
LRP1 Shedding in Ricin-Induced Lung Injury: A Cell-Specific Response to Toxin Exposure


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
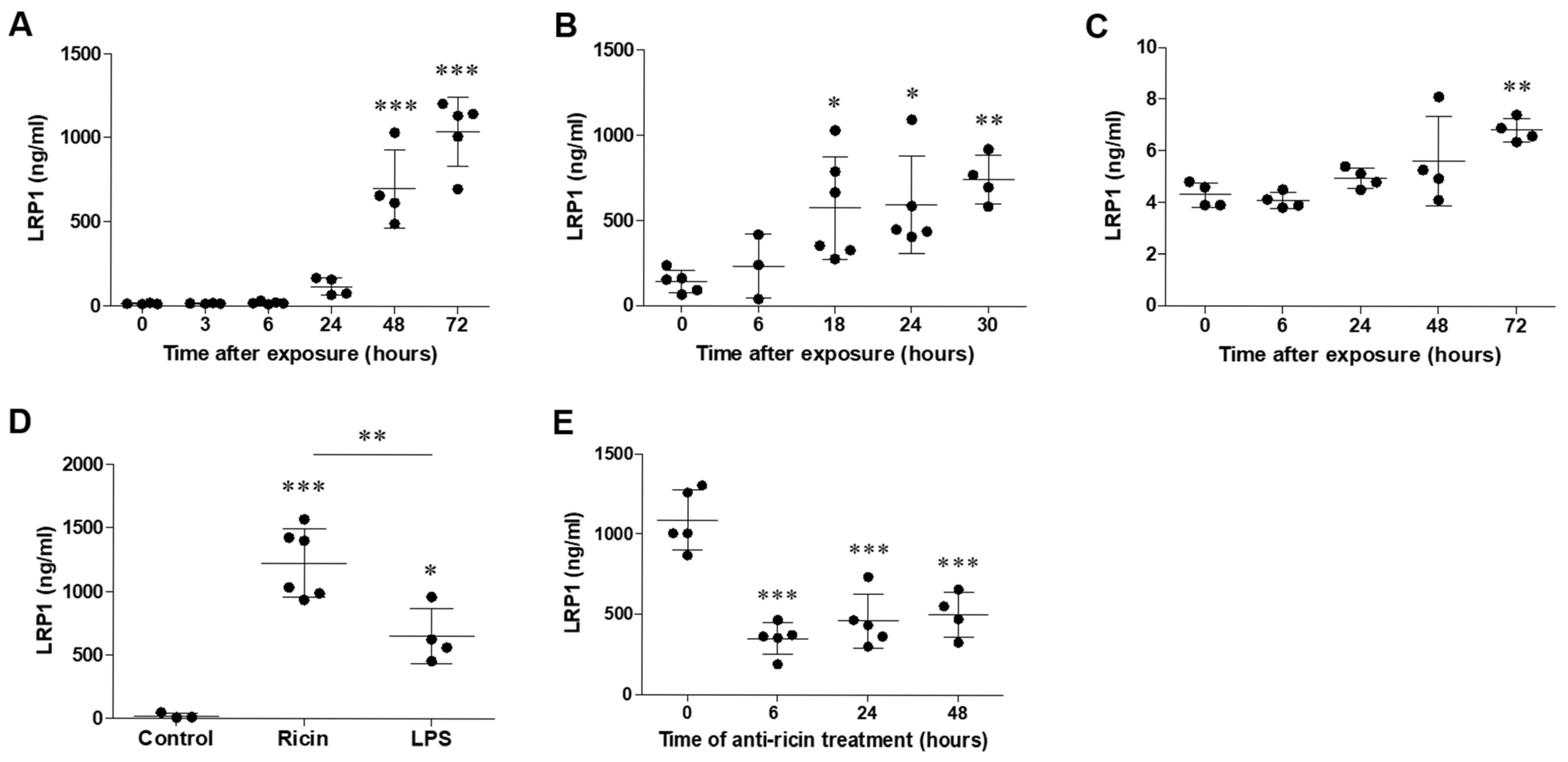
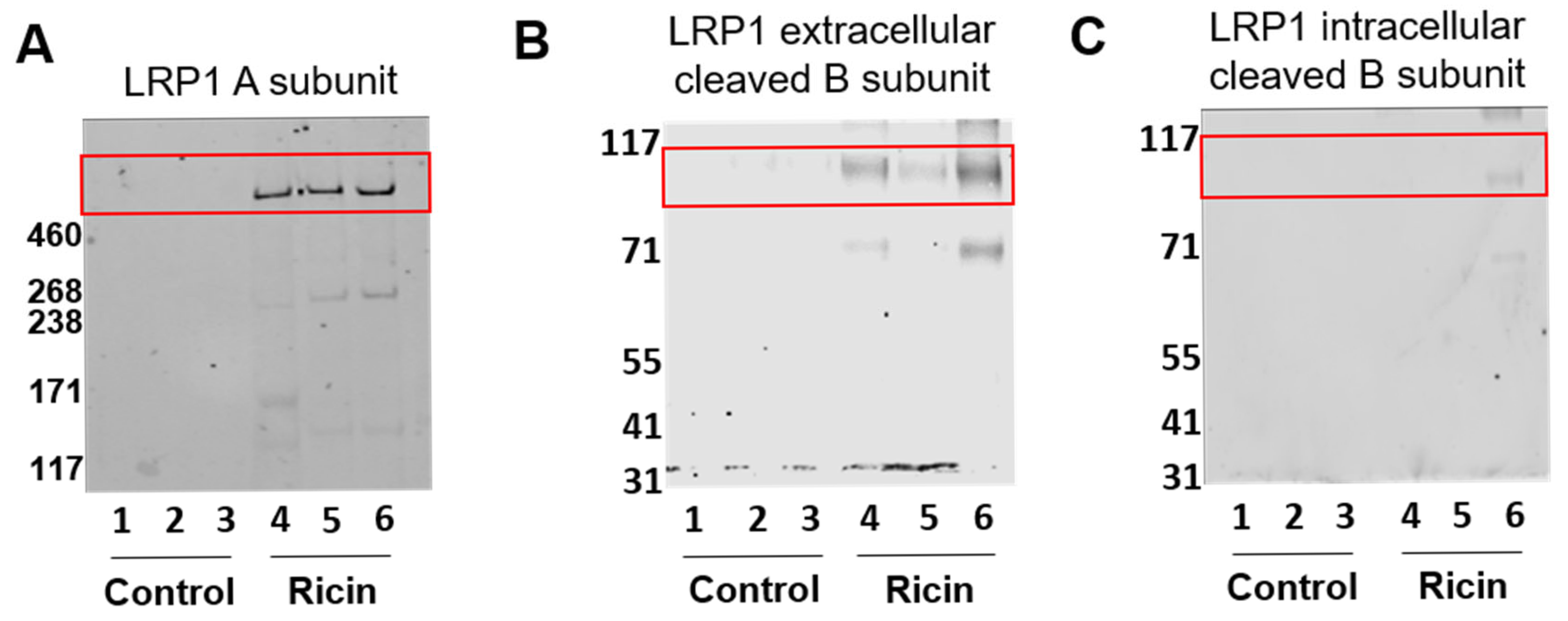
2.1. LRP1 Shedding in the Lungs Following Pulmonary Exposure to Ricin
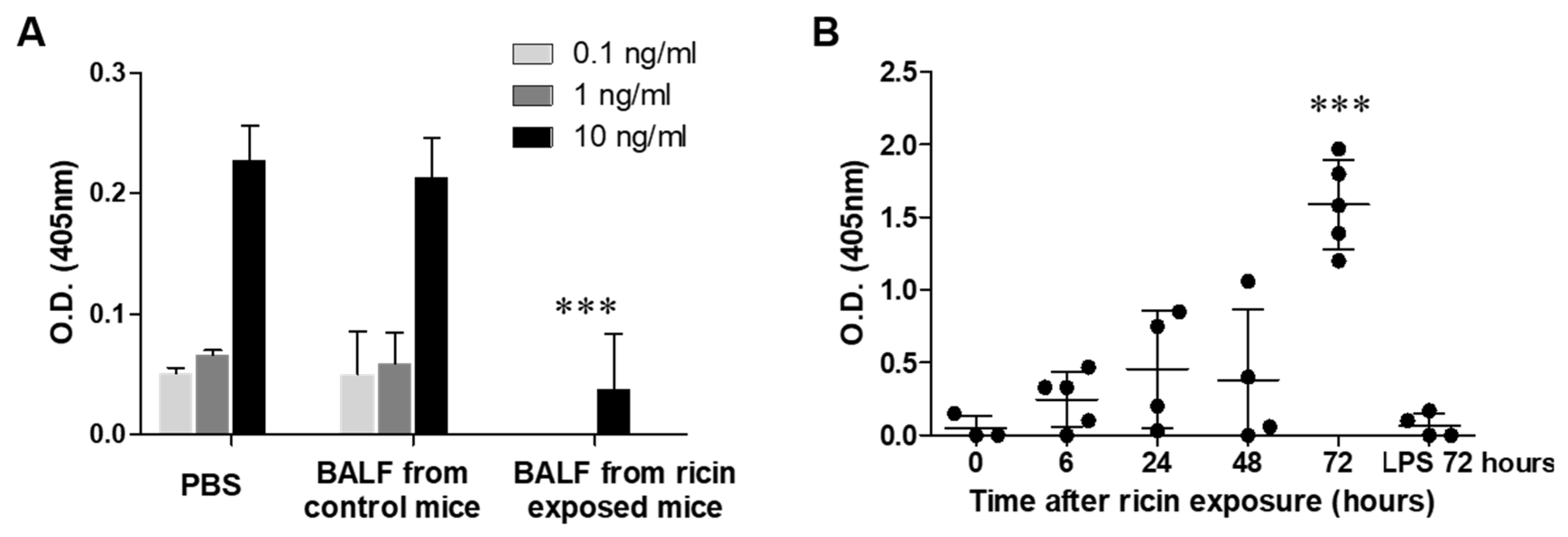
2.2. The Cleaved Configuration of LRP1 Retained the Ability to Bind Ricin
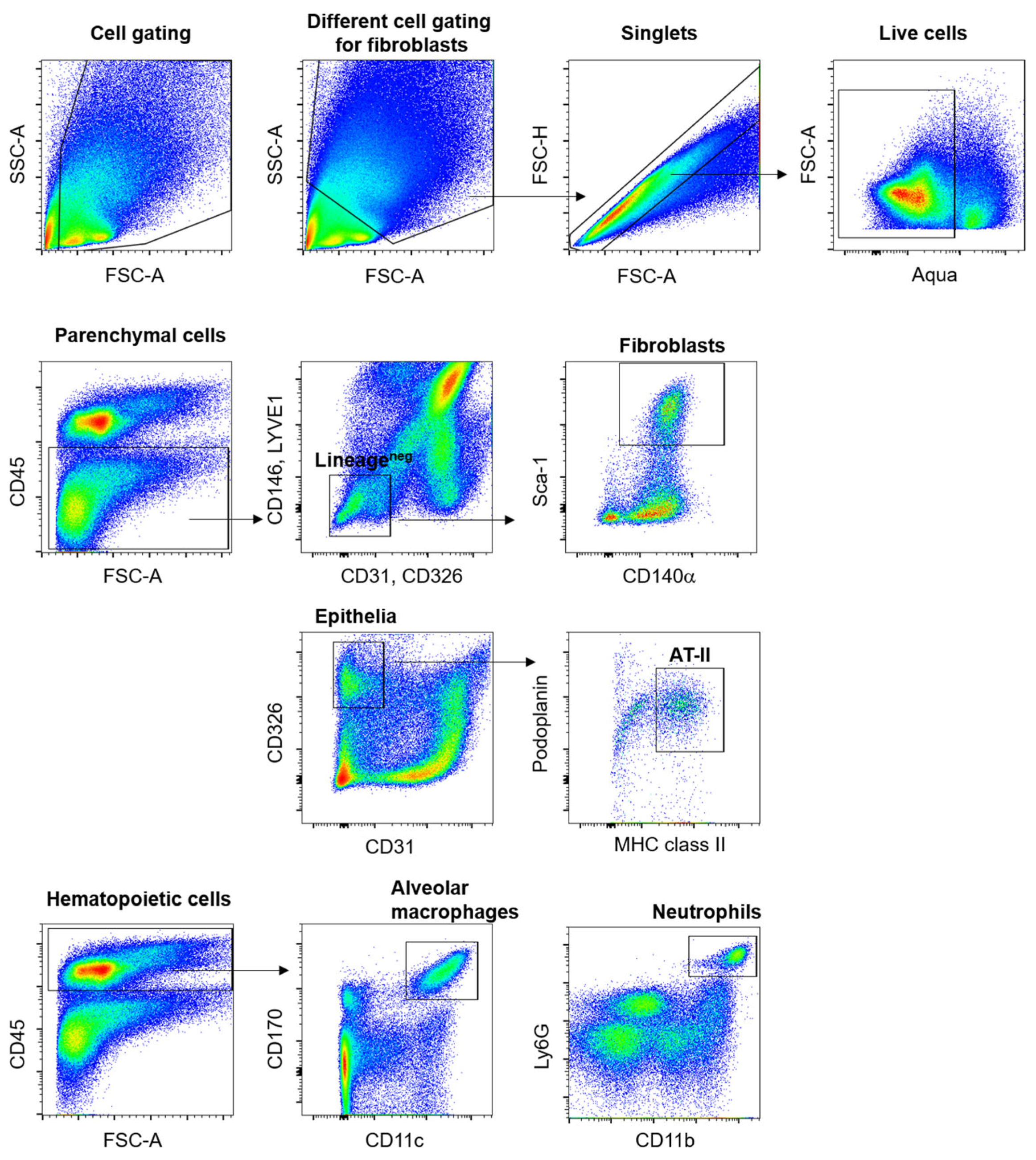
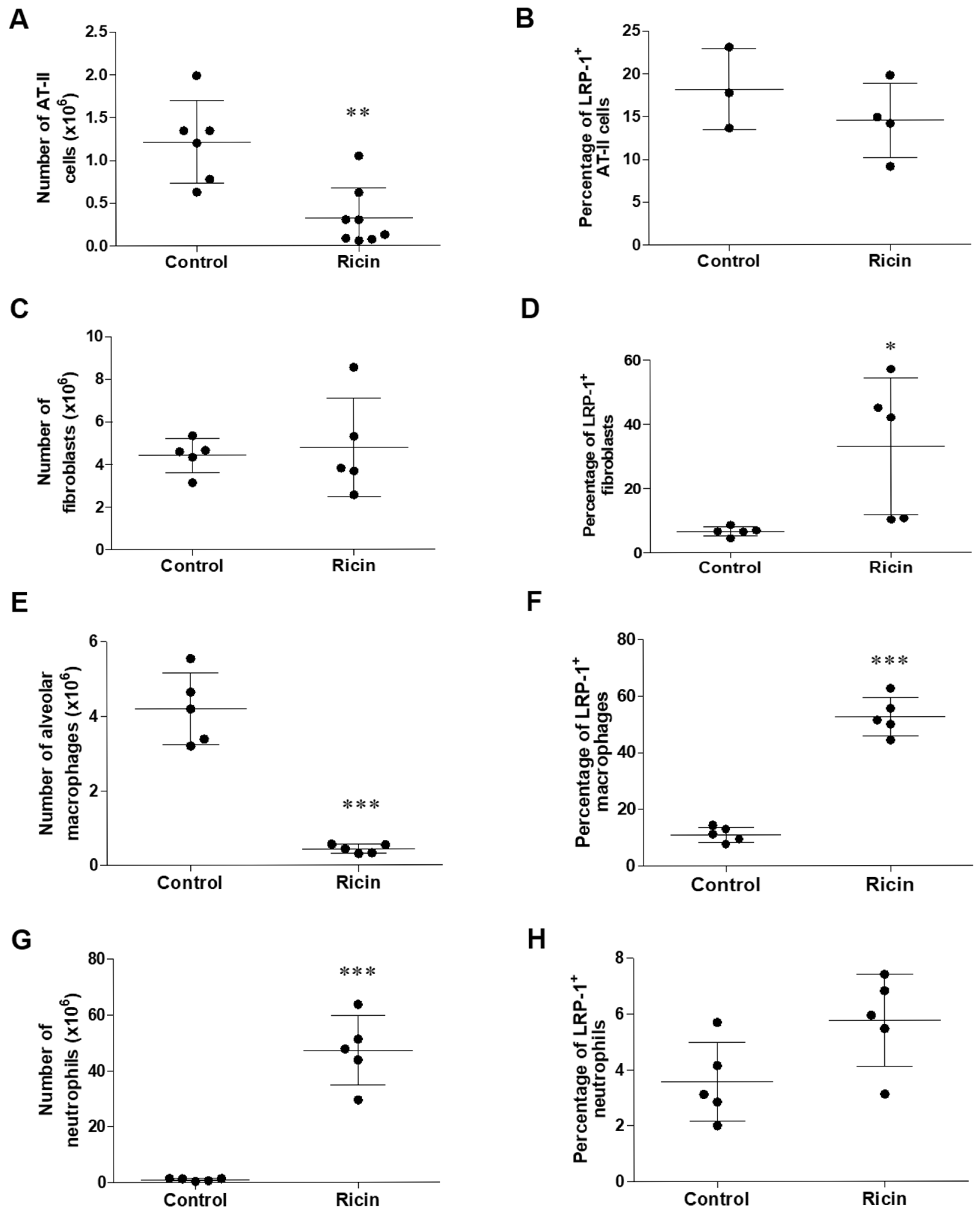
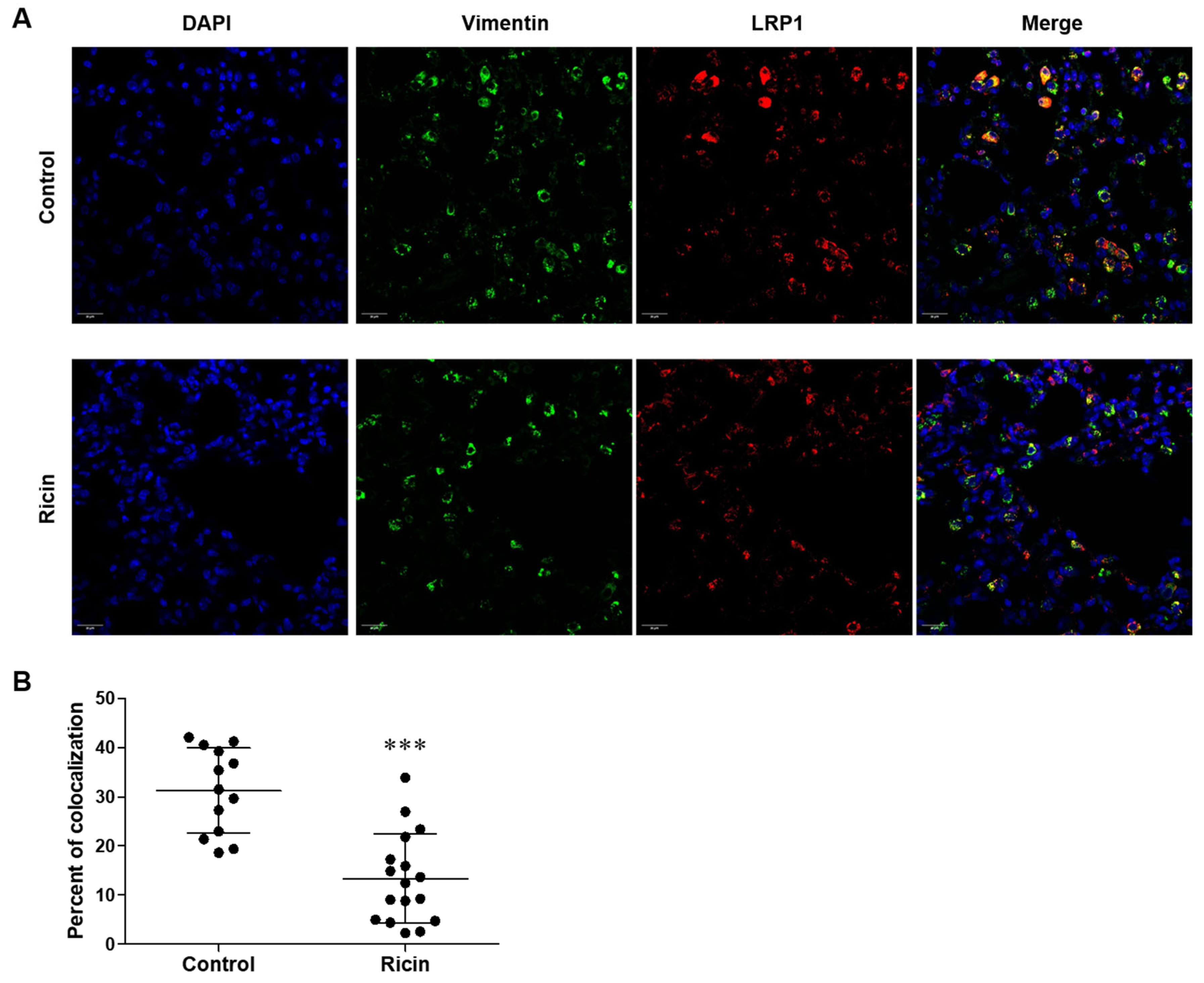
2.3. Differential Shedding of LPR1 in Lung Cell Populations Following Pulmonary Exposure to Ricin
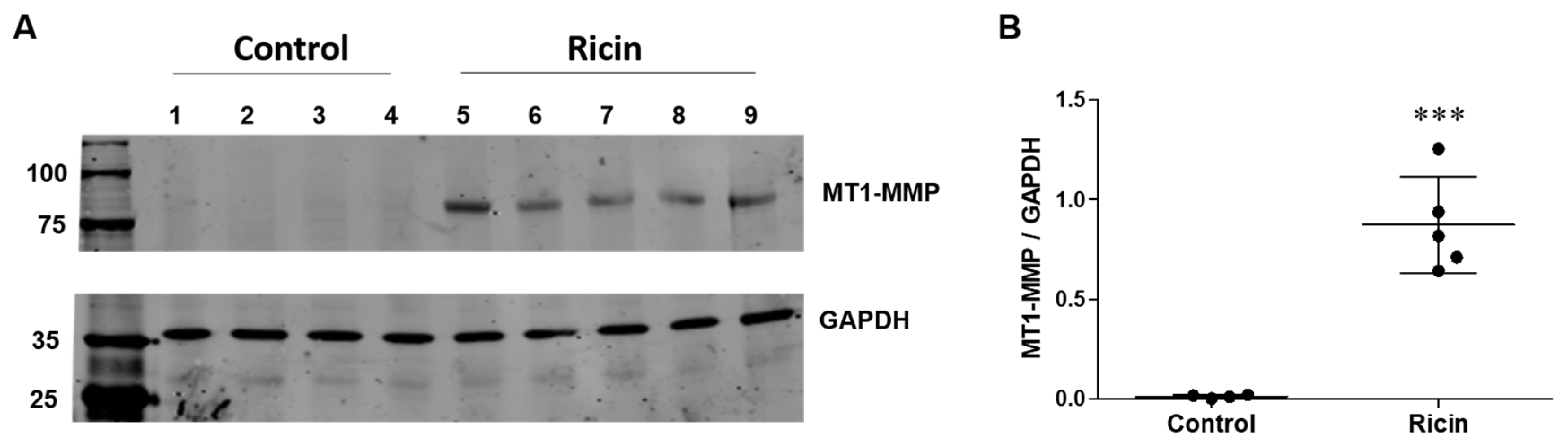
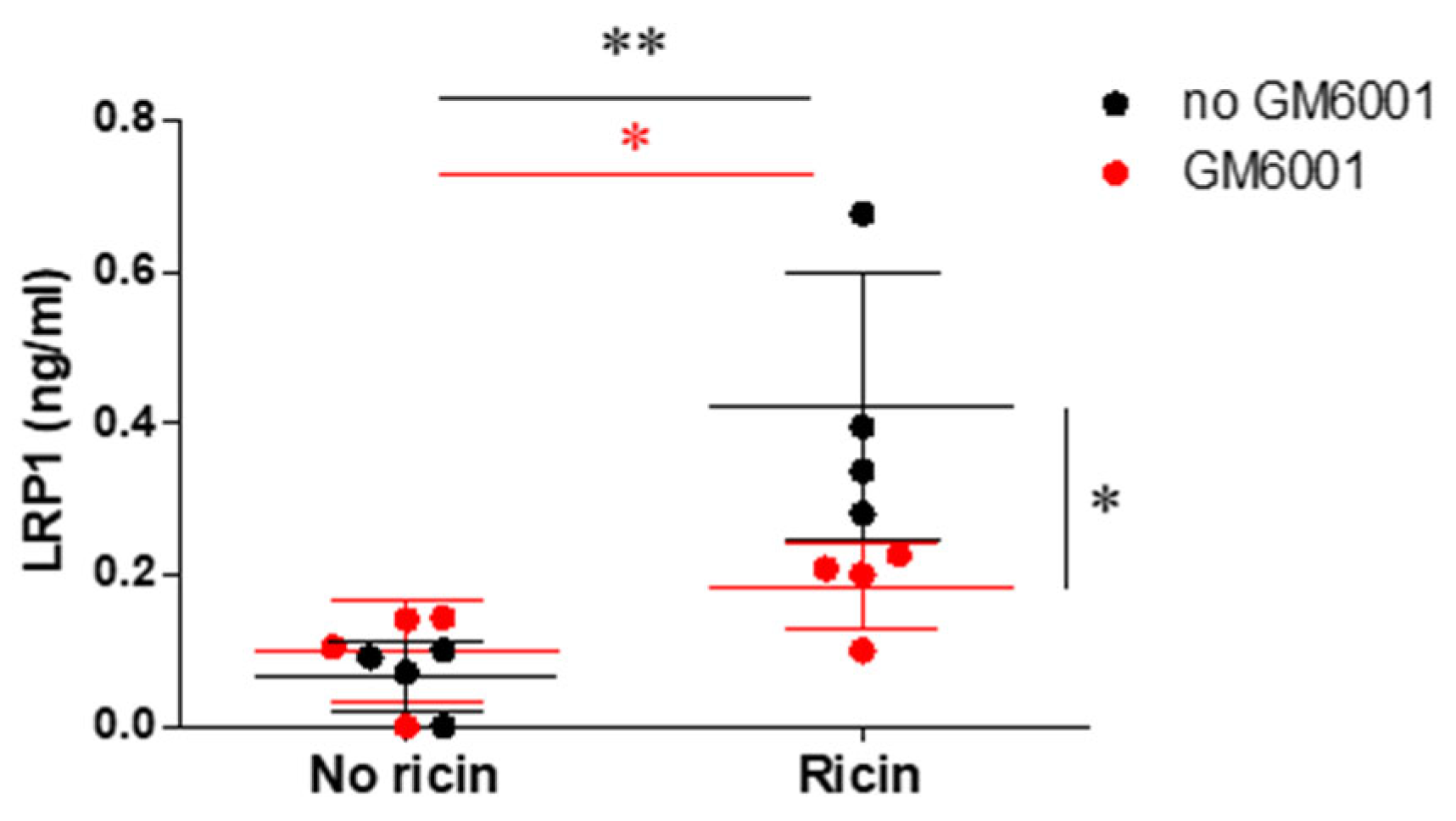
2.4. Evaluation of Metalloproteinase-Mediated LRP1 Shedding in Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Animal Ethics
4.2. Animals
4.3. Ricin Preparation
4.4. Animal Intoxication and Treatment
4.5. BALF Collection
4.6. Western Blot Analysis
4.7. ELISA Analysis
4.7.1. LRP1 ELISA
4.7.2. Ricin ELISA
4.7.3. ELISA for LRP1-Ricin Complexes
4.8. Flow Cytometric Analysis
4.9. Immunohistochemistry
4.10. Mouse Embryonic Fibroblasts
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LRP1 | low-density lipoprotein receptor-related protein 1 |
| AT-II | Alveolar epithelial type II |
| RTA | Ricin toxin A (subunit) |
| RTB | Ricin toxin B (subunit) |
| Gb3 | Globotriaosylceramide |
| TEM8 | Tumor endothelial marker 8 |
| CMG2 | Capillary morphogenesis protein 2 |
| PEA | Pseudomonas aeruginosa exotoxin A |
| kDa | Kilodalton |
| ARDS | Acute respiratory distress syndrome |
| BALF | Bronchoalveolar lavage fluid |
| sLRP1 | Soluble LRP1 |
| LPS | Lipopolysaccharide |
| MMPs | Matrix metalloproteinases |
| MT1-MMP | Membrane type-1 MMP |
| MEF | Mouse Embryonic Fibroblasts |
| ECM | Extracellular matrix |
| uPAR | urokinase receptor |
| uPA | urokinase-type plasminogen activator |
| ADAM | a disintegrin and metalloproteinase |
Appendix A
References
- Greenfield, R.A.; Slater, L.N.; Bronze, M.S.; Brown, B.R.; Jackson, R.; Iandolo, J.J.; Hutchins, J.B. Microbiological, Biological, and Chemical Weapons of Warfare and Terrorism. Am. J. Med. Sci. 2002, 323, 326–340. [Google Scholar] [CrossRef]
- Abbes, M.; Montana, M.; Curti, C.; Vanelle, P. Ricin Poisoning: A Review on Contamination Source, Diagnosis, Treatment, Prevention and Reporting of Ricin Poisoning. Toxicon 2021, 195, 86–92. [Google Scholar] [CrossRef]
