
.jpg)
The Role of the AGPAT2 Gene in Adipose Tissue Biology and Congenital Generalized Lipodystrophy Pathophysiology

 and
and
Abstract
1. Introduction
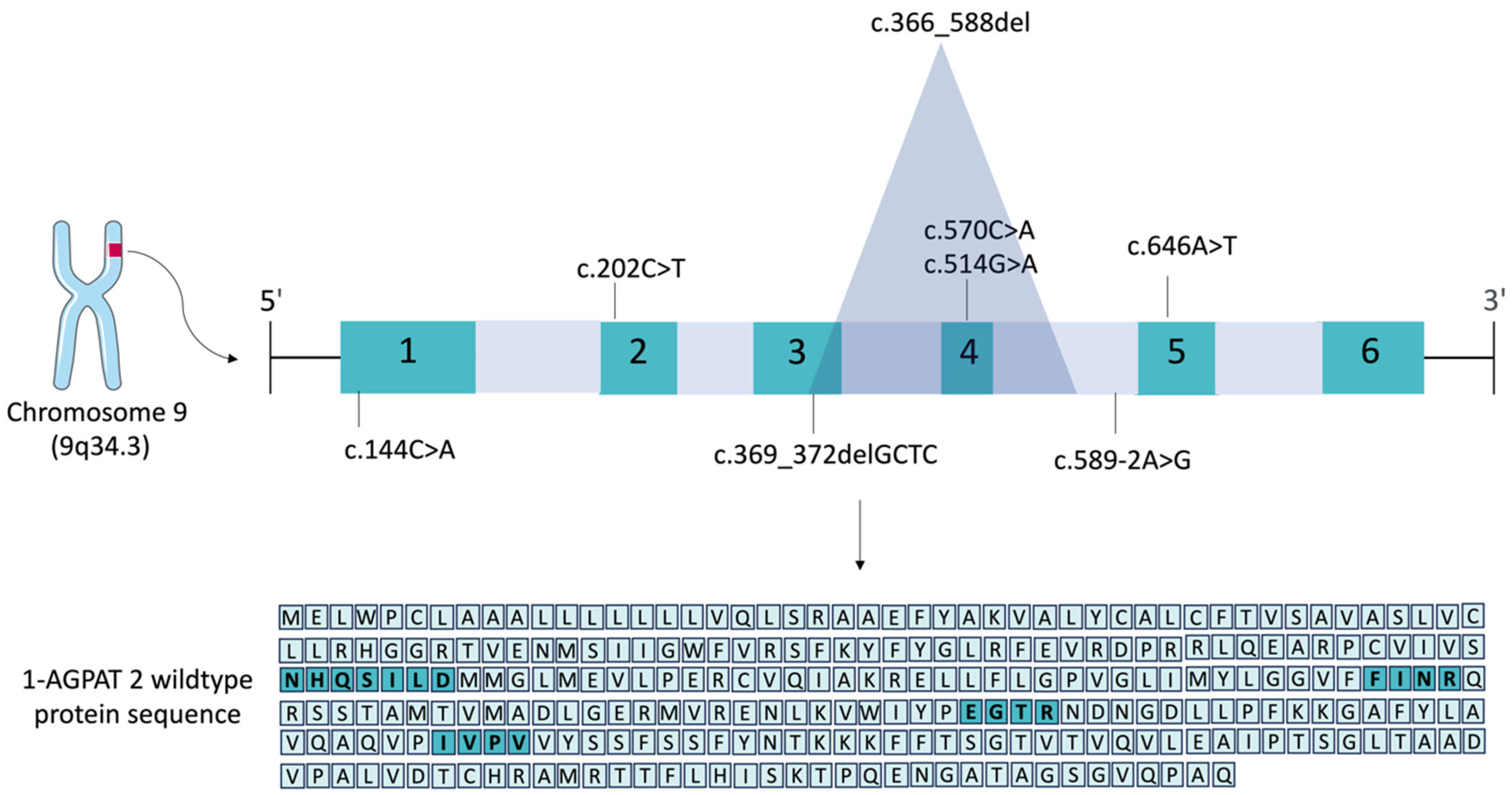
2. The 1-AGPAT 2 Protein and Its Motifs
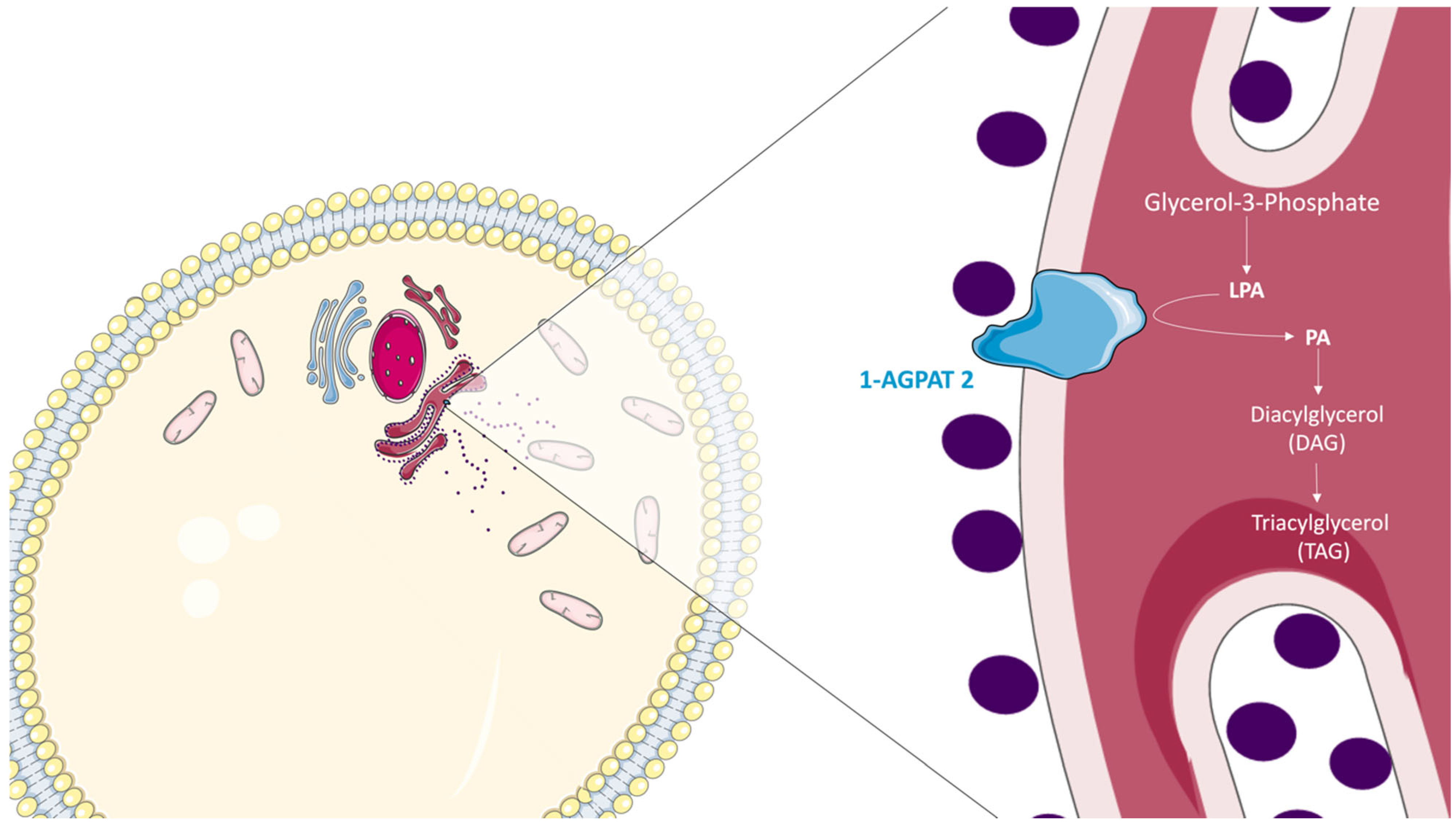
3. The Role of 1-AGPAT 2 in the Biosynthesis of Triacylglycerols
4. The Role of 1-AGPAT 2 in Adipogenesis
5. AGPAT2 and Lipodystrophy
6. Pathogenic Variants in the AGPAT2 Gene

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1-AGPAT 2 (NP_006403.2) | Software | TM1 | TM2 | TM3 | TM4 | N-Terminal | C-Terminal |
|---|---|---|---|---|---|---|---|
| WT | PSIPRED | 14–29 | 33–51 | 123–138 | 190–205 | Cytoplasmic | ER Lumen |
| SOSUI | 2–24 | 30–52 | 58–80 | 122–142 | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | 123–141 | 188–210 | ER Lumen | ER Lumen | |
| T-COFFEE | 31–50 | 59–76 | 123–141 | - | ER Lumen | Cytoplasmic | |
| c.144C>A | PSIPRED | 15–30 | - | - | - | Cytoplasmic | ER Lumen |
| SOSUI | 14–36 | - | - | - | ER Lumen | ER Lumen | |
| TMHMM | 4–21 | 26–45 | - | - | Cytoplasmic | Cytoplasmic | |
| T-COFFEE | 5–21 | 30–46 | - | - | Cytoplasmic | Cytoplasmic | |
| c.202C>T | PSIPRED | 15–30 | 36–51 | - | - | Cytoplasmic | Cytoplasmic |
| SOSUI | 2–24 | 30–52 | - | - | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | - | - | ER Lumen | ER Lumen | |
| T-COFFEE | 4–21 | 30–50 | - | - | Cytoplasmic | Cytoplasmic | |
| c.366-588del | PSIPRED | 12–27 | 31–50 | - | - | Cytoplasmic | Cytoplasmic |
| SOSUI | 2–24 | 30–52 | 58–80 | - | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | - | - | ER Lumen | ER Lumen | |
| T-COFFEE | 31–50 | 59–76 | - | - | ER Lumen | ER Lumen | |
| c.369_372delGCTC | PSIPRED | 13–28 | 32–52 | 61–76 | - | Cytoplasmic | ER Lumen |
| SOSUI | 2–24 | 30–52 | 58–80 | - | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | - | - | ER Lumen | ER Lumen | |
| T-COFFEE | 4–21 | 30–50 | 59–76 | - | Cytoplasmic | ER Lumen | |
| c.514G>A | PSIPRED | 13–28 | 32–51 | 123–138 | 190–205 | Cytoplasmic | Cytoplasmic |
| SOSUI | 2–24 | 30–52 | 58–80 | 122–142 | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | 123–141 | 188–210 | ER Lumen | ER Lumen | |
| T-COFFEE | 32–50 | 59–76 | 123–141 | - | ER Lumen | Cytoplasmic | |
| c.570C>A | PSIPRED | 13–28 | 50–32 | 123–138 | - | Cytoplasmic | ER Lumen |
| SOSUI | 2–24 | 30–52 | 58–80 | 122–142 | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | 123–141 | - | ER Lumen | Cytoplasmic | |
