
Inflammasomes in Cardiovascular Diseases: Current Knowledge and Future Perspectives
 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Inflammasomes
3. Inflammasomes and CVDs
3.1. Atherosclerosis
3.2. Heart Failure
3.3. Atrial Fibrillation
3.4. Chronic Kidney Disease and the NLRP3 Inflammasome
4. Special Focus: Pericarditis and Still’s Disease
5. New Pharmacology Strategies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIM2 | Absent in Melanoma 2 |
| ASC | Apoptosis-associated Speck-like protein containing a CARD |
| AS | Atherosclerosis |
| AOSD | Adult-Onset Still’s Disease |
| CAD | Coronary Artery Disease |
| CARD | Caspase Activation and Recruitment Domain |
| CKD | Chronic Kidney Disease |
| CVD | Cardiovascular Disease |
| DAMPs | Damage-Associated Molecular Patterns |
| ER | Endoplasmic Reticulum |
| FMF | Familial Mediterranean Fever |
| GSDMD | Gasdermin D |
| HF | Heart Failure |
| HFpEF | Heart Failure with Preserved Ejection Fraction |
| HFrEF | Heart Failure with Reduced Ejection Fraction |
| HIF-1α | Hypoxia-Inducible Factor 1-alpha |
| HMGB1 | High-Mobility Group Box 1 |
| IL | Interleukin |
| IR | Ischemia-Reperfusion |
| MAPK | Mitogen-Activated Protein Kinase |
| NACHT | Domain present in NAIP, CIITA, HET-E, and TP1 |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NLRC4 | NOD-Like Receptor Family CARD Domain Containing 4 |
| NLRP3 | NOD-Like Receptor Protein 3 |
| NSAIDs | Non-Steroidal Anti-Inflammatory Drugs |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PPAR-γ | Peroxisome Proliferator-Activated Receptor Gamma |
| PRRs | Pattern Recognition Receptors |
| PYD | Pyrin Domain |
| RAAS | Renin-Angiotensin-Aldosterone System |
| ROS | Reactive Oxygen Species |
| SGLT2 | Sodium-Glucose Cotransporter 2 |
| TGF-β | Transforming Growth Factor Beta |
| TLRs | Toll-Like Receptors |
| TNF | Tumor Necrosis Factor |
| TXNIP | Thioredoxin-Interacting Protein |
| VEGF-A | Vascular Endothelial Growth Factor A |
| VLDL | Very Low-Density Lipoprotein |
References
- Olsen, M.B.; Gregersen, I.; Sandanger, Ø.; Yang, K.; Sokolova, M.; Halvorsen, B.E.; Gullestad, L.; Broch, K.; Aukrust, P.; Louwe, M.C. Targeting the Inflammasome in Cardiovascular Disease. JACC Basic. Transl. Sci. 2022, 7, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-Induced Pyroptotic Cell Death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Arioz, B.I.; Tarakcioglu, E.; Olcum, M.; Genc, S. The Role of Melatonin on NLRP3 Inflammasome Activation in Diseases. Antioxidants 2021, 10, 1020. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Mauro, A.G.; Salloum, F.; Tassell, B.W.; Abbate, A. The Inflammasome in Myocardial Injury and Cardiac Remodeling. Antioxid. Redox Signal. 2015, 22, 1146–1161. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Otsu, K. Mitochondria and Sterile Inflammation in the Heart. Curr. Opin. Physiol. 2018, 1, 68–74. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. AIM2 Inflammasome in Infection, Cancer, and Autoimmunity: Role in DNA Sensing, Inflammation, and Innate Immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E. The AIM2 Inflammasome Is Essential for Host-Defense against Cytosolic Bacteria and DNA Viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef]
- Lammert, C.R.; Frost, E.L.; Bellinger, C.E.; Bolte, A.C.; McKee, C.A.; Hurt, M.E.; Paysour, M.J.; Ennerfelt, H.E.; Lukens, J.R. AIM2 Inflammasome Surveillance of DNA Damage Shapes Neurodevelopment. Nature 2020, 580, 647–652. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; McGeough, M.D.; Peña, C.A.; Marchetti, C.; Sonnino, C.; Tassell, B.W.; Salloum, F.N.; Voelkel, N.F.; Hoffman, H.M. Independent Roles of the Priming and the Triggering of the NLRP3 Inflammasome in the Heart. Cardiovasc. Res. 2015, 105, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, Ø.; Gao, E.; Ranheim, T.; Bliksøen, M.; Kaasbøll, O.J.; Alfsnes, K.; Nymo, S.H.; Rashidi, A.; Ohm, I.K.; Attramadal, H. NLRP3 Inflammasome Activation during Myocardial Ischemia Reperfusion Is Cardioprotective. Biochem. Biophys. Res. Commun. 