

The Problem of Molecular Target Choice for CAR-T Cells in Acute Myeloid Leukemia Therapy

 , ,
, ,
Abstract
1. Introduction
2. Choosing the Molecular Target for CAR-T Cells
3. Targeted Immunotherapy Issues in Acute Myeloid Leukemia
4. Promising Molecular Targets for Acute Myeloid Leukemia
4.1. T-Lymphocyte Antigen—CD7
4.2. Sialic Acid-Binding Immunoglobulin, Siglec-3—CD33
4.3. ADP-Ribosyl Cyclase—CD38
4.4. Cell Adhesion Molecule—CD44v6
4.5. Ligand of the Tumor Necrosis Factor Receptor—CD70
4.6. Leukocyte Immunoglobulin-like Receptor B ILT3—CD85k
4.7. Alpha Chain of the Granulocyte-Macrophage Colony-Stimulating Factor Receptor Complex GM-CSFR—CD116
4.8. Stem Cell Factor Receptor c-Kit—CD117
4.9. Alpha Chain of the IL-3 Receptor Complex—CD123
4.10. Tyrosine Kinase Receptor Flt3—CD135
4.11. NK Cell Ligand B7-H3—CD276
4.12. Inhibitory Protein Siglec-6—CD327
4.13. The Protein Containing Immunoglobulin and Mucin Domains, TIM-3—CD366
4.14. Lectin-like C-Type Domain CLL-1 Protein
4.15. Folate Receptor FRβ
4.16. NKG2D Receptor Ligands
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Wang, M. CAR T-Cell Therapy Effective in B Acute Lymphoblastic Leukaemia. Lancet Oncol. 2017, 18, e314. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.T.; Dholaria, B.; Savani, B.N.; Sengsayadeth, S.; Oluwole, O. Overview of Approved CAR-T Products and Utility in Clinical Practice. Clin. Hematol. Int. 2024, 6, 108–114. [Google Scholar] [CrossRef]
- Ryan, C. FDA Approves Obecabtagene Autoleucel for Adults with Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia; FDA: Silver Spring, MD, USA, 2024.
- Albelda, S.M. CAR T Cell Therapy for Patients with Solid Tumours: Key Lessons to Learn and Unlearn. Nat. Rev. Clin. Oncol. 2024, 21, 47–66. [Google Scholar] [CrossRef]
- Hemminki, K.; Zitricky, F.; Försti, A.; Kontro, M.; Gjertsen, B.T.; Severinsen, M.T.; Juliusson, G. Age-Specific Survival in Acute Myeloid Leukemia in the Nordic Countries through a Half Century. Blood Cancer J. 2024, 14, 44. [Google Scholar] [CrossRef]
- Rao, A.V. Fitness in the Elderly: How to Make Decisions Regarding Acute Myeloid Leukemia Induction. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Zini, G.; Bassan, R. Diagnosis and Subclassification of Acute Lymphoblastic Leukemia. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014073. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.H.; Zandvakili, N.; Bohlander, S.K. Dissecting the Genetic and Non-Genetic Heterogeneity of Acute Myeloid Leukemia Using Next-Generation Sequencing and In Vivo Models. Cancers 2022, 14, 2182. [Google Scholar] [CrossRef]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target Selection for CAR-T Therapy. J. Hematol. Oncol. 2019, 12, 62. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A Biomarker for B Cell Development, Lymphoma Diagnosis and Therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Wudhikarn, K.; Palomba, M.L.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Devlin, S.M.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Shah, G.L.; et al. Infection during the First Year in Patients Treated with CD19 CAR T Cells for Diffuse Large B Cell Lymphoma. Blood Cancer J. 2020, 10, 79. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- França, G.S.; Baron, M.; King, B.R.; Bossowski, J.P.; Bjornberg, A.; Pour, M.; Rao, A.; Patel, A.S.; Misirlioglu, S.; Barkley, D.; et al. Cellular Adaptation to Cancer Therapy along a Resistance Continuum. Nature 2024, 631, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1407222 (accessed on 28 August 2018).
- Ally, F.; Chen, X. Acute Myeloid Leukemia: Diagnosis and Evaluation by Flow Cytometry. Cancers 2024, 16, 3855. [Google Scholar] [CrossRef]
- Zushi, Y.; Sasaki, M.; Mori, A.; Saitoh, T.; Goka, T.; Aoyama, Y.; Goto, Y.; Tsunemine, H.; Kodaka, T.; Takahashi, T. Acute Monocytic Leukemia Diagnosed by Flow Cytometry Includes Acute Myeloid Leukemias with Weakly or Faintly Positive Non-Specific Esterase Staining. Hematol. Rep. 2018, 10, 7435. [Google Scholar] [CrossRef]
- Su, N.; Li, Z.; Yang, J.; Fu, Y.; Zhu, X.; Miao, H.; Yu, Y.; Jiang, W.; Le, J.; Qian, X.; et al. Revealing the Intratumoral Heterogeneity of Non-DS Acute Megakaryoblastic Leukemia in Single-Cell Resolution. Front. Oncol. 2022, 12, 915833. [Google Scholar] [CrossRef]
- Handschuh, L. Not Only Mutations Matter: Molecular Picture of Acute Myeloid Leukemia Emerging from Transcriptome Studies. J. Oncol. 2019, 2019, 7239206. [Google Scholar] [CrossRef]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-Targeted T Cells Mediate Potent Antitumor Effects against Acute Myeloid Leukemia and Multiple Myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and Efficacy of Second Generation CAR T Cell against the LeY Antigen in Acute Myeloid Leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef]
- Russkamp, N.F.; Myburgh, R.; Kiefer, J.D.; Neri, D.; Manz, M.G. Anti-CD117 Immunotherapy to Eliminate Hematopoietic and Leukemia Stem Cells. Exp. Hematol. 2021, 95, 31–45. [Google Scholar] [CrossRef]
- St. Jude Children’s Research Hospital. CD123-Directed Autologous T-Cell Therapy for Acute Myelogenous Leukemia (CATCHAML); clinicaltrials.gov: Bethesda, MD, USA, 2025.