| T-COFFEE | 32–50 | 59–76 | 123–141 | - | ER Lumen | Cytoplasmic | |
| c.589-2A>G | PSIPRED | 13–28 | 32–50 | 123–138 | - | Cytoplasmic | ER Lumen |
| SOSUI | 2–24 | 30–52 | 58–80 | 122–142 | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | 123–141 | - | ER Lumen | Cytoplasmic | |
| T-COFFEE | 32–50 | 59–76 | 123–141 | - | ER Lumen | Cytoplasmic | |
| c.646A>T | PSIPRED | 13–28 | 58–32 | 123–138 | - | Cytoplasmic | ER Lumen |
| SOSUI | 2–24 | 30–52 | 58–80 | 122–142 | ER Lumen | ER Lumen | |
| TMHMM | 7–24 | 28–50 | 123–141 | 188–210 | ER Lumen | ER Lumen | |
| T-COFFEE | 31–50 | 59–76 | 123–141 | 187–207 | ER Lumen | ER Lumen |
| Pathogenic Variant (NM_006412.4) | c.144C>A | c.202C>T | c.366_588del | c.369_372delGCTC | c.514G>A | c.570C>A | c.589-2A>G | c.646A>T |
|---|---|---|---|---|---|---|---|---|
| References | [132] | [132,139] | [157] | [162] | [158] | [126] | [21] | [132] |
| Resulting 1-AGPAT 2 protein (NP_006403.2) | p.Cys48* | p.Arg68* | p.Leu123Cysfs*56 | p.Leu124Serfs*26 | p.Glu172Lys | p.Tyr190* | p.Val197Alafs*19 | p.Lys216* |
| Protein consequence | Smaller and truncated protein with 47 aa | Smaller and truncated protein with 67 aa | Smaller and truncated protein with 177 aa | Smaller and truncated protein with 148 aa | Poorly functional protein with 278 aa | Smaller and truncated protein with 189 aa | Smaller and truncated protein with 214 aa | Smaller and truncated protein with 215 aa |
| Protein domains affected | All domains absent | All domains absent | EGTR, FINR and IVPV domains absent | EGTR, FINR and IVPV domains absent | EGTR domain affected: change from E (glutamate) to K (lysine) aa | IVPV domain absent | IVPV domain absent | All domains preserved |
| Number of patients (n) | 3 | 2 [132]; 2 [139] | 10 | 2 | 2 | 1 | 5 | 1 |
| Age (average in years) | 28 | 13 [132]; 63 [139] | 40 | 8 | 0,3 | 20 | 19 | 25 |
| Generalized lack of subcutaneous WAT (sWAT) | + | + [132]; + [139] | + | + | + | + | + | + |
| Hypertriglyceridemia | + | + [132]; - [139] | + | + | + | + | + | + |
| Diabetes mellitus 2 | + | - [132]; + [139] | + | + | + | + | + | +++ |
| Acanthosis nigricans | + | - [132]; + [139] | + | + | - | + | - | + |
| Insulin resistance | - | +++ [132]; - [139] | +++ | - | + | - | +++ | - |
| Retinopathy | + | - [132]; + [139] | - | - | - | - | - | + |