- Gal, Y.; Mazor, O.; Alcalay, R.; Seliger, N.; Aftalion, M.; Sapoznikov, A.; Falach, R.; Kronman, C.; Sabo, T. Antibody/Doxycycline Combined Therapy for Pulmonary Ricinosis: Attenuation of Inflammation Improves Survival of Ricin-Intoxicated Mice. Toxicol. Rep. 2014, 1, 496–504. [Google Scholar] [CrossRef]
- Wilhelmsen, C.L.; Pitt, M.L.M. Lesions of Acute Inhaled Lethal Ricin Intoxication in Rhesus Monkeys. Vet. Pathol. 1996, 33, 296–302. [Google Scholar] [CrossRef]
- Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Gal, Y.; Seliger, N.; Sabo, T.; Kronman, C. Diverse Profiles of Ricin-Cell Interactions in the Lung Following Intranasal Exposure to Ricin. Toxins 2015, 7, 4817–4831. [Google Scholar] [CrossRef]
- Katalan, S.; Falach, R.; Rosner, A.; Goldvaser, M.; Brosh-Nissimov, T.; Dvir, A.; Mizrachi, A.; Goren, O.; Cohen, B.; Gal, Y. A Novel Swine Model of Ricin-Induced Acute Respiratory Distress Syndrome. Dis. Model. Mech. 2017, 10, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Amukele, T.K.; Roday, S.; Schramm, V.L. Ricin A-Chain Activity on Stem-Loop and Unstructured DNA Substrates. Biochemistry 2005, 44, 4416–4425. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The Mechanism of Action of Ricin and Related Toxic Lectins on Eukaryotic Ribosomes. The Site and the Characteristics of the Modification in 28 S Ribosomal RNA Caused by the Toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [CrossRef] [PubMed]
- Sperti, S.; Montanaro, L.; Mattioli, A.; Testoni, G. Relationship between Elongation Factor I- and Elongation Factor II- Dependent Guanosine Triphosphatase Activities of Ribosomes. Inhibition of Both Activities by Ricin. Biochem. J. 1975, 148, 447–451. [Google Scholar] [CrossRef]
- Olsnes, S.; Fernandez-Puentes, C.; Carrasco, L.; Vazquez, D. Ribosome Inactivation by the Toxic Lectins Abrin and Ricin. Kinetics of the Enzymic Activity of the Toxin A-Chains. Eur. J. Biochem. 1975, 60, 281–288. [Google Scholar] [CrossRef]
- Olmo, N.; Turnay, J.; González de Buitrago, G.; López de Silanes, I.; Gavilanes, J.G.; Lizarbe, M.A. Cytotoxic Mechanism of the Ribotoxin Alpha-Sarcin. Induction of Cell Death via Apoptosis. Eur. J. Biochem. 2001, 268, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Hartley, M.R.; Lord, J.M. Cytotoxic Ribosome-Inactivating Lectins from Plants. Biochim. Biophys. Acta 2004, 1701, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Sarfaty, S.; Chang, T.T.; Palmer, L.M.; Jobling, M.G.; Holmes, R.K.; Hol, W.G. Surprising Leads for a Cholera Toxin Receptor-Binding Antagonist: Crystallographic Studies of CTB Mutants. Structure 1995, 3, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Römer, W. Shiga Toxins—From Cell Biology to Biomedical Applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal Structure of the Holotoxin from Shigella Dysenteriae at 2.5 A Resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef]