- St. Jude Children’s Research Hospital. CAR T-Cell Therapy Directed to CD70 for Pediatric Patients With Hematological Malignancies; clinicaltrials.gov: Bethesda, MD, USA, 2024.
- Bárcena, A.; Muench, M.O.; Galy, A.H.M.; Cupp, J.; Roncarolo, M.G.; Phillips, J.H.; Spits, H. Phenotypic and Functional Analysis of T-Cell Precursors in the Human Fetal Liver and Thymus: CD7 Expression in the Early Stages of T- and Myeloid-Cell Development. Blood 1993, 82, 3401–3414. [Google Scholar] [CrossRef] [PubMed]
- Naeim, F.; Nagesh Rao, P.; Song, S.X.; Phan, R.T. Principles of Immunophenotyping. In Atlas of Hematopathology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 29–56. ISBN 978-0-12-809843-1. [Google Scholar]
- Rabinowich, H.; Pricop, L.; Herberman, R.B.; Whiteside, T.L. Expression and Function of CD7 Molecule on Human Natural Killer Cells. J. Immunol. 1994, 152, 517–526. [Google Scholar] [CrossRef]
- Sheng, B.; Zhang, K.; Tian, S.; Ma, R.; Li, Z.; Wu, H.; Wang, T.; Jiang, L.; You, F.; An, G.; et al. CD7 Protein Plays a Crucial Role in T Cell Infiltration in Tumors. Heliyon 2023, 9, e16961. [Google Scholar] [CrossRef]
- Chang, H.; Yeung, J.; Brandwein, J.; Yi, Q. CD7 Expression Predicts Poor Disease Free Survival and Post-Remission Survival in Patients with Acute Myeloid Leukemia and Normal Karyotype. Leuk. Res. 2007, 31, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Cummins, K.D.; Gill, S. Will CAR T Cell Therapy Have a Role in AML? Promises and Pitfalls. Semin. Hematol. 2019, 56, 155–163. [Google Scholar] [CrossRef]
- Marofi, F.; Rahman, H.S.; Al-Obaidi, Z.M.J.; Jalil, A.T.; Abdelbasset, W.K.; Suksatan, W.; Dorofeev, A.E.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; et al. Novel CAR T Therapy Is a Ray of Hope in the Treatment of Seriously Ill AML Patients. Stem Cell Res. Ther. 2021, 12, 465. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-Shelf” Fratricide-Resistant CAR-T for the Treatment of T Cell Hematologic Malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Atilla, E.; Atilla, P.A.; Mo, F.; Tashiro, H.; Srinivasan, M.; Lulla, P.; Rouce, R.H.; Cabral, J.M.S.; Ramos, C.A.; et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol. Ther. 2019, 27, 272–280. [Google Scholar] [CrossRef]
- Institute of Hematology & Blood Diseases Hospital, China. Efficacy, Safety and PK of CD7 CAR-T in Patients With Relapsed or Refractory CD7+ Hematological Malignancies; clinicaltrials.gov: Bethesda, MD, USA, 2025.
- Crocker, P.R.; Varki, A. Siglecs, Sialic Acids and Innate Immunity. Trends Immunol. 2001, 22, 337–342. [Google Scholar] [CrossRef]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T Cells for Treatment of Relapsed/Refractory Acute Myelogenous Leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef]
- van Houtum, E.J.H.; Büll, C.; Cornelissen, L.A.M.; Adema, G.J. Siglec Signaling in the Tumor Microenvironment. Front. Immunol. 2021, 12, 790317. [Google Scholar] [CrossRef] [PubMed]
- Propris, M.S.D.; Raponi, S.; Diverio, D.; Milani, M.L.; Meloni, G.; Falini, B.; Foà, R.; Guarini, A. High CD33 Expression Levels in Acute Myeloid Leukemia Cells Carrying the Nucleophosmin (NPM1) Mutation. Haematologica 2011, 96, 1548–1551. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Harrington, K.H.; Gudgeon, C.J.; Beddoe, M.E.; Fitzgibbon, M.P.; Ries, R.E.; Lamba, J.K.; McIntosh, M.W.; Meshinchi, S.; Walter, R.B. Expression and Functional Characterization of CD33 Transcript Variants in Human Acute Myeloid Leukemia. Oncotarget 2016, 7, 43281–43294. [Google Scholar] [CrossRef] [PubMed]
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Mele, S.; Devereux, S.; Pepper, A.G.; Infante, E.; Ridley, A.J. Calcium-RasGRP2-Rap1 Signaling Mediates CD38-Induced Migration of Chronic Lymphocytic Leukemia Cells. Blood Adv. 2018, 2, 1551–1561. [Google Scholar] [CrossRef]
- Cui, Q.; Qian, C.; Xu, N.; Kang, L.; Dai, H.; Cui, W.; Song, B.; Yin, J.; Li, Z.; Zhu, X.; et al. CD38-Directed CAR-T Cell Therapy: A Novel Immunotherapy Strategy for Relapsed Acute Myeloid Leukemia after Allogeneic Hematopoietic Stem Cell Transplantation. J. Hematol. Oncol. 2021, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- 920th Hospital of Joint Logistics Support Force of People’s Liberation Army of China. Clinical Study to Evaluate the Safety and Preliminary Efficacy of CLL1 and CD38 Dual CAR-T Injection in the Treatment of Relapsed and Refractory Acute Myeloid Leukemia; clinicaltrials.gov: Bethesda, MD, USA, 2025.