| Diabetic neuropathy | + | - [132]; - [139] | - | - | - | - | - | + |
| Recurrent acute pancreatitis | + | - [132]; - [139] | - | - | - | - | - | - |
| Splenic artery aneurysm | + | - [132]; - [139] | - | - | - | - | - | - |
| Hepatomegaly | - | - [132]; - [139] | + | + | + | - | - | - |
| Bone cysts | + | - [132]; + [139] | - | - | + | - | - | - |
| Polycystic ovary | + | - [132]; - [139] | - | - | + | - | - | - |
| Hypertension | + | - [132]; + [139] | - | - | + | - | - | - |
| Renal failure | + | - [132]; - [139] | - | - | - | - | - | - |
| Muscular hypertrophy | - | - [132]; - [139] | + | + | + | + | - | - |
| Inguinal hernia | - | + [132]; - [139] | - | - | - | - | - | - |
| Umbilical hernia | - | - [132]; - [139] | - | + | - | - | - | - |
| Increased abdominal volume | - | - [132]; - [139] | - | + | - | - | - | - |
| Hepatic steatosis | - | - [132]; - [139] | - | - | - | - | - | - |
| Large ears | - | - [132]; - [139] | - | - | + | - | - | - |
| Genital dysmorphism | - | + [132]; - [139] | - | - | + | - | - | - |
| Acromegaloid dysmorphism | + | + [132]; - [139] | ++ | - | + | - | - | - |
| Hirsutism | - | - [132]; - [139] | + | - | - | - | - | - |
7. Concluding Remarks and Future Directions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ye, G.M.; Chen, C.; Huang, S.; Han, D.D.; Guo, J.H.; Wan, B.; Yu, L. Cloning and characterization a novel human 1-acyl-sn-glycerol-3-phosphate acyltransferase gene AGPAT7. DNA Seq. 2005, 16, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, L.; Beigneux, A.P.; Davis, R.; Watkins, S.M.; Young, S.G.; Reue, K. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J. Lipid Res. 2006, 47, 745–754. [Google Scholar] [CrossRef]
- Beigneux, A.P.; Vergnes, L.; Qiao, X.; Quatela, S.; Davis, R.; Watkins, S.M.; Coleman, R.A.; Walzem, R.L.; Philips, M.; Reue, K.; et al. Agpat6—A novel lipid biosynthetic gene required for triacylglycerol production in mammary epithelium. J. Lipid Res. 2006, 47, 734–744. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Barnes, R.I.; Garg, A. Functional characterization of human 1-acylglycerol-3-phosphate acyltransferase isoform 8: Cloning, tissue distribution, gene structure, and enzymatic activity. Arch. Biochem. Biophys. 2006, 449, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Nagle, C.A.; Vergnes, L.; Dejong, H.; Wang, S.; Lewin, T.M.; Reue, K.; Coleman, R.A. Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4, as the enzyme deficient in Agpat6−/− mice. J. Lipid Res. 2008, 49, 823–831. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Kuo, M.S.; Li, S.; Bui, H.H.; Peake, D.A.; Sanders, P.E.; Thibodeaux, S.J.; Chu, S.; Qian, Y.W.; Zhao, Y.; et al. AGPAT6 Is a Novel Microsomal Glycerol-3-phosphate Acyltransferase. J. Biol. Chem. 2008, 283, 10048–10057. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Garg, A. Enzymatic activity of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 11: Upregulated in breast and cervical cancers. J. Lipid Res. 2010, 51, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Barnes, R.I.; Garg, A.; Agarwal, A.K. Functional characterization of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 10/glycerol-3-phosphate acyltransferase isoform 3. J. Mol. Endocrinol. 2009, 42, 469–478. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Sukumaran, S.; Bartz, R.; Barnes, R.I.; Garg, A. Functional characterization of human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 9: Cloning, tissue distribution, gene structure, and enzymatic activity. J. Endocrinol. 2007, 193, 445–457. [Google Scholar] [CrossRef]
- McMaster, C.R.; Jackson, T.R. 1 Phospholipid synthesis in mammalian cells. In Lipid Metabolism and Membrane Biogenesis; Springer: Berlin/Heidelberg, Germany, 2023; pp. 5–88. [Google Scholar] [CrossRef]
- Coleman, R. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Takeuchi, K.; Reue, K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol.-Endocrinol. Metab. 2009, 296, E1195–E1209. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Garg, A. Congenital generalized lipodystrophy: Significance of triglyceride biosynthetic pathways. Trends Endocrinol. Metab. 2003, 14, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W. The structure and functions of human lysophosphatidic acid acyltransferases. Frontiers in Bioscience 2001, 6, d944. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K. Lysophospholipid acyltransferases: 1-acylglycerol-3-phosphate O-acyltransferases. From discovery to disease. Curr. Opin. Lipidol. 2012, 23, 290–302. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Tompkins, C.K.; Balantac, N.; Nudelman, E.; Meengs, B.; White, T.; Bursten, S.; Coleman, J.; Kumar, A.; Singer, J.W.; et al. Cloning and Expression of Two Human Lysophosphatidic Acid Acyltransferase cDNAs That Enhance Cytokine-Induced Signaling Responses in Cells. DNA Cell Biol. 1997, 16, 691–701. [Google Scholar] [CrossRef]