2016, 469, 1012–1020. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.-Q.; Branton, W.G.; Power, C. Caspase-1 Inhibition Prevents Glial Inflammasome Activation and Pyroptosis in Models of Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 6065–6074. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S.G.D. Gasdermin D in pyroptosis. Cell Rep. 2021, 11, 2768–2782. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J. The NLRP3 Inflammasome Is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Todor, S.B.; Ichim, C.; Boicean, A.; Mihaila, R.G. Cardiovascular Risk in Philadelphia-Negative Myeloproliferative Neoplasms: Mechanisms and Implications—A Narrative Review. Curr. Issues Mol. Biol. 2024, 46, 8407–8423. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, G.; Ding, Q.; Zheng, F.; Shi, X.; Lin, Z.; Liang, Y. The ROS/TXNIP/NLRP3 Pathway Mediates LPS-Induced Microglial Inflammatory Response. Cytokine 2024, 181, 156677. [Google Scholar] [CrossRef]
- Luo, T.; Zhou, X.; Qin, M.; Lin, Y.; Lin, J.; Chen, G.; Liu, A.; Ouyang, D.; Chen, D.; Pan, H. Corilagin Restrains NLRP3 Inflammasome Activation and Pyroptosis through the ROS/TXNIP/NLRP3 Pathway to Prevent Inflammation. Oxid. Med. Cell Longev. 2022, 2022, 1652244. [Google Scholar] [CrossRef]
- Garg, N.J. Inflammasomes in Cardiovascular Diseases. Am. J. Cardiovasc. Dis. 2011, 1, 244–254. [Google Scholar]
- Toldo, S.; Abbate, A. The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.-H.; Qiao, C.; Jiang, C.-Y.; Zhang, K.-Z.; Ding, J.-H.; Hu, G. MicroRNA-7 Targets Nod-like Receptor Protein 3 Inflammasome to Modulate Neuroinflammation in the Pathogenesis of Parkinson’s Disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, T.A.; Kaiser, C.; Lawrence, C.B.; Brough, D.; Allan, S.M.; Green, J.P. The NLRP3 Inflammasome Is Essential for IL-18 Production in a Murine Model of Macrophage Activation Syndrome. Dis. Model. Mech. 2024, 17, dmm050762. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C. The E3 Ubiquitin Ligase TRIM31 Attenuates NLRP3 Inflammasome Activation by Promoting Proteasomal Degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef]
- Stutz, A.; Kolbe, C.-C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 Inflammasome Assembly Is Regulated by Phosphorylation of the Pyrin Domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, S.; Gao, L.; Zhou, Z.; Yang, Z.; Lin, J.; Ren, S.; Xing, H.; Wu, B. Oscillating lncRNA Platr4 Regulates NLRP3 Inflammasome to Ameliorate Nonalcoholic Steatohepatitis in Mice. Theranostics 2021, 11, 426–444. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Srivastava, A.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Mulay, S.R.; Scholze, A. Involvement of Inflammasome Components in Kidney Disease. Antioxidants 2022, 11, 246. [Google Scholar] [CrossRef]
- Vlachakis, P.K.; Theofilis, P.; Kachrimanidis, I.; Giannakopoulos, K.; Drakopoulou, M.; Apostolos, A.; Kordalis, A.; Leontsinis, I.; Tsioufis, K.; Tousoulis, D. The Role of Inflammasomes in Heart Failure. Int. J. Mol. Sci. 2024, 25, 5372. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, X.; Ge, Q.; Zhao, Y.; Mu, H.; Zhang, J. ER Stress Activates the NLRP3 Inflammasome: A Novel Mechanism of Atherosclerosis. Oxid. Med. Cell Longev. 2019, 2019, 3462530. [Google Scholar] [CrossRef] [PubMed]
- Folco, E.J.; Sukhova, G.K.; Quillard, T.; Libby, P. Moderate Hypoxia Potentiates Interleukin-1β Production in Activated Human Macrophages. Circ. Res. 2014, 115, 875–883. [Google Scholar] [CrossRef]
