras, M.; Tatarkova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgova, A. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J. Physiol. Pharmacol. 2014, 65, 15–23. [Google Scholar] [PubMed]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Lombardi, C.; Acanfora, F.; Satta, E.; Cesare, C.M.; Violetti, E.; Romano, M.M.; De Santo, N.G. Possible mechanisms of homocysteine toxicity. Kidney Int. Suppl. 2003, 63, S137–S140. [Google Scholar] [CrossRef] [PubMed]
- Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef] [PubMed]
- Ostrakhovitch, E.A.; Tabibzadeh, S. Homocysteine and age-associated disorders. Ageing Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef]
- Huang, A.; Yang, Y.M.; Feher, A.; Bagi, Z.; Kaley, G.; Sun, D. Exacerbation of endothelial dysfunction during the progression of diabetes: Role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R674–R681. [Google Scholar] [CrossRef]
- Huang, A.; Pinto, J.T.; Froogh, G.; Kandhi, S.; Qin, J.; Wolin, M.S.; Hintze, T.H.; Sun, D. Role of homocysteinylation of ACE in endothelial dysfunction of arteries. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H92–H100. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef]
- Topal, G.; Brunet, A.; Millanvoye, E.; Boucher, J.L.; Rendu, F.; Devynck, M.A.; David-Dufilho, M. Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free Radic. Biol. Med. 2004, 36, 1532–1541. [Google Scholar] [CrossRef]
- Jeremic, N.; Weber, G.J.; Familtseva, A.; Metreveli, N.; Tyagi, S.C. Ablation of Toll-like receptor 4 mitigates central blood pressure response during hyperhomocysteinemia. J. Hypertens. 2017, 35, 2226–2237. [Google Scholar] [CrossRef]
- Jeremic, N.; Weber, G.J.; Tyagi, S.C. Ablation of toll-like receptor 4 mitigates cardiac mitochondrial dysfunction in hyperhomocysteinemia. Can. J. Physiol. Pharmacol. 2017, 95, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Cao, Z.Y.; Wang, M.M.; Liu, X.M.; Gao, T.; Hu, Q.K.; Yuan, W.J.; Lin, L. Up-regulated TLR4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J. Cell. Mol. Med. 2015, 19, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, X.; Pang, J.; Zhang, C.; Ding, J.M.; Peng, X.; Liu, Y.; Cao, J.M. NMDA receptor activation induces mitochondrial dysfunction, oxidative stress and apoptosis in cultured neonatal rat cardiomyocytes. Physiol. Res. 2007, 56, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Soni, C.V.; Tyagi, S.C.; Todnem, N.D.; Givvimani, S.; Pushpakumar, S.B.; Villafane, J.; Maldonado, C. Hyperhomocysteinemia Alters Sinoatrial and Atrioventricular Nodal Function: Role of Magnesium in Attenuating These Effects. Cell Biochem. Biophys. 2016, 74, 59–65. [Google Scholar] [CrossRef]
- Bryushkova, E.A.; Vladychenskaya, E.A.; Stepanova, M.S.; Boldyrev, A.A. Effect of homocysteine on properties of neutrophils activated in vivo. Biochemistry 2011, 76, 467–472. [Google Scholar] [CrossRef]
- Boldyrev, A.; Bryushkova, E.; Mashkina, A.; Vladychenskaya, E. Why is homocysteine toxic for the nervous and immune systems? Curr. Aging Sci. 2013, 6, 29–36. [Google Scholar] [CrossRef]
- Zanin, R.F.; Bergamin, L.S.; Morrone, F.B.; Coutinho-Silva, R.; de Souza Wyse, A.T.; Battastini, A.M. Pathological concentrations of homocysteine increases IL-1β production in macrophages in a P2X7, NF-ĸB, and erk-dependent manner. Purinergic Signal. 2015, 11, 463–470. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, Y.S.; Seo, K.W.; Bae, J.U.; Kim, G.H.; Park, S.Y.; Kim, C.D. Homocysteine enhances MMP-9 production in murine macrophages via ERK and Akt signaling pathways. Toxicol. Appl. Pharmacol. 2012, 260, 89–94. [Google Scholar] [CrossRef]