- Aguado, B.; Campbell, R.D. Characterization of a Human Lysophosphatidic Acid Acyltransferase That Is Encoded by a Gene Located in the Class III Region of the Human Major Histocompatibility Complex. J. Biol. Chem. 1998, 273, 4096–4105. [Google Scholar] [CrossRef]
- Eberhardt, C.; Gray, P.W.; Tjoelker, L.W. cDNA Cloning, Expression and Chromosomal Localization of Two Human Lysophosphatidic Acid Acyltransferases. In Eicosanoids and Other Bioactive Lipids in Cancer, Inflammation, and Radiation Injury; Springer: Boston, MA, USA, 1999; pp. 351–356. [Google Scholar] [CrossRef]
- Nigam, S.; Honn, K.V.; Marnett, L.J.; Walden, T.L. (Eds.) Eicosanoids and other bioactive lipids in cancer, inflammation and radiation injury. In Proceedings of the 2nd International Conference Klinikum Steglitz, Berlin, Germany, 17–21 September 1991; Abstracts. Eicosanoids 4 Suppl. Springer: New York, NY, USA, 1991; pp. S1–S29. [Google Scholar]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Arioglu, E.; De Almeida, S.; Akkoc, N.; Taylor, S.I.; Bowcock, A.M.; Barnes, R.I.; Garg, A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet. 2002, 31, 21–23. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Garg, A. Genetic Disorders of Adipose Tissue Development, Differentiation, and Death. Annu. Rev. Genom. Hum. Genet. 2006, 7, 175–199. [Google Scholar] [CrossRef]
- Nilay, G.; Kutlu, T.; Tekant, G.T.; Eroğlu, A.G.; Üstündağ, N.Ç.; Öztürk, B.; Onay, H.; Tüysüz, B. Congenital generalized lipodystrophy: The evaluation of clinical follow-up findings in a series of five patients with type 1 and two patients with type 4. Eur. J. Med. Genet. 2020, 63, 103819. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Barnes, R.I.; Garg, A. Genetic basis of congenital generalized lipodystrophy. Int. J. Obes. 2004, 28, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.E.; Kropman, E.; Kranendonk, M.E.; Koppen, A.; Hamers, N.; Stroes, E.S.; Kalkhoven, E.; Monajemi, H. Characterisation of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPARγ mutation (Y151C). Diabetologia 2011, 54, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Broekema, M.F.; Massink, M.P.G.; De Ligt, J.; Stigter, E.C.A.; Monajemi, H.; De Ridder, J.; Burgering, B.M.T.; van Haaften, G.W.; Kalkhoven, E. A Single Complex Agpat2 Allele in a Patient With Partial Lipodystrophy. Front. Physiol. 2018, 9, 1363. [Google Scholar] [CrossRef] [PubMed]