- Zöller, M. CD44, Hyaluronan, the Hematopoietic Stem Cell, and Leukemia-Initiating Cells. Front. Immunol. 2015, 6, 235. [Google Scholar] [CrossRef]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 Eradicates Human Acute Myeloid Leukemic Stem Cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Tang, L.; Huang, H.; Tang, Y.; Li, Q.; Wang, J.; Li, D.; Zhong, Z.; Zou, P.; You, Y.; Cao, Y.; et al. CD44v6 Chimeric Antigen Receptor T Cell Specificity towards AML with FLT3 or DNMT3A Mutations. Clin. Transl. Med. 2022, 12, e1043. [Google Scholar] [CrossRef]
- AGC Biologics S.p.A. A Phase I-IIa Trial to Assess the Safety and Antitumor Activity of Autologous CD44v6 CAR T-Cells in Acute Myeloid Leukemia and Multiple Myeloma Expressing CD44v6; clinicaltrials.gov: Bethesda, MD, USA, 2021.
- Nolte, M.A.; van Olffen, R.W.; van Gisbergen, K.P.J.M.; van Lier, R.A.W. Timing and Tuning of CD27-CD70 Interactions: The Impact of Signal Strength in Setting the Balance between Adaptive Responses and Immunopathology. Immunol. Rev. 2009, 229, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Riether, C.; Schürch, C.M.; Bührer, E.D.; Hinterbrandner, M.; Huguenin, A.-L.; Hoepner, S.; Zlobec, I.; Pabst, T.; Radpour, R.; Ochsenbein, A.F. CD70/CD27 Signaling Promotes Blast Stemness and Is a Viable Therapeutic Target in Acute Myeloid Leukemia. J. Exp. Med. 2017, 214, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Müller-Tidow, C.; et al. CD70-Specific CAR T Cells Have Potent Activity against Acute Myeloid Leukemia without HSC Toxicity. Blood 2021, 138, 318–330. [Google Scholar] [CrossRef]
- Huang, H. Clinical Trial for the Safety and Efficacy of CD 70 CAR T for Patients With CD70 Positive Malignant Hematologic Diseases; clinicaltrials.gov: Bethesda, MD, USA, 2021.
- MD Anderson Cancer Center. Phase I/II Study of CAR.70- Engineered IL15-Transduced Cord Blood-Derived NK Cells in Conjunction With Lymphodepleting Chemotherapy for the Management of Relapse/Refractory Hematological Malignances; clinicaltrials.gov: Bethesda, MD, USA, 2025.
- Cella, M.; Döhring, C.; Samaridis, J.; Dessing, M.; Brockhaus, M.; Lanzavecchia, A.; Colonna, M. A Novel Inhibitory Receptor (ILT3) Expressed on Monocytes, Macrophages, and Dendritic Cells Involved in Antigen Processing. J. Exp. Med. 1997, 185, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Kim-Schulze, S.; Seki, T.; Vlad, G.; Scotto, L.; Fan, J.; Colombo, P.C.; Liu, J.; Cortesini, R.; Suciu-Foca, N. Regulation of ILT3 Gene Expression by Processing of Precursor Transcripts in Human Endothelial Cells. Am. J. Transplant. 2006, 6, 76–82. [Google Scholar] [CrossRef]
- Mori, Y.; Tsuji, S.; Inui, M.; Sakamoto, Y.; Endo, S.; Ito, Y.; Fujimura, S.; Koga, T.; Nakamura, A.; Takayanagi, H.; et al. Inhibitory Immunoglobulin-Like Receptors LILRB and PIR-B Negatively Regulate Osteoclast Development1. J. Immunol. 2008, 181, 4742–4751. [Google Scholar] [CrossRef]
- Li, Z.; Deng, M.; Huang, F.; Jin, C.; Sun, S.; Chen, H.; Liu, X.; He, L.; Sadek, A.H.; Zhang, C.C. LILRB4 ITIMs Mediate the T Cell Suppression and Infiltration of Acute Myeloid Leukemia Cells. Cell Mol. Immunol. 2020, 17, 272–282. [Google Scholar] [CrossRef]
- Marques-Piubelli, M.L.; Kumar, B.; Basar, R.; Panowski, S.; Srinivasan, S.; Norwood, K.; Prashad, S.; Szenes, V.; Balakumaran, A.; Arandhya, A.; et al. Increased Expression of CD70 in Relapsed Acute Myeloid Leukemia after Hypomethylating Agents. Virchows Arch. 2024, 485, 937–941. [Google Scholar] [CrossRef]
- Dobrowolska, H.; Gill, K.Z.; Serban, G.; Ivan, E.; Li, Q.; Qiao, P.; Suciu-Foca, N.; Savage, D.; Alobeid, B.; Bhagat, G.; et al. Expression of Immune Inhibitory Receptor ILT3 in Acute Myeloid Leukemia with Monocytic Differentiation. Cytom. Part B Clin. Cytom. 2013, 84B, 21–29. [Google Scholar] [CrossRef]
- Carbiogene Therapeutics Co., Ltd. Clinical Study of Autologous T Cells Modified With ILT3 Chimeric Antigen Receptor for Relapsed/Refractory Acute Myeloid Leukemia (M4/M5); clinicaltrials.gov: Bethesda, MD, USA, 2021.