- Tsarouhas, K.; Tsitsimpikou, C.; Apostolakis, S.; Haliassos, A.; Tzardi, M.; Panagiotou, M.; Tsatsakis, A.; Spandidos, D.A. Homocysteine and metalloprotease-3 and -9 in patients with ascending aorta aneurysms. Thromb. Res. 2011, 128, e95–e99. [Google Scholar] [CrossRef]
- Vacek, T.P.; Vacek, J.C.; Tyagi, S.C. Mitochondrial mitophagic mechanisms of myocardial matrix metabolism and remodelling. Arch. Physiol. Biochem. 2012, 118, 31–42. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef]
- CARSON, N.A.; DENT, C.E.; FIELD, C.M.; GAULL, G.E. HOMOCYSTINURIA: CLINICAL AND PATHOLOGICAL REVIEW OF TEN CASES. J. Pediatr. 1965, 66, 565–583. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef] [PubMed]
- Harker, L.A.; Slichter, S.J.; Scott, C.R.; Ross, R. Homocystinemia. Vascular injury and arterial thrombosis. N. Engl. J. Med. 1974, 291, 537–543. [Google Scholar] [CrossRef]
- Peng, Y.P.; Huang, M.Y.; Xue, Y.J.; Pan, J.L.; Lin, C. Association of Hyperhomocysteinemia with Increased Coronary Microcirculatory Resistance and Poor Short-Term Prognosis of Patients with Acute Myocardial Infarction after Elective Percutaneous Coronary Intervention. BioMed Res. Int. 2020, 2020, 1710452. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Nurk, E.; Smith, A.D.; Ueland, P.M.; Gjesdal, C.G.; Bjelland, I.; Tverdal, A.; Tell, G.S.; Nygård, O.; Vollset, S.E. The Hordaland Homocysteine Study: A community-based study of homocysteine, its determinants, and associations with disease. J. Nutr. 2006, 136, 1731S–1740S. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cai, Y.; Adachi, M.T.; Oshiro, S.; Aso, T.; Kaufman, R.J.; Kitajima, S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001, 276, 35867–35874. [Google Scholar] [CrossRef]
- Aitken, R.J.; Flanagan, H.M.; Connaughton, H.; Whiting, S.; Hedges, A.; Baker, M.A. Involvement of homocysteine, homocysteine thiolactone, and paraoxonase type 1 (PON-1) in the etiology of defective human sperm function. Andrology 2016, 4, 345–360. [Google Scholar] [CrossRef]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef]
- Chen, S.M.; Tang, X.Q. Homocysteinylation and Sulfhydration in Diseases. Curr. Neuropharmacol. 2022, 20, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.J. Consequences of homocysteine export and oxidation in the vascular system. Semin. Thromb. Hemost. 2000, 26, 227–232. [Google Scholar] [CrossRef]
- Sass, J.O.; Nakanishi, T.; Sato, T.; Sperl, W.; Shimizu, A. S-homocysteinylation of transthyretin is detected in plasma and serum of humans with different types of hyperhomocysteinemia. Biochem. Biophys. Res. Commun. 2003, 310, 242–246. [Google Scholar] [CrossRef]
- Kang, S.S.; Wong, P.W.; Becker, N. Protein-bound homocyst(e)ine in normal subjects and in patients with homocystinuria. Pediatr. Res. 1979, 13, 1141–1143. [Google Scholar] [CrossRef]
- Glushchenko, A.V.; Jacobsen, D.W. Molecular targeting of proteins by L-homocysteine: Mechanistic implications for vascular disease. Antioxid. Redox Signal. 2007, 9, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Marczak, L.; Twardowski, T.; Stobiecki, M.; Jakubowski, H. Direct monitoring of albumin lysine-525 N-homocysteinylation in human serum by liquid chromatography/mass spectrometry. Anal. Biochem. 2010, 405, 132–134. [Google Scholar] [CrossRef]
- Perła-Kajan, J.; Utyro, O.; Rusek, M.; Malinowska, A.; Sitkiewicz, E.; Jakubowski, H. N-Homocysteinylation impairs collagen cross-linking in cystathionine β-synthase-deficient mice: A novel mechanism of connective tissue abnormalities. FASEB J. 2016, 30, 3810–3821. [Google Scholar] [CrossRef]
- Jakubowski, H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [CrossRef]
- Jakubowski, H. Aminoacyl-tRNA synthetases and the evolution of coded peptide synthesis: The Thioester World. FEBS Lett. 2016, 590, 469–481. [Google Scholar] [CrossRef]