- Chiquette, E.; Oral, E.A.; Garg, A.; Araújo-Vilar, D.; Dhankhar, P. Estimating the prevalence of generalized and partial lipodystrophy: Findings and challenges. Diabetes Metab. Syndr. Obes. 2017, 10, 375–383. [Google Scholar] [CrossRef]
- de Azevedo Medeiros, L.B.; Cândido Dantas, V.K.; Craveiro Sarmento, A.S.; Agnez-Lima, L.F.; Meireles, A.L.; Xavier Nobre, T.T.; de Lima, J.G.; de Melo Campos, J.T.A. High prevalence of Berardinelli-Seip Congenital Lipodystrophy in Rio Grande do Norte State, Northeast Brazil. Diabetol. Metab. Syndr. 2017, 9, 80. [Google Scholar] [CrossRef]
- Lima, J.G.; Nobrega, L.H.C.; Lima, N.N.; Dos Santos, M.C.F.; Silva, P.H.D.; Baracho, M.F.P.; Lima, D.N.; de Melo Campos, J.T.A.; Ferreira, L.C.; Freire Neto, F.P.; et al. Causes of death in patients with Berardinelli-Seip congenital generalized lipodystrophy. PLoS ONE 2018, 13, e0199052. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef]
- Karagiota, A.; Chachami, G.; Paraskeva, E. Lipid Metabolism in Cancer: The Role of Acylglycerolphosphate Acyltransferases (AGPATs). Cancers 2022, 14, 228. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Lefter, M.; Vis, J.K.; Vermaat, M.; den Dunnen, J.T.; Taschner, P.E.M.; Laros, J.F.J. Mutalyzer 2: Next generation HGVS nomenclature checker. Bioinformatics 2021, 37, 2811–2817. [Google Scholar] [CrossRef]
- Bradley, R.M.; Duncan, R.E. The lysophosphatidic acid acyltransferases (acylglycerophosphate acyltransferases) family: One reaction, five enzymes, many roles. Curr. Opin. Lipidol. 2018, 29, 110–115. [Google Scholar] [CrossRef]
- Prasad, S.S.; Garg, A.; Agarwal, A.K. Enzymatic activities of the human AGPAT isoform 3 and isoform 5: Localization of AGPAT5 to mitochondria. J. Lipid Res. 2011, 52, 451–462. [Google Scholar] [CrossRef]
- Cao, J.; Shan, D.; Revett, T.; Li, D.; Wu, L.; Liu, W.; Tobin, J.F.; Gimeno, R.E. Molecular Identification of a Novel Mammalian Brain Isoform of Acyl-CoA:Lysophospholipid Acyltransferase with Prominent Ethanolamine Lysophospholipid Acylating Activity, LPEAT2. J. Biol. Chem. 2008, 283, 19049–19057. [Google Scholar] [CrossRef]
- Mak, H.Y.; Ouyang, Q.; Tumanov, S.; Xu, J.; Rong, P.; Dong, F.; Lam, S.M.; Wang, X.; Lukmantara, I.; Du, X.; et al. AGPAT2 interaction with CDP-diacylglycerol synthases promotes the flux of fatty acids through the CDP-diacylglycerol pathway. Nat. Commun. 2021, 12, 6877. [Google Scholar] [CrossRef]