- Testa, U.; Fossati, C.; Samoggia, P.; Masciulli, R.; Mariani, G.; Hassan, H.J.; Sposi, N.M.; Guerriero, R.; Rosato, V.; Gabbianelli, M.; et al. Expression of Growth Factor Receptors in Unilineage Differentiation Culture of Purified Hematopoietic Progenitors. Blood 1996, 88, 3391–3406. [Google Scholar] [CrossRef]
- Chen, B.D.; Clark, C.R.; Chou, T.H. Granulocyte/Macrophage Colony-Stimulating Factor Stimulates Monocyte and Tissue Macrophage Proliferation and Enhances Their Responsiveness to Macrophage Colony-Stimulating Factor. Blood 1988, 71, 997–1002. [Google Scholar] [CrossRef]
- Lanza, F.; Castagnari, B.; Rigolin, G.; Moretti, S.; Latorraca, A.; Ferrari, L.; Bardi, A.; Castoldi, G. Flow Cytometry Measurement of GM-CSF Receptors in Acute Leukemic Blasts, and Normal Hemopoietic Cells. Leukemia 1997, 11, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Hecht, K.; Reif, S.; Pelka-Fleischer, R.; Pfister, K.; Schmetzer, H. Expression and Prognostic Value of Hemopoietic Cytokine Receptors in Acute Myeloid Leukemia (AML): Implications for Future Therapeutical Strategies. Eur. J. Haematol. 2004, 72, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Riccioni, R.; Diverio, D.; Riti, V.; Buffolino, S.; Mariani, G.; Boe, A.; Cedrone, M.; Ottone, T.; Foà, R.; Testa, U. Interleukin (IL)-3/Granulocyte Macrophage-Colony Stimulating Factor/IL-5 Receptor Alpha and Beta Chains Are Preferentially Expressed in Acute Myeloid Leukaemias with Mutated FMS-Related Tyrosine Kinase 3 Receptor. Br. J. Haematol. 2009, 144, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, A.; Saito, S.; Narimatsu, S.; Nakano, S.; Nagai, M.; Ohnota, H.; Inada, Y.; Morokawa, H.; Nakashima, I.; Morita, D.; et al. Mutated GM-CSF-Based CAR-T Cells Targeting CD116/CD131 Complexes Exhibit Enhanced Anti-Tumor Effects against Acute Myeloid Leukaemia. Clin. Transl. Immunol. 2021, 10, e1282. [Google Scholar] [CrossRef]
- Kimura, Y.; Ding, B.; Imai, N.; Nolan, D.J.; Butler, J.M.; Rafii, S. C-Kit-Mediated Functional Positioning of Stem Cells to Their Niches Is Essential for Maintenance and Regeneration of Adult Hematopoiesis. PLoS ONE 2011, 6, e26918. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem Cell Factor Receptor/c-Kit: From Basic Science to Clinical Implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- Gao, X.; Lin, J.; Gao, L.; Deng, A.; Lu, X.; Li, Y.; Wang, L.; Yu, L. High Expression of C-Kit mRNA Predicts Unfavorable Outcome in Adult Patients with t(8;21) Acute Myeloid Leukemia. PLoS ONE 2015, 10, e0124241. [Google Scholar] [CrossRef]
- Heo, S.-K.; Noh, E.-K.; Kim, J.Y.; Jeong, Y.K.; Jo, J.-C.; Choi, Y.; Koh, S.; Baek, J.H.; Min, Y.J.; Kim, H. Targeting C-KIT (CD117) by Dasatinib and Radotinib Promotes Acute Myeloid Leukemia Cell Death. Sci. Rep. 2017, 7, 15278. [Google Scholar] [CrossRef]
- Woźniak, J.; Kopeć-Szlęzak, J. C-Kit Receptor (CD117) Expression on Myeloblasts and White Blood Cell Counts in Acute Myeloid Leukemia. Cytom. Part B Clin. Cytom. 2004, 58B, 9–16. [Google Scholar] [CrossRef]
- Myburgh, R.; Kiefer, J.D.; Russkamp, N.F.; Magnani, C.F.; Nuñez, N.; Simonis, A.; Pfister, S.; Wilk, C.M.; McHugh, D.; Friemel, J.; et al. Anti-Human CD117 CAR T-Cells Efficiently Eliminate Healthy and Malignant CD117-Expressing Hematopoietic Cells. Leukemia 2020, 34, 2688–2703. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and Levels of Cell Surface Expression of CD33 and CD123 in Acute Myeloid Leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed]
- Reddy, E.P.; Korapati, A.; Chaturvedi, P.; Rane, S. IL-3 Signaling and the Role of Src Kinases, JAKs and STATs: A Covert Liaison Unveiled. Oncogene 2000, 19, 2532–2547. [Google Scholar] [CrossRef] [PubMed]
- Vergez, F.; Green, A.S.; Tamburini, J.; Sarry, J.-E.; Gaillard, B.; Cornillet-Lefebvre, P.; Pannetier, M.; Neyret, A.; Chapuis, N.; Ifrah, N.; et al. High Levels of CD34+CD38low/−CD123+ Blasts Are Predictive of an Adverse Outcome in Acute Myeloid Leukemia: A Groupe Ouest-Est Des Leucémies Aiguës et Maladies Du Sang (GOELAMS) Study. Haematologica 2011, 96, 1792–1798. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Mughal, T.I.; Brooks, C.; Lindsay, R.; Pemmaraju, N. Targeting CD123 in Hematologic Malignancies: Identifying Suitable Patients for Targeted Therapy. Leuk. Lymphoma 2021, 62, 2568–2586. [Google Scholar] [CrossRef]
- El Achi, H.; Dupont, E.; Paul, S.; Khoury, J.D. CD123 as a Biomarker in Hematolymphoid Malignancies: Principles of Detection and Targeted Therapies. Cancers 2020, 12, 3087. [Google Scholar] [CrossRef]
- Beijing Immunochina Medical Science & Technology Co., Ltd. Safety and Efficacy Evaluation of IM23 CAR-T Cells On CD123+ AML Patients; clinicaltrials.gov: Bethesda, MD, USA, 2023.