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures. Possible mechanism for pathological consequences of elevated homocysteine levels. J. Biol. Chem. 1997, 272, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Translational accuracy of aminoacyl-tRNA synthetases: Implications for atherosclerosis. J. Nutr. 2001, 131, 2983S–2987S. [Google Scholar] [CrossRef] [PubMed]
- Borowczyk, K.; Suliburska, J.; Jakubowski, H. Demethylation of methionine and keratin damage in human hair. Amino Acids 2018, 50, 537–546. [Google Scholar] [CrossRef]
- Undas, A.; Perła, J.; Lacinski, M.; Trzeciak, W.; Kaźmierski, R.; Jakubowski, H. Autoantibodies against N-homocysteinylated proteins in humans: Implications for atherosclerosis. Stroke 2004, 35, 1299–1304. [Google Scholar] [CrossRef]
- Leri, M.; Rebuzzini, P.; Caselli, A.; Luti, S.; Natalello, A.; Giorgetti, S.; Marchese, L.; Garagna, S.; Stefani, M.; Paoli, P.; et al. S-Homocysteinylation effects on transthyretin: Worsening of cardiomyopathy onset. Biochim. Et Biophys. Acta Gen. Subj. 2020, 1864, 129453. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Marczak, Ł.; Kaján, L.; Skowronek, P.; Twardowski, T.; Jakubowski, H. Modification by homocysteine thiolactone affects redox status of cytochrome C. Biochemistry 2007, 46, 6225–6231. [Google Scholar] [CrossRef]
- Hultberg, B.; Andersson, A.; Isaksson, A. Protein binding of homocysteine and other thiols in HeLa cell cultures after addition of homocysteine and copper ions. Clin. Chim. Acta 1998, 269, 175–184. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Vilar, R.; Fish, R.J.; Casini, A.; Neerman-Arbez, M. Fibrin(ogen) in human disease: Both friend and foe. Haematologica 2020, 105, 284–296. [Google Scholar] [CrossRef]
- Kaido, T.; Yoda, M.; Kamijo, T.; Arai, S.; Taira, C.; Higuchi, Y.; Okumura, N. A Novel Amino Acid Substitution, Fibrinogen Bβp.Pro234Leu, Associated with Hypofibrinogenemia Causing Impairment of Fibrinogen Assembly and Secretion. Int. J. Mol. Sci. 2020, 21, 9422. [Google Scholar] [CrossRef] [PubMed]
- Piechocka, I.K.; Kurniawan, N.A.; Grimbergen, J.; Koopman, J.; Koenderink, G.H. Recombinant fibrinogen reveals the differential roles of α- and γ-chain cross-linking and molecular heterogeneity in fibrin clot strain-stiffening. J. Thromb. Haemost. 2017, 15, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Weisel, J.W.; Ischiropoulos, H. Functional impact of oxidative posttranslational modifications on fibrinogen and fibrin clots. Free Radic. Biol. Med. 2013, 65, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Medved, L.; Weisel, J.W.; Haemostasis, F.a.F.X.S.o.S.S.C.o.I.S.o.T.a. Recommendations for nomenclature on fibrinogen and fibrin. J. Thromb. Haemost. 2009, 7, 355–359. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef]
- Wolberg, A.S. Thrombin generation and fibrin clot structure. Blood Rev. 2007, 21, 131–142. [Google Scholar] [CrossRef]
- Luyendyk, J.P.; Schoenecker, J.G.; Flick, M.J. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood 2019, 133, 511–520. [Google Scholar] [CrossRef]
- Weisel, J.W. Structure of fibrin: Impact on clot stability. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 116–124. [Google Scholar] [CrossRef]
- Doolittle, R.F. The conversion of fibrinogen to fibrin: A brief history of some key events. Matrix Biol. 2017, 60–61, 5–7. [Google Scholar] [CrossRef]
- Risman, R.A.; Belcher, H.A.; Ramanujam, R.K.; Weisel, J.W.; Hudson, N.E.; Tutwiler, V. Comprehensive Analysis of the Role of Fibrinogen and Thrombin in Clot Formation and Structure for Plasma and Purified Fibrinogen. Biomolecules 2024, 14, 230. [Google Scholar] [CrossRef]