- van der Goot, G.; Young, J.A.T. Receptors of Anthrax Toxin and Cell Entry. Mol. Aspects Med. 2009, 30, 406–412. [Google Scholar] [CrossRef]
- Lord, M.J.; Jolliffe, N.A.; Marsden, C.J.; Pateman, C.S.; Smith, D.C.; Spooner, R.A.; Watson, P.D.; Roberts, L.M. Ricin. Mechanisms of Cytotoxicity. Toxicol. Rev. 2003, 22, 53–64. [Google Scholar] [CrossRef]
- Brandt, N.N.; Chikishev, A.Y.; Sotnikov, A.I.; Savochkina, Y.A.; Agapov, I.I.; Tonevitsky, A.G. Ricin, Ricin Agglutinin, and the Ricin Binding Subunit Structural Comparison by Raman Spectroscopy. J. Mol. Struct. 2005, 735–736, 293–298. [Google Scholar] [CrossRef]
- Olsnes, S.; Kozlov, J.V. Ricin. Toxicon 2001, 39, 1723–1728. [Google Scholar] [CrossRef]
- Nicolson, G.L.; Blaustein, J.; Etzler, M.E. Characterization of Two Plant Lectins from Ricinus Communis and Their Quantitative Interaction with a Murine Lymphoma. Biochemistry 1974, 13, 196–204. [Google Scholar] [CrossRef]
- Falach, R.; Sapoznikov, A.; Gal, Y.; Israeli, O.; Leitner, M.; Seliger, N.; Ehrlich, S.; Kronman, C.; Sabo, T. Quantitative Profiling of the In Vivo Enzymatic Activity of Ricin Reveals Disparate Depurination of Different Pulmonary Cell Types. Toxicol. Lett. 2016, 258, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Falach, R.; Sapoznikov, A.; Gal, Y.; Elhanany, E.; Evgy, Y.; Shifman, O.; Aftalion, M.; Ehrlich, S.; Lazar, S.; Sabo, T.; et al. The Low Density Receptor-Related Protein 1 Plays a Significant Role in Ricin-Mediated Intoxication of Lung Cells. Sci. Rep. 2020, 10, 9007. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V.; Hanson, A.J.; Knisely, J.; Bu, G.; Fitzgerald, D.J. LRP 1 B Functions as a Receptor for Pseudomonas Exotoxin. Biochim. Biophys. Acta 2005, 1741, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. LDL Receptor-Related Protein 1: Unique Tissue-Specific Functions Revealed by Selective Gene Knockout Studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef]
- Hussain, M.M. Structural, Biochemical and Signaling Properties of the Low-Density Lipoprotein Receptor Gene Family. Front. Biosci. 2001, 6, D417–D428. [Google Scholar] [CrossRef]