- Corliss, B.A.; Azimi, M.S.; Munson, J.M.; Peirce, S.M.; Murfee, W.L. Macrophages: An Inflammatory Link Between Angiogenesis and Lymphangiogenesis. Microcirculation 2016, 23, 95–121. [Google Scholar] [CrossRef]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H. CD163+ Macrophages Promote Angiogenesis and Vascular Permeability Accompanied by Inflammation in Atherosclerosis. J. Clin. Investig. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Yang, K.; Jiang, Q.; Wang, Z.; Li, M.; Zhang, Q.; Lu, W.; Wang, J. Mutual Inhibitory Mechanisms between PPARγ and Hif-1α: Implication in Pulmonary Hypertension. Recept. Clin. Investig. 2015, 2, e626. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Choi, M.-J.; Lee, E.-J.; Park, J.-S.; Kim, S.-N.; Park, E.-M.; Kim, H.-S. Anti-Inflammatory Mechanism of Galangin in Lipopolysaccharide-Stimulated Microglia: Critical Role of PPAR-γ Signaling Pathway. Biochem. Pharmacol. 2017, 144, 120–131. [Google Scholar] [CrossRef]
- Wolfson, R.K.; Chiang, E.T.; Garcia, J.G.N. HMGB1 Induces Human Lung Endothelial Cell Cytoskeletal Rearrangement and Barrier Disruption. Microvasc. Res. 2011, 81, 189–197. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, S. Hypoxia Inducible Factor-1α/Vascular Endothelial Growth Factor Signaling Activation Correlates with Response to Radiotherapy and Its Inhibition Reduces Hypoxia-Induced Angiogenesis in Lung Cancer. J. Cell Biochem. 2018, 119, 7707–7718. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yang, Y.; Hou, J.; Shu, Q.; Yin, Y.; Fu, W.; Han, F.; Hou, T.; Zeng, C.; Nemeth, E. Increased Gene Copy Number of DEFA1/DEFA3 Worsens Sepsis by Inducing Endothelial Pyroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 3161–3170. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T. CD36 Coordinates NLRP3 Inflammasome Activation by Facilitating Intracellular Nucleation of Soluble Ligands into Particulate Ligands in Sterile Inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.; Hamilton, C.; Mortensen, R.; Jeyaprakash, A.A.; Ghosh, S.; Anand, P.K. Trafficking of Cholesterol to the ER Is Required for NLRP3 Inflammasome Activation. J. Cell Biol. 2018, 217, 3560–3576. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M. NLRP3 Inflamasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals That Form Early in Disease. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Altaf, A.; Qu, P.; Zhao, Y.; Wang, H.; Lou, D.; Niu, N. NLRP3 Inflammasome in Peripheral Blood Monocytes of Acute Coronary Syndrome Patients and Its Relationship with Statins. Coron. Artery Dis. 2015, 26, 409–421. [Google Scholar] [CrossRef]
- Khair, M.; Khair, M.; Vangaveti, V.N.; Malabu, U.H. The Role of the NLRP3 Inflammasome in Atherosclerotic Disease. Syst. Rev. Meta-Anal. J. Cardiol. 2024, 84, 14–21. [Google Scholar] [CrossRef]
- Afrasyab, A.; Qu, P.; Zhao, Y.; Peng, K.; Wang, H.; Lou, D.; Niu, N.; Yuan, D. Correlation of NLRP3 with Severity and Prognosis of Coronary Atherosclerosis in Acute Coronary Syndrome Patients. Heart Vessel. 2016, 31, 1218–1229. [Google Scholar] [CrossRef]
- Shi, X.; Xie, W.-L.; Kong, W.-W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Orecchioni, M.; Kobiyama, K.; Winkels, H.; Ghosheh, Y.; McArdle, S.; Mikulski, Z.; Kiosses, W.B.; Fan, Z.; Wen, L.; Jung, Y. Olfactory Receptor 2 in Vascular Macrophages Drives Atherosclerosis by NLRP3-Dependent IL-1 Production. Science 2022, 375, 214–221. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Borborema, M.E.d.A.; Crovella, S.; Oliveira, D.; Azevêdo Silva, J. Inflammasome Activation by NLRP1 and NLRC4 in Patients with Coronary Stenosis. Immunobiology 2020, 225, 151940. [Google Scholar] [CrossRef] [PubMed]
- Bleda, S.; Haro, J.; Varela, C.; Ferruelo, A.; Acin, F. Elevated Levels of Triglycerides and Vldl-Cholesterol Provoke Activation of Nlrp1 Inflammasome in Endothelial Cells. Int. J. Cardiol. 2016, 220, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Bleda, S.; Haro, J.; Varela, C.; Esparza, L.; Ferruelo, A.; Acin, F. NLRP1 inflammasome, and not NLRP3, Is the Key in the Shift to Proinflammatory State on Endothelial Cells in Peripheral Arterial Disease. Int. J. Cardiol. 2014, 172, 282–284. [Google Scholar] [CrossRef]