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef]
- Ozeki, K.; Kiyoi, H.; Hirose, Y.; Iwai, M.; Ninomiya, M.; Kodera, Y.; Miyawaki, S.; Kuriyama, K.; Shimazaki, C.; Akiyama, H.; et al. Biologic and Clinical Significance of the FLT3 Transcript Level in Acute Myeloid Leukemia. Blood 2004, 103, 1901–1908. [Google Scholar] [CrossRef]
- The First Affiliated Hospital of Soochow University. Pilot Study of the Safety and Efficacy of Anti-FLT3 Chimeric Antigen Receptor Engineered T-Cells in the Treatment of Relapsed or Refractory Acute Myeloid Leukemia (AML); clinicaltrials.gov: Bethesda, MD, USA, 2021.
- PersonGen BioTherapeutics (Suzhou) Co., Ltd. TAA05 Cell Injection in the Treatment of Recurrent/Refractory Acute Myeloid Leukemia; clinicaltrials.gov: Bethesda, MD, USA, 2021.
- PersonGen BioTherapeutics (Suzhou) Co., Ltd. Clinical Study of TAA05 Injection in the Treatment of Adult Patients With FLT3-Positive Relapsed/Refractory Acute Myeloid Leukemia; clinicaltrials.gov: Bethesda, MD, USA, 2022.
- Heng, M. Safety and Efficacy of Anti-FLT3 CAR- T Cell (TAA05 Cell Injection) in the Treatment of Relapsed/Refractory Acute Myeloid Leukemia; clinicaltrials.gov: Bethesda, MD, USA, 2022.
- Tan, X.; Zhao, X. B7-H3 in Acute Myeloid Leukemia: From Prognostic Biomarker to Immunotherapeutic Target. Chin. Med. J. 2024, 137, 2540–2551. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, L.; Qian, J.; Lin, J.; Chen, Q.; Yuan, Q.; Zhou, J.; Zhang, T.; Shi, J.; Zhou, H. Expression Characteristic of 4Ig B7-H3 and 2Ig B7-H3 in Acute Myeloid Leukemia. Bioengineered 2021, 12, 11987–12002. [Google Scholar] [CrossRef]
- Guéry, T.; Roumier, C.; Berthon, C.; Lepelley, P.; Renneville, A.; Nibourel, O.; Dumezy, F.; Soenen, V.; Roche, C.; Preudhomme, C.; et al. The B7-H3 Protein In Acute Myeloid Leukemia. Blood 2013, 122, 2620. [Google Scholar] [CrossRef]
- Kirkey, D.C.; Blankenfeld, M.; Hylkema, T.; Loo, D.; Ward, A.; Robinson, L.; Peplinski, J.H.; Wallace, L.K.; Pardo, L.; Menssen, A.J.; et al. CD276 (B7-H3) Is an Immunotherapeutic Target in Acute Myeloid Leukemia with Preclinical Efficacy of Vobramitamab Duocarmazine, an Investigational CD276 Antibody-Drug Conjugate. Blood 2023, 142, 5958. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, C.; Liu, Z.; Yang, M.; Tang, X.; Wang, Y.; Zheng, M.; Huang, J.; Zhong, K.; Zhao, S.; et al. B7-H3-Targeted CAR-T Cells Exhibit Potent Antitumor Effects on Hematologic and Solid Tumors. Mol. Ther. Oncolytics 2020, 17, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, E.I.; Du, H.; Shou, P.; Song, F.; Suzuki, K.; Ahn, S.; Li, G.; Ferrone, S.; Su, L.; Savoldo, B.; et al. Preclinical Evaluation of B7-H3–Specific Chimeric Antigen Receptor T Cells for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 3141–3153. [Google Scholar] [CrossRef]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T Cells Targeting CD70 and B7-H3 Exhibit Potent Preclinical Activity against Multiple Solid Tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef] [PubMed]
- Jetani, H.; Navarro-Bailón, A.; Maucher, M.; Frenz, S.; Verbruggen, C.; Yeguas, A.; Vidriales, M.B.; González, M.; Rial Saborido, J.; Kraus, S.; et al. Siglec-6 Is a Novel Target for CAR T-Cell Therapy in Acute Myeloid Leukemia. Blood 2021, 138, 1830–1842. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.L. Application of Anti-Siglec-6 CAR-T Cell Therapy in Relapsed and Refractory Acute Myeloid Leukemia (rr/AML); clinicaltrials.gov: Bethesda, MD, USA, 2022.
- Han, G.; Chen, G.; Shen, B.; Li, Y. Tim-3: An Activation Marker and Activation Limiter of Innate Immune Cells. Front. Immunol. 2013, 4, 449. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Wang, M.; Zhang, L.; Yu, L. One Stone, Two Birds: The Roles of Tim-3 in Acute Myeloid Leukemia. Front. Immunol. 2021, 12, 618710. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Yu, X.; Zhu, Y.; Ma, C.; Xia, R.; Ma, J.; Gu, C.; Ye, L.; Wu, D. Tim-3 Is Highly Expressed in T Cells in Acute Myeloid Leukemia and Associated with Clinicopathological Prognostic Stratification. Int. J. Clin. Exp. Pathol. 2014, 7, 6880–6888. [Google Scholar]
- American Association for Cancer Research. Effective Killing of Acute Myeloid Leukemia by TIM-3 Targeted Chimeric Antigen Receptor T Cells. Molecular Cancer Therapeutics. Available online: https://aacrjournals.org/mct/article/20/9/1702/673365/Effective-Killing-of-Acute-Myeloid-Leukemia-by-TIM (accessed on 8 September 2022).
- Xu, K.L. Application of Anti Tim-3/CD123 CAR-T Cell Therapy in Relapsed and Refractory Acute Myeloid Leukemia (rr/AML); clinicaltrials.gov: Bethesda, MD, USA, 2023.