- Nykjaer, A.; Petersen, C.M.; Møller, B.; Jensen, P.H.; Moestrup, S.K.; Holtet, T.L.; Etzerodt, M.; Thøgersen, H.C.; Munch, M.; Andreasen, P.A. Purified Alpha 2-Macroglobulin Receptor/LDL Receptor-Related Protein Binds Urokinase. Plasminogen Activator Inhibitor Type-1 Complex. Evidence That the Alpha 2-Macroglobulin Receptor Mediates Cellular Degradation of Urokinase Receptor-Bound Complexes. J. Biol. Chem. 1992, 267, 14543–14546. [Google Scholar] [CrossRef]
- Yamamoto, K.; Scilabra, S.D.; Bonelli, S.; Jensen, A.; Scavenius, C.; Enghild, J.J.; Strickland, D.K. Novel Insights into the Multifaceted and Tissue-Specific Roles of the Endocytic Receptor LRP1. J. Biol. Chem. 2024, 300, 107521. [Google Scholar] [CrossRef]
- Wujak, L.; Schnieder, J.; Schaefer, L.; Wygrecka, M. LRP1: A Chameleon Receptor of Lung Inflammation and Repair. Matrix Biol. 2018, 68–69, 366–381. [Google Scholar] [CrossRef]
- May, P.; Herz, J.; Bock, H.H. Molecular Mechanisms of Lipoprotein Receptor Signalling. Cell. Mol. Life Sci. 2005, 62, 2325–2338. [Google Scholar] [CrossRef]
- Garcia-Arcos, I.; Park, S.S.; Mai, M.; Alvarez-Buve, R.; Chow, L.; Cai, H.; Baumlin-Schmid, N.; Agudelo, C.W.; Martinez, J.; Kim, M.D.; et al. LRP1 Loss in Airway Epithelium Exacerbates Smoke-Induced Oxidative Damage and Airway Remodeling. J. Lipid Res. 2022, 63, 100185. [Google Scholar] [CrossRef]
- Nykjaer, A.; Kjøller, L.; Cohen, R.L.; Lawrence, D.A.; Garni-Wagner, B.A.; Todd, R.F., 3rd; van Zonneveld, A.J.; Gliemann, J.; Andreasen, P.A. Regions Involved in Binding of Urokinase-Type-1 Inhibitor Complex and pro-Urokinase to the Endocytic Alpha 2-Macroglobulin Receptor/Low Density Lipoprotein Receptor-Related Protein. Evidence That the Urokinase Receptor Protects pro-Urokinase against Bind. J. Biol. Chem. 1994, 269, 25668–25676. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; van Den Berg, B.M.; Lookene, A.; Olivecrona, G.; Pannekoek, H.; van Zonneveld, A.J. The Second and Fourth Cluster of Class A Cysteine-Rich Repeats of the Low Density Lipoprotein Receptor-Related Protein Share Ligand-Binding Properties. J. Biol. Chem. 1999, 274, 31305–31311. [Google Scholar] [CrossRef] [PubMed]