- Paulin, N.; Viola, J.R.; Maas, S.L.; Jong, R.; Fernandes-Alnemri, T.; Weber, C.; Drechsler, M.; Döring, Y.; Soehnlein, O. Double-Strand DNA Sensing Aim2 Inflammasome Regulates Atherosclerotic Plaque Vulnerability. Circulation 2018, 138, 321–323. [Google Scholar] [CrossRef]
- Hoes, A.W.; Mosterd, A.; Grobbee, D.E. An Epidemic of Heart Failure? Recent Evidence from Europe. Eur. Heart J. 1998, 19 (Suppl. L), 2–9. [Google Scholar]
- Yan, T.; Zhu, S.; Yin, X.; Xie, C.; Xue, J.; Zhu, M.; Weng, F.; Zhu, S.; Xiang, B.; Zhou, X. Burden, Trends, and Inequalities of Heart Failure Globally, 1990 to 2019: A Secondary Analysis Based on the Global Burden of Disease 2019 Study. J. Am. Heart Assoc. 2023, 12, e027852. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal Activation in Heart Failure with Reduced Ejection Fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef]
- Kurmani, S.; Squire, I. Acute Heart Failure: Definition, Classification and Epidemiology. Curr. Heart Fail. Rep. 2017, 14, 385–392. [Google Scholar] [CrossRef]
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar] [CrossRef]
- Golla, M.S.G.; Hajouli, S.; Ludhwani, D. Heart Failure and Ejection Fraction. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Sharma, K.; Kass, D.A. Heart Failure with Preserved Ejection Fraction: Mechanisms, Clinical Features, and Therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef]
- Cianci, R.; Franza, L.; Borriello, R.; Pagliari, D.; Gasbarrini, A.; Gambassi, G. The Role of Gut Microbiota in Heart Failure: When Friends Become Enemies. Biomedicines 2022, 10, 2712. [Google Scholar] [CrossRef]
- Onódi, Z.; Ruppert, M.; Kucsera, D.; Sayour, A.A.; Tóth, V.E.; Koncsos, G.; Novák, J.; Brenner, G.B.; Makkos, A.; Baranyai, T. AIM2-Driven Inflammasome Activation in Heart Failure. Cardiovasc. Res. 2021, 117, 2639–2651. [Google Scholar] [CrossRef]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [PubMed]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the Role of Inflammation in Heart Failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The Pathogenesis of Cardiac Fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S. NLRP3 Inflammasome-Mediated Pyroptosis Contributes to the Pathogenesis of Non-Ischemic Dilated Cardiomyopathy. Redox Biol. 2020, 34, 101523. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Pan, L.; Zhao, S.; Dai, F.; Chao, M.; Jiang, H.; Li, X.; Lin, Z.; Huang, Z.; Meng, G. SNO-MLP (S-Nitrosylation of Muscle LIM Protein) Facilitates Myocardial Hypertrophy Through TLR3 (Toll-Like Receptor 3)-Mediated RIP3 (Receptor-Interacting Protein Kinase 3) and NLRP3 (NOD-Like Receptor Pyrin Domain Containing 3) Inflammasome Activation. Circulation 2020, 141, 984–1000. [Google Scholar] [CrossRef]
- Toldo, S.; Kannan, H.; Bussani, R.; Anzini, M.; Sonnino, C.; Sinagra, G.; Merlo, M.; Mezzaroma, E.; De-Giorgio, F.; Silvestri, F. Formation of the Inflammasome in Acute Myocarditis. Int. J. Cardiol. 2014, 171, 119–121. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, K.; Zhu, L. Emerging Roles of Inflammasomes in Cardiovascular Diseases. Front. Immunol. 2022, 13, 834289. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H.; Yang, L.; Yong, H.; Qin, Q.; Tan, M.; Xu, L.; Liang, K.; Zong, J.; Qian, W. NLRP3 Deficiency Accelerates Pressure Overload-Induced Cardiac Remodeling via Increased TLR4 Expression. J. Mol. Med. 2018, 96, 1189–1202. [Google Scholar] [CrossRef]
- Buckley, L.F.; Abbate, A. Interleukin-1 Blockade in Cardiovascular Diseases: A Clinical Update. Eur. Heart J. 2018, 39, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Li, H.; Wang, J.-J.; Zhang, J.-S.; Shen, J.; An, X.-B.; Zhang, C.-C.; Wu, J.-M.; Song, Y.; Wang, X.-Y. IL-18 Cleavage Triggers Cardiac Inflammation and Fibrosis upon β-Adrenergic Insult. Eur. Heart J. 2018, 39, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Heijman, J.; Hiram, R.; Li, N.; Nattel, S. Inflammatory Signalling in Atrial Cardiomyocytes: A Novel Unifying Principle in Atrial Fibrillation Pathophysiology. Nat. Rev. Cardiol. 2023, 20, 145–167. [Google Scholar] [CrossRef]