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for Acute Myeloid Leukemia Therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Xiao, W.; Li, W.; Wang, L.; Yang, S.; Wang, W.; Xu, L.; Liao, S.; Liu, W.; et al. CAR-T Cells Targeting CLL-1 as an Approach to Treat Acute Myeloid Leukemia. J. Hematol. Oncol. 2018, 11, 7. Available online: https://link.springer.com/article/10.1186/s13045-017-0553-5#Sec2 (accessed on 29 May 2024). [CrossRef] [PubMed]
- Bakker, A.B.H.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.C.; Visser, T.J.; Bijl, N.; Geuijen, C.A.W.; et al. C-Type Lectin-like Molecule-1: A Novel Myeloid Cell Surface Marker Associated with Acute Myeloid Leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Chen, H.; Li, W.; Huang, T.; Zhang, W.; Ling, W.; Lai, P.; Wang, Y.; Geng, S.; et al. C-Type Lectin-Like Molecule-1 as a Biomarker for Diagnosis and Prognosis in Acute Myeloid Leukemia: A Preliminary Study. Biomed. Res. Int. 2021, 2021, 6643948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gan, W.-T.; Hao, W.-G.; Wang, P.-F.; Li, Z.-Y.; Chang, L.-J. Successful Anti-CLL1 CAR T-Cell Therapy in Secondary Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 685. [Google Scholar] [CrossRef]
- Hill, L. Chimeric Antigen Receptor T-Cells for the Treatment of Acute Myeloid Leukemia Expressing CLL-1 Antigen; clinicaltrials.gov: Bethesda, MD, USA, 2024.
- Kite, a Gilead Company. A Phase 1 Open-Label, Multicenter Study Evaluating the Safety of KITE-222, an Autologous Anti-CLL-1 CAR T-Cell Therapy, in Subjects With Relapsed/Refractory Acute Myeloid Leukemia; clinicaltrials.gov: Bethesda, MD, USA, 2024.
- Reddy, J.A.; Haneline, L.S.; Srour, E.F.; Antony, A.C.; Clapp, D.W.; Low, P.S. Expression and Functional Characterization of the Beta-Isoform of the Folate Receptor on CD34(+) Cells. Blood 1999, 93, 3940–3948. [Google Scholar] [CrossRef]
- Zhang, F.; Huang, B.; Utturkar, S.M.; Luo, W.; Cresswell, G.; Herr, S.A.; Zheng, S.; Napoleon, J.V.; Jiang, R.; Zhang, B.; et al. Tumor-Specific Activation of Folate Receptor Beta Enables Reprogramming of Immune Cells in the Tumor Microenvironment. Front. Immunol. 2024, 15, 1354735. [Google Scholar] [CrossRef]
- Lynn, R.C.; Poussin, M.; Kalota, A.; Feng, Y.; Low, P.S.; Dimitrov, D.S.; Powell, D.J. Targeting of Folate Receptor β on Acute Myeloid Leukemia Blasts with Chimeric Antigen Receptor-Expressing T Cells. Blood 2015, 125, 3466–3476. [Google Scholar] [CrossRef]
- Lynn, R.C.; Feng, Y.; Schutsky, K.; Poussin, M.; Kalota, A.; Dimitrov, D.S.; Powell, D.J. High-Affinity FRβ-Specific CAR T Cells Eradicate AML and Normal Myeloid Lineage without HSC Toxicity. Leukemia 2016, 30, 1355–1364. [Google Scholar] [CrossRef]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Hilpert, J.; Grosse-Hovest, L.; Grünebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive Analysis of NKG2D Ligand Expression and Release in Leukemia: Implications for NKG2D-Mediated NK Cell Responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef]
- Celyad Oncology SA. A Phase 1 Study of Chimeric Antigen Receptor Modified T-Cells Targeting NKG2D-Ligands in Patients With Acute Myeloid Leukemia (AML)/Advanced Myelodysplastic Syndrome (MDS-RAEB) and Multiple Myeloma; clinicaltrials.gov: Bethesda, MD, USA, 2018.
- Celyad Oncology SA. Open-Label, Phase I, Multi-Center Study to Determine in Relapsed/Refractory Acute Myeloid Leukemia or Myelodysplastic Syndrome Patients the Recommended Dose of CYAD-02 After a Non-Myeloablative Preconditioning Chemotherapy Followed by a Potential Consolidation Cycle; clinicaltrials.gov: Bethesda, MD, USA, 2020.
- Nkarta, Inc. A Phase 1 Study of NKX101, an Activating Chimeric Receptor Natural Killer Cell Therapy, in Subjects With Hematological Malignancies or Dysplasias; clinicaltrials.gov: Bethesda, MD, USA, 2024.
- Huang, H. Clinical Trial for the Safety and Efficacy of NKG2D CAR-T Cell Therapy for Patients With Relapsed and/or Refractory Acute Myeloid Leukemia; clinicaltrials.gov: Bethesda, MD, USA, 2020.