- LU, B.; Jiang, Y.J.; Zhou, Y.; Xu, F.Y.; Hatch, G.M.; Choy, P.C. Cloning and characterization of murine 1-acyl-sn-glycerol 3-phosphate acyltransferases and their regulation by PPARα in murine heart. Biochem. J. 2005, 385, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Hayashi, Y.; Matsumoto, N.; Nemoto-Sasaki, Y.; Oka, S.; Tanikawa, T.; Sugiura, T. Glycerophosphate/Acylglycerophosphate Acyltransferases. Biology 2014, 3, 801–830. [Google Scholar] [CrossRef]
- Yamashita, A.; Hayashi, Y.; Nemoto-Sasaki, Y.; Ito, M.; Oka, S.; Tanikawa, T.; Waku, K.; Sugiura, T. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid Res. 2014, 53, 18–81. [Google Scholar] [CrossRef]
- Haque, W.; Garg, A.; Agarwal, A.K. Enzymatic activity of naturally occurring 1-acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital generalized lipodystrophy. Biochem. Biophys. Res. Commun. 2005, 327, 446–453. [Google Scholar] [CrossRef]
- Akita, Y. Protein Kinase C- (PKC-): Its Unique Structure and Function. J. Biochem. 2002, 132, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Pagès, C.; Simon, M.-F.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat. 2001, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Lee, M.-J.; Ancellin, N.; Paik, J.H.; Kluk, M.J. Lysophospholipids--Receptor Revelations. Science (1979) 2001, 294, 1875–1878. [Google Scholar] [CrossRef]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic Acid-Mediated Mitogenic Activation of mTOR Signaling. Science (1979) 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Sprong, H.; van der Sluijs, P.; van Meer, G. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell Biol. 2001, 2, 504–513. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Capeau, J.; Magré, J.; Caron-Debarle, M.; Lagathu, C.; Antoine, B.; Béréziat, V.R.; Lascols, O.; Bastard, J.P.; Vigouroux, C. Human Lipodystrophies: Genetic and Acquired Diseases of Adipose Tissue. Endocr. Dev. 2010, 19, 1–20. [Google Scholar] [CrossRef]
- Capeau, J.; Magré, J.; Lascols, O.; Caron, M.; Béréziat, V.; Vigouroux, C. Les lipodystrophies primitives. Ann Endocrinol. 2007, 68, 10–20. [Google Scholar] [CrossRef]