- He, S.; Wang, Y.; Yao, Y.; Cao, Z.; Yin, J.; Zi, L.; Chen, H.; Fu, Y.; Wang, X.; Zhao, Q. Inhibition of KCa3.1 Channels Suppresses Atrial Fibrillation via the Attenuation of Macrophage Pro-Inflammatory Polarization in a Canine Model with Prolonged Rapid Atrial Pacing. Front. Cardiovasc. Med. 2021, 8, 656631. [Google Scholar] [CrossRef] [PubMed]
- Kugler, S.; Onódi, Z.; Ruppert, M.; Sayour, A.A.; Oláh, A.; Benke, K.; Ferdinandy, P.; Merkely, B.; Radovits, T.; Varga, Z.V. Inflammasome Activation in End-Stage Heart Failure-Associated Atrial Fibrillation. ESC Heart Fail. 2022, 9, 2747–2752. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef]
- Turner, N.A.; Das, A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Interleukin-1alpha Stimulates Proinflammatory Cytokine Expression in Human Cardiac Myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, 1117–1127. [Google Scholar] [CrossRef]
- Poppenborg, T.; Saljic, A.; Bruns, F.; Abu-Taha, I.; Dobrev, D.; Fender, A.C. A Short History of the Atrial NLRP3 Inflammasome and Its Distinct Role in Atrial Fibrillation. J. Mol. Cell. Cardiol. 2025, 202, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Yang, J.; Liu, Z.; Yu, Y.; Zhang, C.; Guo, Y.; Yu, F.; Zhou, Y.; Song, Z.; Shi, J. Inhibition of the P2X7 Receptor Prevents Atrial Proarrhythmic Remodeling in Experimental Post-Operative Atrial Fibrillation. Int. Immunopharmacol. 2024, 129, 111536. [Google Scholar] [CrossRef] [PubMed]
- Mitrokhin, V.M.; Makarenko, E.Y.; Abramochkin, D.V.; Sutyagin, P.V.; Kamkin, A.G. Effects of Interleukin-18 on Bioelectric Activity of Rat Atrial Cardiomyocytes under Normal Conditions and during Gradual Stretching of the Tissue. Bull. Exp. Biol. Med. 2014, 157, 409–412. [Google Scholar] [CrossRef]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef]
- Komada, T.; Muruve, D.A. The Role of Inflammasomes in Kidney Disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ricart, M.; Torramade-Moix, S.; Pascual, G.; Palomo, M.; Moreno-Castaño, A.B.; Martinez-Sanchez, J.; Vera, M.; Cases, A.; Escolar, G. Endothelial Damage, Inflammation and Immunity in Chronic Kidney Disease. Toxins 2020, 12, 361. [Google Scholar] [CrossRef]
- Imazio, M.; Spodick, D.H.; Brucato, A.; Trinchero, R.; Markel, G.; Adler, Y. Diagnostic Issues in the Clinical Management of Pericarditis. Int. J. Clin. Pract. 2010, 64, 1384–1392. [Google Scholar] [CrossRef]
- Dababneh, E.; Siddique, M.S. Pericarditis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Lazarou, E.; Tsioufis, P.; Vlachopoulos, C.; Tsioufis, C.; Lazaros, G. Acute Pericarditis: Update. Curr. Cardiol. Rep. 2022, 24, 905. [Google Scholar] [CrossRef]
- Peet, C.J.; Rowczenio, D.; Omoyinmi, E.; Papadopoulou, C.; Mapalo, B.R.R.; Wood, M.R.; Capon, F.; Lachmann, H.J. Pericarditis and Autoinflammation: A Clinical and Genetic Analysis of Patients with Idiopathic Recurrent Pericarditis and Monogenic Autoinflammatory Diseases at a National Referral Center. J. Am. Heart Assoc. 2022, 11, e024931. [Google Scholar] [CrossRef]
- Vecchié, A.; Bonaventura, A.; Golino, M.; Thomas, G.; Abbate, A. Novel Therapeutic Insights into the Treatment of Pericarditis: Targeting the Innate Immune System. J. Cardiovasc. Pharmacol. 2024, 83, 377–383. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine–Update on Mechanisms of Action and Therapeutic Uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Cola, I.; Vitale, A.; Caggiano, V.; Palumbo, P.; Cesare, E.; Torres-Ruiz, J.; Guaracha-Basañez, G.A.; Martín-Nares, E.; Ciccia, F. The Evaluation of Myocarditis in Patients with Still’s Disease; Clinical Findings from the Multicentre International AIDA Network Still’s Disease Registry. J. Rheumatol. 2025, 52, 226–233. [Google Scholar] [CrossRef]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H. Increased Neutrophil Extracellular Traps Activate NLRP3 and Inflammatory Macrophages in Adult-Onset Still’s Disease. Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef]