- Wolberg, A.S.; Campbell, R.A. Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci. 2008, 38, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cesarman-Maus, G.; Hajjar, K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005, 129, 307–321. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Pieters, M.; de Lange-Loots, Z.; Weisel, J.W. Fibrinogen and Fibrin. Subcell. Biochem. 2021, 96, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S. Fibrinogen and fibrin: Synthesis, structure, and function in health and disease. J. Thromb. Haemost. 2023, 21, 3005–3015. [Google Scholar] [CrossRef]
- de Vries, J.J.; Snoek, C.J.M.; Rijken, D.C.; de Maat, M.P.M. Effects of Post-Translational Modifications of Fibrinogen on Clot Formation, Clot Structure, and Fibrinolysis: A Systematic Review. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 554–569. [Google Scholar] [CrossRef]
- Tenopoulou, M. Fibrinogen post-translational modifications are biochemical determinants of fibrin clot properties and interactions. FEBS J. 2025, 292, 11–27. [Google Scholar] [CrossRef]
- Nencini, F.; Bettiol, A.; Argento, F.R.; Borghi, S.; Giurranna, E.; Emmi, G.; Prisco, D.; Taddei, N.; Fiorillo, C.; Becatti, M. Post-translational modifications of fibrinogen: Implications for clotting, fibrin structure and degradation. Mol. Biomed. 2024, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Sovova, Z.; Suttnar, J.; Dyr, J.E. Molecular Dynamic Simulations Suggest That Metabolite-Induced Post-Translational Modifications Alter the Behavior of the Fibrinogen Coiled-Coil Domain. Metabolites 2021, 11, 307. [Google Scholar] [CrossRef]
- Ząbczyk, M.; Ariëns, R.A.S.; Undas, A. Fibrin clot properties in cardiovascular disease: From basic mechanisms to clinical practice. Cardiovasc. Res. 2023, 119, 94–111. [Google Scholar] [CrossRef]
- Konieczyńska, M.; Natorska, J.; Undas, A. Thrombosis and Aging: Fibrin Clot Properties and Oxidative Stress. Antioxid. Redox Signal. 2024, 41, 233–254. [Google Scholar] [CrossRef]
- Hugenholtz, G.C.; Macrae, F.; Adelmeijer, J.; Dulfer, S.; Porte, R.J.; Lisman, T.; Ariëns, R.A. Procoagulant changes in fibrin clot structure in patients with cirrhosis are associated with oxidative modifications of fibrinogen. J. Thromb. Haemost. 2016, 14, 1054–1066. [Google Scholar] [CrossRef]
- Rosenfeld, M.A.; Yurina, L.V.; Gavrilina, E.S.; Vasilyeva, A.D. Post-Translational Oxidative Modifications of Hemostasis Proteins: Structure, Function, and Regulation. Biochemistry 2024, 89, S14–S33. [Google Scholar] [CrossRef]
- Azizova, O.A.; Piryazev, A.P.; Aseychev, A.V.; Shvachko, A.G. Oxidative modification of fibrinogen inhibits its transformation into fibrin under the effect of thrombin. Bull. Exp. Biol. Med. 2009, 147, 201–203. [Google Scholar] [CrossRef]
- Piryazev, A.P.; Aseichev, A.V.; Azizova, O.A. Effect of oxidation-modified fibrinogen on the formation and lysis of fibrin clot in the plasma. Bull. Exp. Biol. Med. 2009, 148, 881–885. [Google Scholar] [CrossRef]
- Sovová, Ž.; Štikarová, J.; Kaufmanová, J.; Májek, P.; Suttnar, J.; Šácha, P.; Malý, M.; Dyr, J.E. Impact of posttranslational modifications on atomistic structure of fibrinogen. PLoS ONE 2020, 15, e0227543. [Google Scholar] [CrossRef]
- Tadeusiewicz, J.; Nowak, P. The role of post-translational modification of fibrinogen in the pathogenesis of thrombosis. Pol. Merkur. Lek. 2015, 38, 107–112. [Google Scholar]
- Fini, E.; Argento, F.R.; Borghi, S.; Giurranna, E.; Nencini, F.; Cirillo, M.; Fatini, C.; Taddei, N.; Coccia, M.E.; Fiorillo, C.; et al. Fibrinogen Structural Changes and Their Potential Role in Endometriosis-Related Thrombosis. Antioxidants 2024, 13, 1456. [Google Scholar] [CrossRef]