- Potere, N.; Del Buono, M.G.; Mauro, A.G.; Abbate, A.; Toldo, S. Low Density Lipoprotein Receptor-Related Protein-1 in Cardiac Inflammation and Infarct Healing. Front. Cardiovasc. Med. 2019, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Brifault, C.; Gilder, A.S.; Laudati, E.; Banki, M.; Gonias, S.L. Shedding of Membrane-Associated LDL Receptor-Related Protein-1 from Microglia Amplifies and Sustains Neuroinflammation. J. Biol. Chem. 2017, 292, 18699–18712. [Google Scholar] [CrossRef]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the Pathogenesis of Alzheimer’s Disease: Evidence from Clinical and Preclinical Studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef]
- de Gonzalo-Calvo, D.; Elosua, R.; Vea, A.; Subirana, I.; Sayols-Baixeras, S.; Marrugat, J.; Llorente-Cortés, V. Soluble Low-Density Lipoprotein Receptor-Related Protein 1 as a Biomarker of Coronary Risk: Predictive Capacity and Association with Clinical Events. Atherosclerosis 2019, 287, 93–99. [Google Scholar] [CrossRef]
- Wygrecka, M.; Wilhelm, J.; Jablonska, E.; Zakrzewicz, D.; Preissner, K.T.; Seeger, W.; Guenther, A.; Markart, P. Shedding of Low-Density Lipoprotein Receptor–Related Protein-1 in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2011, 184, 438–448. [Google Scholar] [CrossRef]
- Gorovoy, M.; Gaultier, A.; Campana, W.M.; Firestein, G.S.; Gonias, S.L. Inflammatory Mediators Promote Production of Shed LRP1/CD91, Which Regulates Cell Signaling and Cytokine Expression by Macrophages. J. Leukoc. Biol. 2010, 88, 769–778. [Google Scholar] [CrossRef]
- Quinn, K.A.; Pye, V.J.; Dai, Y.P.; Chesterman, C.N.; Owensby, D.A. Characterization of the Soluble Form of the Low Density Lipoprotein Receptor-Related Protein (LRP). Exp. Cell Res. 1999, 251, 433–441. [Google Scholar] [CrossRef]
- Grimsley, P.G.; Quinn, K.A.; Owensby, D.A. Soluble Low-Density Lipoprotein Receptor-Related Protein. Trends Cardiovasc. Med. 1998, 8, 363–368. [Google Scholar] [CrossRef]
- Falach, R.; Sapoznikov, A.; Alcalay, R.; Aftalion, M.; Ehrlich, S.; Makovitzki, A.; Agami, A.; Mimran, A.; Rosner, A.; Sabo, T.; et al. Generation of Highly Efficient Equine-Derived Antibodies for Post-Exposure Treatment of Ricin Intoxications by Vaccination with Monomerized Ricin. Toxins 2018, 10, 466. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.R.; Riordan, J.F. Membrane Proteins with Soluble Counterparts: Role of Proteolysis in the Release of Transmembrane Proteins. Biochemistry 1991, 30, 10065–10074. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Heinrich, P.C. Soluble Receptors for Cytokines and Growth Factors: Generation and Biological Function. Biochem. J. 1994, 300 Pt 2, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Maliszewski, C.R.; Sato, T.A.; Davison, B.; Jacobs, C.A.; Finkelman, F.D.; Fanslow, W.C. In Vivo Biological Effects of Recombinant Soluble Interleukin-4 Receptor. Proc. Soc. Exp. Biol. Med. 1994, 206, 233–237. [Google Scholar] [CrossRef]