- Poggi, A.; Catellani, S.; Garuti, A.; Pierri, I.; Gobbi, M.; Zocchi, M.R. Effective in Vivo Induction of NKG2D Ligands in Acute Myeloid Leukaemias by All-Trans-Retinoic Acid or Sodium Valproate. Leukemia 2009, 23, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Tokaz, M.C.; Baldomero, H.; Cowan, A.J.; Saber, W.; Greinix, H.; Koh, M.B.C.; Kröger, N.; Mohty, M.; Galeano, S.; Okamoto, S.; et al. An Analysis of the Worldwide Utilization of Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia. Transplant. Cell. Ther. 2023, 29, 279.e1–279.e10. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Ma, H. Targeting CD38 for Acute Leukemia. Front. Oncol. 2022, 12, 1007783. [Google Scholar] [CrossRef]
- Sentman, C.L.; Meehan, K.R. NKG2D CARs as Cell Therapy for Cancer. Cancer J. 2014, 20, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Maiorova, V.; Mollaev, M.D.; Vikhreva, P.; Kulakovskaya, E.; Pershin, D.; Chudakov, D.M.; Kibardin, A.; Maschan, M.A.; Larin, S. Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines. Vaccines 2021, 9, 1238. [Google Scholar] [CrossRef]
- Zoine, J.T.; Prince, C.; Story, J.Y.; Branella, G.M.; Lytle, A.M.; Fedanov, A.; Alexander, J.S.; Porter, C.C.; Doering, C.B.; Spencer, H.T.; et al. Thrombopoietin-Based CAR-T Cells Demonstrate in Vitro and in Vivo Cytotoxicity to MPL Positive Acute Myelogenous Leukemia and Hematopoietic Stem Cells. Gene Ther. 2021, 29, 1–12. [Google Scholar] [CrossRef]
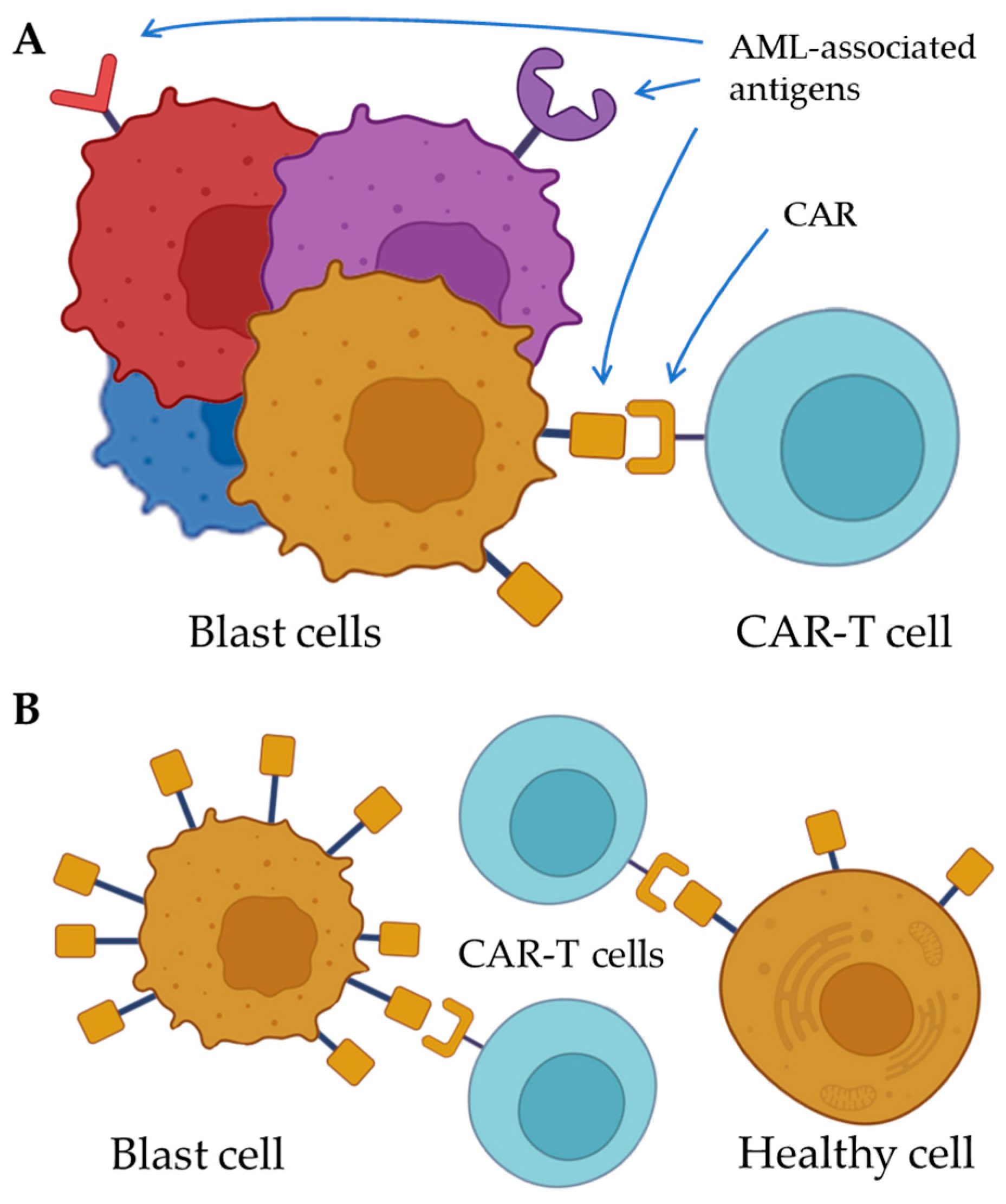
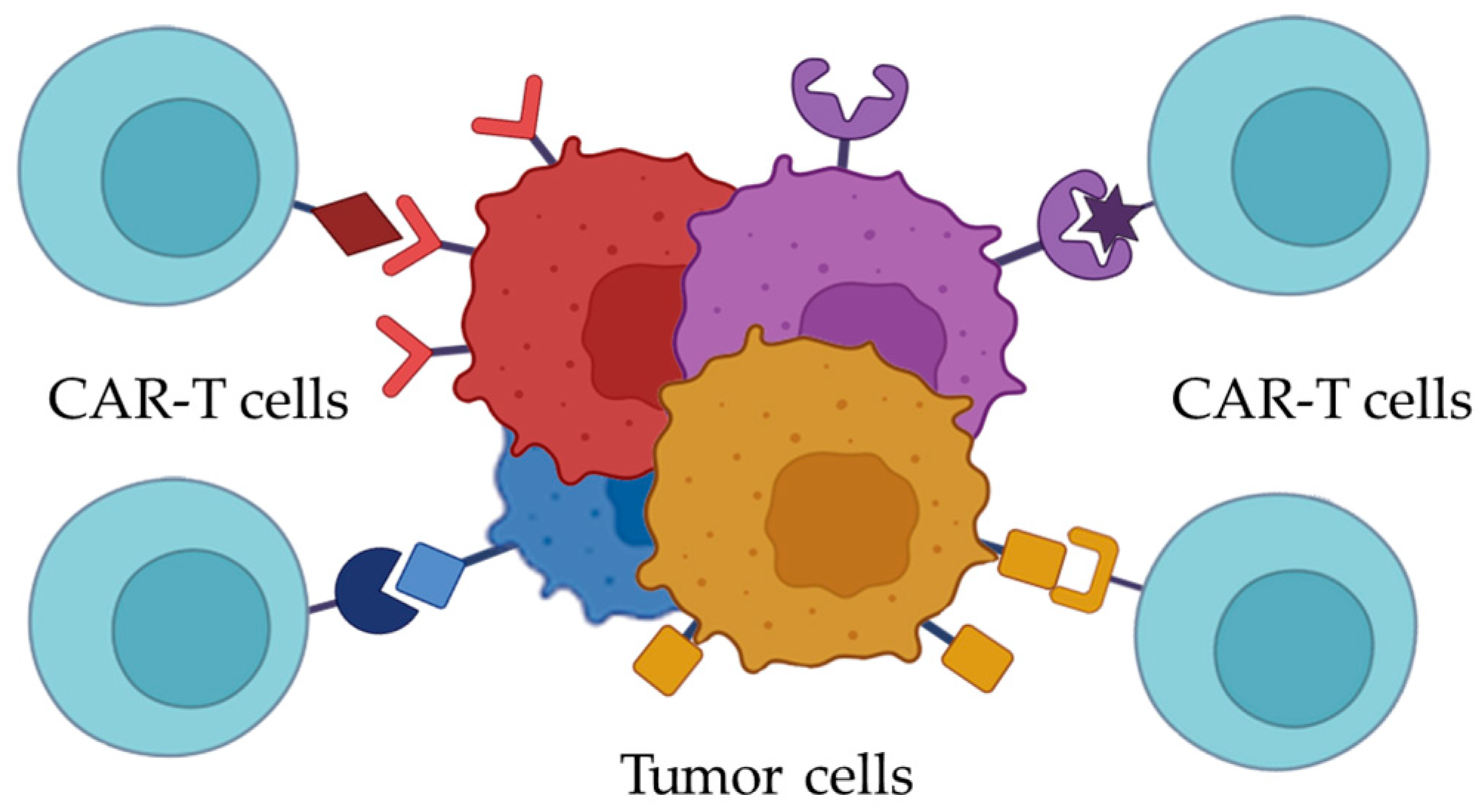

{kind=link}
{kind=link}
| Antigen | Frequency of Expression on the Blast Cells in AML | Presence on All the Blast Cells | Potential on-Target Off-Tumor Toxicity | Biological Function in AML |
|---|---|---|---|---|
| CD7 | 30% of cases | T cells, NK cells | Metastasis | |
| CD33 | 85–90% of cases | Neurotoxicity | ||
| CD38 | 50–90% of cases | Antigen density may vary (300–6000 protein/cell) [118] | Diverse | Migration |
| CD44v6 | 60% of cases | Diverse | Homing, migration, and proliferation | |
| CD70 | 34–39% of cases | No | Monocytes | Blast cells’ stemness |