- Becatti, M.; Emmi, G.; Silvestri, E.; Bruschi, G.; Ciucciarelli, L.; Squatrito, D.; Vaglio, A.; Taddei, N.; Abbate, R.; Emmi, L.; et al. Neutrophil Activation Promotes Fibrinogen Oxidation and Thrombus Formation in Behçet Disease. Circulation 2016, 133, 302–311. [Google Scholar] [CrossRef]
- Gitto, S.; Fiorillo, C.; Argento, F.R.; Fini, E.; Borghi, S.; Falcini, M.; Roccarina, D.; La Delfa, R.; Lillo, L.; Zurli, T.; et al. Oxidative stress-induced fibrinogen modifications in liver transplant recipients: Unraveling a novel potential mechanism for cardiovascular risk. Res. Pract. Thromb. Haemost. 2024, 8, 102555. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Marcucci, R.; Bruschi, G.; Taddei, N.; Bani, D.; Gori, A.M.; Giusti, B.; Gensini, G.F.; Abbate, R.; Fiorillo, C. Oxidative modification of fibrinogen is associated with altered function and structure in the subacute phase of myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Risman, R.A.; Sen, M.; Tutwiler, V.; Hudson, N.E. Deconstructing fibrin(ogen) structure. J. Thromb. Haemost. 2024, 23, 368–380. [Google Scholar] [CrossRef]
- Mihalko, E.; Brown, A.C. Clot Structure and Implications for Bleeding and Thrombosis. Semin. Thromb. Hemost. 2020, 46, 96–104. [Google Scholar] [CrossRef]
- Zeng, Z.; Fagnon, M.; Nallan Chakravarthula, T.; Alves, N.J. Fibrin clot formation under diverse clotting conditions: Comparing turbidimetry and thromboelastography. Thromb. Res. 2020, 187, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: A potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef]
- Sauls, D.L.; Wolberg, A.S.; Hoffman, M. Elevated plasma homocysteine leads to alterations in fibrin clot structure and stability: Implications for the mechanism of thrombosis in hyperhomocysteinemia. J. Thromb. Haemost. 2003, 1, 300–306. [Google Scholar] [CrossRef]
- Malinowska, J.; Olas, B. Homocysteine and its thiolactone-mediated modification of fibrinogen affect blood platelet adhesion. Platelets 2012, 23, 409–412. [Google Scholar] [CrossRef]
- Malinowska, J.; Tomczynska, M.; Olas, B. Changes of blood platelet adhesion to collagen and fibrinogen induced by homocysteine and its thiolactone. Clin. Biochem. 2012, 45, 1225–1228. [Google Scholar] [CrossRef]
- Lauricella, A.M.; Quintana, I.L.; Kordich, L.C. Effects of homocysteine thiol group on fibrin networks: Another possible mechanism of harm. Thromb. Res. 2002, 107, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Lauricella, A.M.; Quintana, I.; Castañon, M.; Sassetti, B.; Kordich, L. Influence of homocysteine on fibrin network lysis. Blood Coagul. Fibrinolysis 2006, 17, 181–186. [Google Scholar] [CrossRef]
- Undas, A.; Brozek, J.; Jankowski, M.; Siudak, Z.; Szczeklik, A.; Jakubowski, H. Plasma homocysteine affects fibrin clot permeability and resistance to lysis in human subjects. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Marchi, R.; Carvajal, Z.; Weisel, J.W. Comparison of the effect of different homocysteine concentrations on clot formation using human plasma and purified fibrinogen. Thromb. Haemost. 2008, 99, 451–452. [Google Scholar] [CrossRef]
- Sauls, D.L.; Warren, M.; Hoffman, M. Homocysteinylated fibrinogen forms disulfide-linked complexes with albumin. Thromb. Res. 2011, 127, 576–581. [Google Scholar] [CrossRef]
- Malinowska, J.; Olas, B. Analysis of biological properties of selected elements of haemostasis after treatment with the oxidized form of homocysteine in vitro. Platelets 2011, 22, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Genoud, V.; Lauricella, A.M.; Kordich, L.C.; Quintana, I. Impact of homocysteine-thiolactone on plasma fibrin networks. J. Thromb. Thrombolysis 2014, 38, 540–545. [Google Scholar] [CrossRef]