- Quinn, K.A.; Grimsley, P.G.; Dai, Y.P.; Tapner, M.; Chesterman, C.N.; Owensby, D.A. Soluble Low Density Lipoprotein Receptor-Related Protein (LRP) Circulates in Human Plasma. J. Biol. Chem. 1997, 272, 23946–23951. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, J.; Tran, H.; Verbeek, M.M.; Reiss, K.; Estus, S.; Bu, G. LRP1 Shedding in Human Brain: Roles of ADAM10 and ADAM17. Mol. Neurodegener. 2009, 4, 17. [Google Scholar] [CrossRef]
- Yamamoto, K.; Santamaria, S.; Botkjaer, K.A.; Dudhia, J.; Troeberg, L.; Itoh, Y.; Murphy, G.; Nagase, H. Inhibition of Shedding of Low-Density Lipoprotein Receptor-Related Protein 1 Reverses Cartilage Matrix Degradation in Osteoarthritis. Arthritis Rheumatol. 2017, 69, 1246–1256. [Google Scholar] [CrossRef]
- Scilabra, S.D.; Troeberg, L.; Yamamoto, K.; Emonard, H.; Thøgersen, I.; Enghild, J.J.; Strickland, D.K.; Nagase, H. Differential Regulation of Extracellular Tissue Inhibitor of Metalloproteinases-3 Levels by Cell Membrane-Bound and Shed Low Density Lipoprotein Receptor-Related Protein 1. J. Biol. Chem. 2013, 288, 332–342. [Google Scholar] [CrossRef]
- Yamamoto, K.; Okano, H.; Miyagawa, W.; Visse, R.; Shitomi, Y.; Santamaria, S.; Dudhia, J.; Troeberg, L.; Strickland, D.K.; Hirohata, S.; et al. MMP-13 Is Constitutively Produced in Human Chondrocytes and Co-Endocytosed with ADAMTS-5 and TIMP-3 by the Endocytic Receptor LRP1. Matrix Biol. 2016, 56, 57–73. [Google Scholar] [CrossRef]
- Gaultier, A.; Hollister, M.; Reynolds, I.; Hsieh, E.; Gonias, S.L. LRP1 Regulates Remodeling of the Extracellular Matrix by Fibroblasts. Matrix Biol. 2010, 29, 22–30. [Google Scholar] [CrossRef]
- Lin, L.; Hu, K. LRP-1: Functions, Signaling and Implications in Kidney and Other Diseases. Int. J. Mol. Sci. 2014, 15, 22887–22901. [Google Scholar] [CrossRef] [PubMed]
- Schnieder, J.; Mamazhakypov, A.; Birnhuber, A.; Wilhelm, J.; Kwapiszewska, G.; Ruppert, C.; Markart, P.; Wujak, L.; Rubio, K.; Barreto, G.; et al. Loss of LRP1 Promotes Acquisition of Contractile-Myofibroblast Phenotype and Release of Active TGF-Β1 from ECM Stores. Matrix Biol. 2020, 88, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Sizova, O.; John, L.S.; Ma, Q.; Molldrem, J.J. Multi-Faceted Role of LRP1 in the Immune System. Front. Immunol. 2023, 14, 1166189. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Bock, H.H.; Nofer, J.-R. Low Density Receptor-Related Protein 1 (LRP1) Promotes Anti-Inflammatory Phenotype in Murine Macrophages. Cell Tissue Res. 2013, 354, 887–889. [Google Scholar] [CrossRef]