- Ruscitti, P.; Cantarini, L.; Nigrovic, P.A.; McGonagle, D.; Giacomelli, R. Recent Advances and Evolving Concepts in Still’s Disease. Nat. Rev. Rheumatol. 2024, 20, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Sfriso, P.; Bindoli, S.; Galozzi, P. Adult-Onset Still’s Disease: Molecular Pathophysiology and Therapeutic Advances. Drugs 2018, 78, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Delplanque, M.; Aouba, A.; Hirsch, P.; Fenaux, P.; Graveleau, J.; Malard, F.; Roos-Weil, D.; Belfeki, N.; Drevon, L.; Oganesyan, A. USAID Associated with Myeloid Neoplasm and VEXAS Syndrome: Two Differential Diagnoses of Suspected Adult Onset Still’s Disease in Elderly Patients. J. Clin. Med. 2021, 10, 5586. [Google Scholar] [CrossRef]
- Bindoli, S.; Baggio, C.; Doria, A.; Sfriso, P. Adult-Onset Still’s Disease (AOSD): Advances in Understanding Pathophysiology, Genetics and Emerging Treatment Options. Drugs 2024, 84, 257–274. [Google Scholar] [CrossRef]
- Song, Y.H.; Jun, J.B.; Chung, W.T.; Choe, J.Y.; Kim, T.G.; Yoo, D.H. Association between HLA-DR B1 and Clinical Features of Adult Onset Still’s Disease in Korea. Clin. Exp. Rheumatol. 2003, 21, 489–492. [Google Scholar]
- Castillo, R.L.; Farías, J.; Sandoval, C.; González-Candia, A.; Figueroa, E.; Quezada, M.; Cruz, G.; Llanos, P.; Jorquera, G.; Kostin, S. Role of NLRP3 Inflammasome in Heart Failure Patients Undergoing Cardiac Surgery as a Potential Determinant of Postoperative Atrial Fibrillation and Remodeling: Is SGLT2 Cotransporter Inhibition an Alternative for Cardioprotection? Antioxidants 2024, 13, 1388. [Google Scholar] [CrossRef]
- Rroji, M.; Spahia, N.; Figurek, A.; Spasovski, G. Targeting Diabetic Atherosclerosis: The Role of GLP-1 Receptor Agonists, SGLT2 Inhibitors, and Nonsteroidal Mineralocorticoid Receptor Antagonists in Vascular Protection and Disease Modulation. Biomedicines 2025, 13, 728. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Zhuravlev, A.D.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Mitochondria-Mediated Cardiovascular Benefits of Sodium-Glucose Co-Transporter 2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 5371. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.C.; Rothman, A.M.K.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W. The Effect of Interleukin-1 Receptor Antagonist Therapy on Markers of Inflammation in Non-ST Elevation Acute Coronary Syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients with ST-Segment-Elevation Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D. ESC Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, R.C.; Borisy, G.G.; Taylor, E.W. The Colchicine-Binding Protein of Mammalian Brain and Its Relation to Microtubules. Biochemistry 1968, 7, 4466–4479. [Google Scholar] [CrossRef]
- Imazio, M.; Agrimi, C.; Cescon, L.; Panzolli, G.; Collini, V.; Sinagra, G. Colchicine for the Treatment of the Spectrum of Cardiovascular Diseases: Current Evidence and Ongoing Perspectives. J. Cardiovasc. Med. 2024, 25, 653–663. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Duan, J.; Gong, C.; Feng, Y.; Hu, J.; Gu, R.; Xu, B. Colchicine Ameliorates Dilated Cardiomyopathy Via SIRT2-Mediated Suppression of NLRP3 Inflammasome Activation. J. Am. Heart Assoc. 2022, 11, e025266. [Google Scholar] [CrossRef]
- Tengesdal, I.W.; Banks, M.; Dinarello, C.A.; Marchetti, C. Screening NLRP3 Drug Candidates in Clinical Development: Lessons from Existing and Emerging Technologies. Front. Immunol. 2024, 15, 1422249. [Google Scholar] [CrossRef]
- Lilly, L.S. Treatment of Acute and Recurrent Idiopathic Pericarditis. Circulation 2013, 127, 1723–1726. [Google Scholar] [CrossRef]
- Imazio, M.; Brucato, A.; Barbieri, A.; Ferroni, F.; Maestroni, S.; Ligabue, G.; Chinaglia, A.; Cumetti, D.; Casa, G.D.; Bonomi, F. Good Prognosis for Pericarditis with and Without Myocardial Involvement. Circulation 2013, 128, 42–49. [Google Scholar] [CrossRef]