- Cellai, A.P.; Lami, D.; Antonucci, E.; Liotta, A.A.; Rogolino, A.; Fedi, S.; Fiorillo, C.; Becatti, M.; Cenci, C.; Marcucci, R.; et al. Hyperhomocysteinemia in patients with pulmonary embolism is associated with impaired plasma fibrinolytic capacity. J. Thromb. Thrombolysis 2014, 38, 45–49. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, Ł.; Kubalska, J.; Graban, A.; Jakubowski, H. Identification of N-homocysteinylation sites in plasma proteins. Amino Acids 2014, 46, 235–244. [Google Scholar] [CrossRef]
- Lentz, S.R. Mechanisms of thrombosis in hyperhomocysteinemia. Curr. Opin. Hematol. 1998, 5, 343–349. [Google Scholar] [CrossRef]
- Aronow, W.S.; Ahn, C.; Gutstein, H. Increased plasma homocysteine is an independent predictor of new atherothrombotic brain infarction in older persons. Am. J. Cardiol. 2000, 86, 585–586, A150. [Google Scholar] [CrossRef]
- Genoud, V.; Quintana, P.G.; Gionco, S.; Baldessari, A.; Quintana, I. Structural changes of fibrinogen molecule mediated by the N-homocysteinylation reaction. J. Thromb. Thrombolysis 2018, 45, 66–76. [Google Scholar] [CrossRef]
- Acevedo, M.; Pearce, G.L.; Kottke-Marchant, K.; Sprecher, D.L. Elevated fibrinogen and homocysteine levels enhance the risk of mortality in patients from a high-risk preventive cardiology clinic. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1042–1045. [Google Scholar] [CrossRef]
- Sikora, M.; Bretes, E.; Perła-Kaján, J.; Utyro, O.; Borowczyk, K.; Piechocka, J.; Głowacki, R.; Wojtasz, I.; Kaźmierski, R.; Jakubowski, H. Homocysteine thiolactone and other sulfur-containing amino acid metabolites are associated with fibrin clot properties and the risk of ischemic stroke. Sci. Rep. 2024, 14, 11222. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Skrzydlewski, P.; Perła-Kaján, J.; Jakubowski, H. Homocysteine thiolactone contributes to the prognostic value of fibrin clot structure/function in coronary artery disease. PLoS ONE 2022, 17, e0275956. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Sharma, P.K.; Chatterjee, B.; Bissa, B.; Nattarayan, V.; Ramasamy, S.; Bhat, A.; Lal, M.; Samaddar, S.; Banerjee, S.; et al. Defective quality control autophagy in Hyperhomocysteinemia promotes ER stress and consequent neuronal apoptosis through proteotoxicity. Cell Commun. Signal. 2023, 21, 258. [Google Scholar] [CrossRef]
- Luzzi, S.; Papiri, G.; Viticchi, G.; Baldinelli, S.; Fiori, C.; Silvestrini, M.; Toraldo, A. Association between homocysteine levels and cognitive profile in Alzheimer’s Disease. J. Clin. Neurosci. 2021, 94, 250–256. [Google Scholar] [CrossRef]
- Zuliani, G.; Brombo, G.; Polastri, M.; Romagnoli, T.; Mola, G.; Riccetti, R.; Seripa, D.; Trentini, A.; Cervellati, C. High plasma homocysteine levels predict the progression from mild cognitive impairment to dementia. Neurochem. Int. 2024, 177, 105763. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, L.; Li, H.; Chen, G.; Qi, G.; Ma, X.; Jin, Y. Role of homocysteine in the development and progression of Parkinson’s disease. Ann. Clin. Transl. Neurol. 2020, 7, 2332–2338. [Google Scholar] [CrossRef]
- Du, X.; Xiao, L.; Sun, R.; Li, K.; Liang, L.; Song, L.; Liu, Z. A prospective cohort study of MTHFR C677T gene polymorphism and its influence on the therapeutic effect of homocysteine in stroke patients with hyperhomocysteinemia. BMC Neurol. 2020, 20, 128. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, R.X.; He, Z.Y.; Liu, X.; Liu, H.N. Association of MTHFR C677T with total homocysteine plasma levels and susceptibility to Parkinson’s disease: A meta-analysis. Neurol. Sci. 2015, 36, 945–951. [Google Scholar] [CrossRef]