- Mantuano, E.; Brifault, C.; Lam, M.S.; Azmoon, P.; Gilder, A.S.; Gonias, S.L. LDL Receptor-Related Protein-1 Regulates NFκB and MicroRNA-155 in Macrophages to Control the Inflammatory Response. Proc. Natl. Acad. Sci. USA 2016, 113, 1369–1374. [Google Scholar] [CrossRef]
- Emonard, H.; Marbaix, E. Low-Density Lipoprotein Receptor-Related Protein in Metalloproteinase-Mediated Pathologies: Recent Insights. Met. Med. 2015, 9, 9–18. [Google Scholar] [CrossRef]
- Gaultier, A.; Arandjelovic, S.; Niessen, S.; Overton, C.D.; Linton, M.F.; Fazio, S.; Campana, W.M.; Cravatt, B.F., 3rd; Gonias, S.L. Regulation of Tumor Necrosis Factor Receptor-1 and the IKK-NF-KappaB Pathway by LDL Receptor-Related Protein Explains the Antiinflammatory Activity of This Receptor. Blood 2008, 111, 5316–5325. [Google Scholar] [CrossRef]
- Overton, C.D.; Yancey, P.G.; Major, A.S.; Linton, M.F.; Fazio, S. Deletion of Macrophage LDL Receptor-Related Protein Increases Atherogenesis in the Mouse. Circ. Res. 2007, 100, 670–677. [Google Scholar] [CrossRef]
- Torii, K.; Iida, K.; Miyazaki, Y.; Saga, S.; Kondoh, Y.; Taniguchi, H.; Taki, F.; Takagi, K.; Matsuyama, M.; Suzuki, R. Higher Concentrations of Matrix Metalloproteinases in Bronchoalveolar Lavage Fluid of Patients with Adult Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 1997, 155, 43–46. [Google Scholar] [CrossRef]
- Fligiel, S.E.G.; Standiford, T.; Fligiel, H.M.; Tashkin, D.; Strieter, R.M.; Warner, R.L.; Johnson, K.J.; Varani, J. Matrix Metalloproteinases and Matrix Metalloproteinase Inhibitors in Acute Lung Injury. Hum. Pathol. 2006, 37, 422–430. [Google Scholar] [CrossRef]
- Ricou, B.; Nicod, L.; Lacraz, S.; Welgus, H.G.; Suter, P.M.; Dayer, J.M. Matrix Metalloproteinases and TIMP in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 1996, 154, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Sapoznikov, A.; Gal, Y.; Falach, R.; Sagi, I.; Ehrlich, S.; Lerer, E.; Makovitzki, A.; Aloshin, A.; Kronman, C.; Sabo, T. Early Disruption of the Alveolar-Capillary Barrier in a Ricin-Induced Ards Mouse Model: Neutrophil-Dependent and-Independent Impairment of Junction Proteins. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L255–L268. [Google Scholar] [CrossRef] [PubMed]