- Adler, Y.; Charron, P.; Imazio, M.; Badano, L.; Barón-Esquivias, G.; Bogaert, J.; Brucato, A.; Gueret, P.; Klingel, K.; Lionis, C.; et al. 2015 ESC Guidelines for the diagnosis and management of pericardial diseases: The Task Force for the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology (ESC)Endorsed by: The European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. Oxf. Acad. 2015, 36, 2921–2964. [Google Scholar]
- Imazio, M.; Gaita, F. Diagnosis and Treatment of Pericarditis. Heart 2015, 101, 1159–1168. [Google Scholar] [CrossRef]
- Imazio, M.; Brucato, A.; Mayosi, B.M.; Derosa, F.G.; Lestuzzi, C.; Macor, A.; Trinchero, R.; Spodick, D.H.; Adler, Y. Medical Therapy of Pericardial Diseases: Part I: Idiopathic and Infectious Pericarditis. J. Cardiovasc. Med. 2010, 11, 712–722. [Google Scholar] [CrossRef]
- Abadie, B.Q.; Cremer, P.C. Interleukin-1 Antagonists for the Treatment of Recurrent Pericarditis. BioDrugs 2022, 36, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Lazzara, A.; Paglialonga, L.; Viafora, F.; Aurelio, C.; Esposito, S. Recurrent Pericarditis and Interleukin (IL)-1 Inhibitors. Int. Immunopharmacol. 2024, 141, 113017. [Google Scholar] [CrossRef] [PubMed]
- Lazaros, G.; Antonopoulos, A.S.; Antonatou, K.; Skendros, P.; Ritis, K.; Hadziyannis, E.; Lazarou, E.; Leontsinis, I.; Simantiris, S.; Vlachopoulos, C. Hydroxychloroquine for Colchicine-Resistant Glucocorticoid-Dependent Idiopathic Recurrent Pericarditis: A Pilot Observational Prospective Study. Int. J. Cardiol. 2020, 311, 77–82. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (Resatorvid), a Small-Molecule Inhibitor of Toll-like Receptor (TLR) 4 Signaling, Binds Selectively to TLR4 and Interferes with Interactions between TLR4 and Its Adaptor Molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Y.; Sun, Y.; Gao, Q.; Li, H.; Yang, Z.; Wang, Y.; Jiang, X.; Yu, B. Target of MCC950 in Inhibition of NLRP3 Inflammasome Activation: A Literature Review. Inflammation 2020, 43, 17–23. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, M.; Yu, J.; Xu, Y.; Zhang, J.; Liu, J.; Zheng, Z.; Ye, J.; Wang, Z.; Ye, D. MCC950, a Selective NLRP3 Inhibitor, Attenuates Adverse Cardiac Remodeling Following Heart Failure Through Improving the Cardiometabolic Dysfunction in Obese Mice. Front. Cardiovasc. Med. 2022, 9, 727474. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W. Oridonin Is a Covalent NLRP3 Inhibitor with Strong Anti-Inflammasome Activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Zheng, Y.; Yu, Q.; Zeng, M.; Bai, L.; Yang, L.; Guo, M.; Jiang, X.; Gan, J. Inhibitors of the NLRP3 Inflammasome Pathway as Promising Therapeutic Candidates for Inflammatory Diseases. Int. J. Mol. Med. 2023, 51, 35. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Tassell, B.W.; Mezzaroma, E.; Del Buono, M.G.; Prestamburgo, A.; Potere, N.; Abbate, A. The NLRP3 Inflammasome Inhibitor, OLT1177 (Dapansutrile), Reduces Infarct Size and Preserves Contractile Function After Ischemia Reperfusion Injury in the Mouse. J. Cardiovasc. Pharmacol. 2019, 73, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Traughber, C.A.; Timinski, K.; Prince, A.; Bhandari, N.; Neupane, K.; Khan, M.R.; Opoku, E.; Opoku, E.; Brubaker, G.; Shin, J. Disulfiram Reduces Atherosclerosis and Enhances Efferocytosis, Autophagy, and Atheroprotective Gut Microbiota in Hyperlipidemic Mice. J. Am. Heart Assoc. 2024, 13, e033881. [Google Scholar] [CrossRef]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J. microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel Inhibitors of Cytokine-Induced IkappaBalpha Phosphorylation and Endothelial Cell Adhesion Molecule Expression Show Anti-Inflammatory Effects in Vivo. J. Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef]