| ILT3 (CD85k) | Mostly M4 and M5 AML | Dendritic cells, monocytes, osteoclasts, endothelial cells | Malignant cell infiltration and evading immune surveillance | |
| CD116 | 63–78% of cases (mostly M4 and M5 AML) | No | Granulocytic and monocytic lineage, myeloblasts | Leukocyte expansion, proliferation, survival |
| CD117 | 80–90% of cases | No | HSC | Proliferation, survival |
| CD123 | 70–80% of cases | No | Myeloid progenitor cells | Proliferation, survival |
| Flt3 (CD135) | >80% of cases | Dendritic cells | Proliferation, survival | |
| B7-H3 (CD276) | 37% of cases | Proliferation, migration, and inhibiting tumor microenvironment | ||
| Siglec-6 (CD327) | 60% of cases | No | B cells, mast cells, placenta | |
| TIM-3 (CD366) | No | T-cell subtypes, macrophages | Inhibiting tumor microenvironment | |
| CLL-1 (CD371) | >80% of cases | No | Granulocytic and monocytic lineage | |
| FRb | 70% of cases | Monocytic lineage | Leukocyte expansion | |
| NKG2D Ligands | 75% of cases | Damaged or infected cells | Evading immune surveillance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiorova, V.; Mollaev, M.D.; Vikhreva, P.; Kibardin, A.; Maschan, M.A.; Larin, S.S. The Problem of Molecular Target Choice for CAR-T Cells in Acute Myeloid Leukemia Therapy. Int. J. Mol. Sci. 2025, 26, 5428. https://doi.org/10.3390/ijms26125428
Maiorova V, Mollaev MD, Vikhreva P, Kibardin A, Maschan MA, Larin SS. The Problem of Molecular Target Choice for CAR-T Cells in Acute Myeloid Leukemia Therapy. International Journal of Molecular Sciences. 2025; 26(12):5428. https://doi.org/10.3390/ijms26125428
Chicago/Turabian StyleMaiorova, Varvara, Murad D. Mollaev, Polina Vikhreva, Alexey Kibardin, Michael A. Maschan, and Sergey S. Larin. 2025. "The Problem of Molecular Target Choice for CAR-T Cells in Acute Myeloid Leukemia Therapy" International Journal of Molecular Sciences 26, no. 12: 5428. https://doi.org/10.3390/ijms26125428
APA StyleMaiorova, V., Mollaev, M. D., Vikhreva, P., Kibardin, A., Maschan, M. A., & Larin, S. S. (2025). The Problem of Molecular Target Choice for CAR-T Cells in Acute Myeloid Leukemia Therapy. International Journal of Molecular Sciences, 26(12), 5428. https://doi.org/10.3390/ijms26125428