- Warner, R.L.; Lewis, C.S.; Beltran, L.; Younkin, E.M.; Varani, J.; Johnson, K.J. The Role of Metalloelastase in Immune Complex-Induced Acute Lung Injury. Am. J. Pathol. 2001, 158, 2139–2144. [Google Scholar] [CrossRef]
- Warner, R.L.; Beltran, L.; Younkin, E.M.; Lewis, C.S.; Weiss, S.J.; Varani, J.; Johnson, K.J. Role of Stromelysin 1 and Gelatinase B in Experimental Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2001, 24, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Delclaux, C.; d’Ortho, M.P.; Delacourt, C.; Lebargy, F.; Brun-Buisson, C.; Brochard, L.; Lemaire, F.; Lafuma, C.; Harf, A. Gelatinases in Epithelial Lining Fluid of Patients with Adult Respiratory Distress Syndrome. Am. J. Physiol. 1997, 272, L442–L451. [Google Scholar] [CrossRef]
- Idell, S.; Maunder, R.; Fein, A.M.; Switalska, H.I.; Tuszynski, G.P.; McLarty, J.; Niewiarowski, S. Platelet-Specific Alpha-Granule Proteins and Thrombospondin in Bronchoalveolar Lavage in the Adult Respiratory Distress Syndrome. Chest 1989, 96, 1125–1132. [Google Scholar] [CrossRef]
- Salicioni, A.M.; Mizelle, K.S.; Loukinova, E.; Mikhailenko, I.; Strickland, D.K.; Gonias, S.L. The Low Density Lipoprotein Receptor-Related Protein Mediates Fibronectin Catabolism and Inhibits Fibronectin Accumulation on Cell Surfaces. J. Biol. Chem. 2002, 277, 16160–16166. [Google Scholar] [CrossRef]
- Modig, J.; Borg, T. Biochemical Markers in a Porcine Model of Adult Respiratory Distress Syndrome Induced by Endotoxemia. Resuscitation 1986, 14, 225–236. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapoznikov, A.; Evgy, Y.; Aftalion, M.; Falach, R. LRP1 Shedding in Ricin-Induced Lung Injury: A Cell-Specific Response to Toxin Exposure. Int. J. Mol. Sci. 2025, 26, 5448. https://doi.org/10.3390/ijms26125448
Sapoznikov A, Evgy Y, Aftalion M, Falach R. LRP1 Shedding in Ricin-Induced Lung Injury: A Cell-Specific Response to Toxin Exposure. International Journal of Molecular Sciences. 2025; 26(12):5448. https://doi.org/10.3390/ijms26125448
Chicago/Turabian StyleSapoznikov, Anita, Yentl Evgy, Moshe Aftalion, and Reut Falach. 2025. "LRP1 Shedding in Ricin-Induced Lung Injury: A Cell-Specific Response to Toxin Exposure" International Journal of Molecular Sciences 26, no. 12: 5448. https://doi.org/10.3390/ijms26125448
APA StyleSapoznikov, A., Evgy, Y., Aftalion, M., & Falach, R. (2025). LRP1 Shedding in Ricin-Induced Lung Injury: A Cell-Specific Response to Toxin Exposure. International Journal of Molecular Sciences, 26(12), 5448. https://doi.org/10.3390/ijms26125448