- Irrera, N.; Vaccaro, M.; Bitto, A.; Pallio, G.; Pizzino, G.; Lentini, M.; Arcoraci, V.; Minutoli, L.; Scuruchi, M.; Cutroneo, G. BAY 11-7082 Inhibits the NF-κB and NLRP3 Inflammasome Pathways and Protects against IMQ-Induced Psoriasis. Clin. Sci. 2017, 131, 487–498. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Yao, N.; Lin, Z. Immunoproteasome Modulates NLRP3 Inflammasome-Mediated Neuroinflammation under Cerebral Ischaemia and Reperfusion Conditions. J. Cell Mol. Med. 2022, 26, 462–474. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, J.S.; Kwon, J.S.; Jeong, M.H.; Cho, J.G.; Park, J.C.; Kang, J.C.; Ahn, Y. BAY 11-7082, a Nuclear Factor-κB Inhibitor, Reduces Inflammation and Apoptosis in a Rat Cardiac Ischemia-Reperfusion Injury Model. Int. Heart J. 2010, 51, 348–353. [Google Scholar] [CrossRef]
- Putnam, C.D.; Broderick, L.; Hoffman, H.M. The Discovery of NLRP3 and Its Function in Cryopyrin-Associated Periodic Syndromes and Innate Immunity. Immunol. Rev. 2024, 322, 259–282. [Google Scholar] [CrossRef] [PubMed]
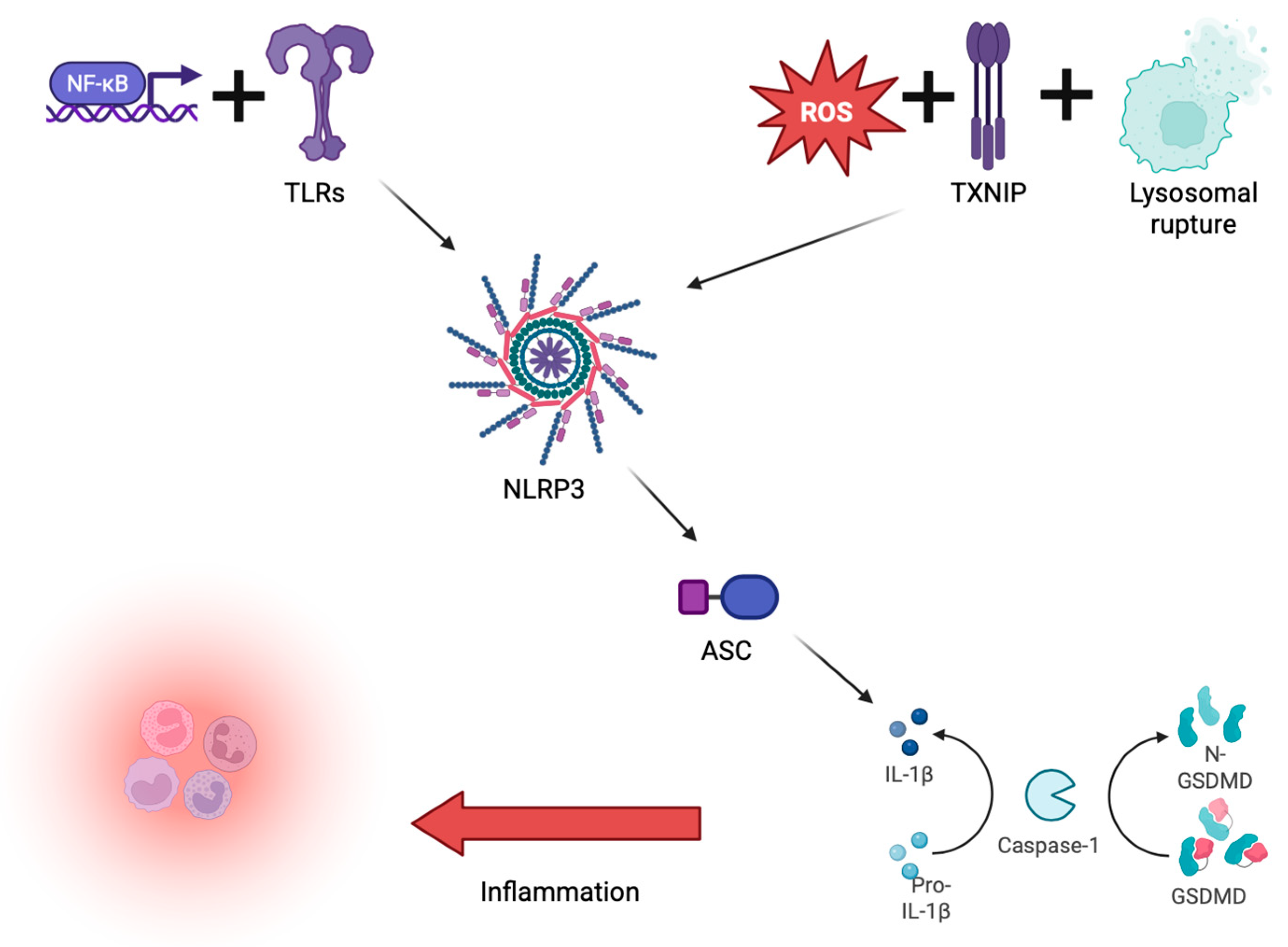


{kind=link}
{kind=link}
| Trigger | Sensor/Pathway | Key Molecules | Downstream Effect | Associated Conditions | References |
|---|---|---|---|---|---|
| Cholesterol crystals | Lysosomal rupture | NLRP3, ASC, caspase-1 | IL-1β, IL-18 release, pyroptosis | Atherosclerosis | [44,46] |
| ROS/oxidative stress | ROS–TXNIP–NLRP3 | TXNIP, NLRP3, caspase-1 | Inflammation, fibrosis | Heart failure, CKD | [19,20] |
| Mitochondrial dysfunction | mtROS, mtDNA | NLRP3, GSDMD | Pyroptosis, cytokine secretion | AF, HF, CKD | [19] |
| ER stress | Ca2+ signaling | NLRP3 | IL-1β production, cell damage | CKD, myocardial injury | [23,24] |
| Uric acid/ATP | P2X7 receptor activation | K+ efflux, NLRP3 | Inflammasome priming/activation | Pericarditis, CKD | [19,88] |
| Mechanical stretch | Ion channels, ROS | NLRP3, NF-κB | Fibrosis, arrhythmias | Atrial fibrillation | [76,79,81] |
| Molecule | Target | Type of Study | References |
|---|---|---|---|
| Anti-SGLT-2 | Sodium–glucose cotransporter-2 | Clinical | [101] |
| Anakinra, Canakinumab | IL-1 | Clinical | [104,105] |
| Colchicine | Interacts with tubulin and thereby inhibits microtubule-dependent functions in rapidly proliferating cells | Preclinical in vitro | [108] |
| TAK-242 (resatorvid) | TLR4 | Preclinical | [121] |
| MCC950 | NACHT domain of NLRP3. | Review of preclinical studies | [123] |
| Oridonin | NACHT domain of NLRP3 | Preclinical | [124] |
| OLT1177 | Inhibits ASC oligomerization | Preclinical | [126] |
| Disulfiram | Causes autophagy in a variety of macrophages, smooth muscle cells, endothelial cells, hepatocytes, and tissue samples from atherosclerotic plaques | Preclinical | [127] |
| BAY 11-7082 | Inhibits NF-κB | Preclinical | [132] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caldarelli, M.; Franza, L.; Cutrupi, S.; Menegolo, M.; Franceschi, F.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Inflammasomes in Cardiovascular Diseases: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2025, 26, 5439. https://doi.org/10.3390/ijms26125439
Caldarelli M, Franza L, Cutrupi S, Menegolo M, Franceschi F, Gasbarrini A, Gambassi G, Cianci R. Inflammasomes in Cardiovascular Diseases: Current Knowledge and Future Perspectives. International Journal of Molecular Sciences. 2025; 26(12):5439. https://doi.org/10.3390/ijms26125439
Chicago/Turabian StyleCaldarelli, Mario, Laura Franza, Sebastiano Cutrupi, Martina Menegolo, Francesco Franceschi, Antonio Gasbarrini, Giovanni Gambassi, and Rossella Cianci. 2025. "Inflammasomes in Cardiovascular Diseases: Current Knowledge and Future Perspectives" International Journal of Molecular Sciences 26, no. 12: 5439. https://doi.org/10.3390/ijms26125439
APA StyleCaldarelli, M., Franza, L., Cutrupi, S., Menegolo, M., Franceschi, F., Gasbarrini, A., Gambassi, G., & Cianci, R. (2025). Inflammasomes in Cardiovascular Diseases: Current Knowledge and Future Perspectives. International Journal of Molecular Sciences, 26(12), 5439. https://doi.org/10.3390/ijms26125439