- Lupi-Herrera, E.; Soto-López, M.E.; Lugo-Dimas, A.J.; Núñez-Martínez, M.E.; Gamboa, R.; Huesca-Gómez, C.; Sierra-Galán, L.M.; Guarner-Lans, V. Polymorphisms C677T and A1298C of MTHFR Gene: Homocysteine Levels and Prothrombotic Biomarkers in Coronary and Pulmonary Thromboembolic Disease. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029618780344. [Google Scholar] [CrossRef]
- Cho, S.E.; Hong, K.S.; Shin, G.J.; Chung, W.S. The methylenetetrahydrofolate reductase C677T gene mutation is associated with hyperhomocysteinemia, cardiovascular disease and plasma B-type natriuretic peptide levels in Korea. Clin. Chem. Lab. Med. 2006, 44, 1070–1075. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Fibrinogen Analysis | ||||
|---|---|---|---|---|
| Author | Model | Method | Polymerization | Fibrinolysis |
| Lauricella et al. (2002) [118] | In vitro | Plasma + 300 μM Hcys, Cys, Hcyst | = | Probably - |
| Sauls et al. (2003) [115] | In vivo (rabbit model) | Plasma or fibrinogen | − with reptilase + with thrombin | _ |
| Lauricella et al. (2006) [119] | In vitro | Plasma + 500 μM Hcys | = + Max Abs | _ |
| Sauls et al. (2006) [114] | In vitro | Fibrinogen + 300 μM HTL | nd | _ |
| Undas et al. (2006) [120] | In vitro (human) | Plasma + Hcy | nd | _ |
| Marchi et al. (2008) [121] | In vitro | Plasma + 13, 19, 52 μM Hcy | − + Lag phase − Max Abs | _ |
| Marchi et al. (2008) [121] | In vitro | Plasma + 251 μM Hcy | = = Lag phase = Max Abs | nd |
| Marchi et al. (2008) [121] | In vitro | Fibrinogen + 408 μM Hcy | − + Lag phase − Max Abs | nd |
| Sauls et al. (2011) [122] | In vitro | Fibrinogen + 300 μM HTL | nd | _ |
| Malinowska et al. (2011) [123] | In vitro | Plasma + 0.1–1 mM HTL | + | _ |
| Genoud et al. (2014) [124] | In vitro | Fibrinogen + 100, 500 and 1000 μmol/L HTL | − + Lag phase − Max Abs | nd |
| Cellai et al. (2014) [125] | Clinical trial—patients with a history of PE | Plasma | nd | _ |
| Clot Analysis | ||
|---|---|---|
| Author | Method | Clot Analysis |
| Lauricella et al. (2002) [118] | Analysis of fibrin networks by electronic microscopy | + fiber diameter + density + branched + shorter |
| Sauls et al. (2003) [115] | SEM of plasma clots | − fiber diameter + shorter + density |
| Lauricella et al. (2006) [119] | Electron microscopy | + fiber diameter + density |
| Sauls et al. (2006) [114] | Mass spectrometric analysis | − fiber diameter + density |
| Undas et al. (2006) [120] | Fibrin permeation analysis | + density |
| Marchi et al. (2008) [121] | SEM | = fiber diameter |
| Genoud et al. (2014) [124] | SEM | − fiber diameter + density |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giurranna, E.; Nencini, F.; Borghi, S.; Barbaro, I.; Taddei, N.; Fiorillo, C.; Becatti, M. Homocysteinylation of Fibrinogen: A Post-Translational Link to Thrombosis. Int. J. Mol. Sci. 2025, 26, 5471. https://doi.org/10.3390/ijms26125471
Giurranna E, Nencini F, Borghi S, Barbaro I, Taddei N, Fiorillo C, Becatti M. Homocysteinylation of Fibrinogen: A Post-Translational Link to Thrombosis. International Journal of Molecular Sciences. 2025; 26(12):5471. https://doi.org/10.3390/ijms26125471
Chicago/Turabian StyleGiurranna, Elvira, Francesca Nencini, Serena Borghi, Ilenia Barbaro, Niccolò Taddei, Claudia Fiorillo, and Matteo Becatti. 2025. "Homocysteinylation of Fibrinogen: A Post-Translational Link to Thrombosis" International Journal of Molecular Sciences 26, no. 12: 5471. https://doi.org/10.3390/ijms26125471
APA StyleGiurranna, E., Nencini, F., Borghi, S., Barbaro, I., Taddei, N., Fiorillo, C., & Becatti, M. (2025). Homocysteinylation of Fibrinogen: A Post-Translational Link to Thrombosis. International Journal of Molecular Sciences, 26(12), 5471. https://doi.org/10.3390/ijms26125471