- Capeau, J.; Vigouroux, C.; Magré, J.; Lascols, O.; Caron, M.; Bastard, J.P. Les syndromes lipodystrophiques: Des adipopathies congénitales ou acquises. C R. Biol. 2006, 329, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Capeau, J.; Magré, J.; Lascols, O.; Caron, M.; Béréziat, V.; Vigouroux, C.; Bastard, J.P. Diseases of adipose tissue: Genetic and acquired lipodystrophies. Biochem. Soc. Trans. 2005, 33, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Agarwal, A.K. Lipodystrophies: Disorders of adipose tissue biology. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 507–513. [Google Scholar] [CrossRef]
- Garg, A. Lipodystrophies: Genetic and Acquired Body Fat Disorders. J. Clin. Endocrinol. Metab. 2011, 96, 3313–3325. [Google Scholar] [CrossRef]
- Subauste, A.R.; Elliott, B.; Das, A.K.; Burant, C.F. A role for 1-acylglycerol-3-phosphate-O-acyltransferase-1 in myoblast differentiation. Differentiation 2010, 80, 140–146. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Tunison, K.; Vale, G.; McDonald, J.G.; Li, X.; Horton, J.D.; Garg, A. Adipose-specific overexpression of human AGPAT2 in mice causes increased adiposity and mild hepatic dysfunction. iScience 2024, 27, 108653. [Google Scholar] [CrossRef]
- Ruan, H.; Pownall, H.J. Overexpression of 1-Acyl-Glycerol-3-Phosphate Acyltransferase-αEnhances Lipid Storage in Cellular Models of Adipose Tissue and Skeletal Muscle. Diabetes 2001, 50, 233–240. [Google Scholar] [CrossRef]
- Sankella, S.; Garg, A.; Horton, J.D.; Agarwal, A.K. Hepatic Gluconeogenesis Is Enhanced by Phosphatidic Acid Which Remains Uninhibited by Insulin in Lipodystrophic Agpat2−/− Mice. J. Biol. Chem. 2014, 289, 4762–4777. [Google Scholar] [CrossRef]
- Cautivo, K.M.; Lizama, C.O.; Tapia, P.J.; Agarwal, A.K.; Garg, A.; Horton, J.D.; Cortés, V.A. AGPAT2 is essential for postnatal development and maintenance of white and brown adipose tissue. Mol. Metab. 2016, 5, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, I.; Gaspar, R.C.; Luukkonen, P.K.; Kahn, M.; Zhang, D.; Zhang, X.; Murray, S.; Golla, J.P.; Vatner, D.F.; Samuel, V.T.; et al. Lysophosphatidic acid triggers inflammation in the liver and white adipose tissue in rat models of 1-acyl-sn-glycerol-3-phosphate acyltransferase 2 deficiency and overnutrition. Proc. Natl. Acad. Sci. USA 2023, 120, e2312666120. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.E.; Frolov, A.; Han, X.; Bickel, P.E.; Cao, L.; Bowcock, A.; Schaffer, J.E.; Ory, D.S. A Regulatory Role for 1-Acylglycerol-3-phosphate-O-acyltransferase 2 in Adipocyte Differentiation. J. Biol. Chem. 2006, 281, 11082–11089. [Google Scholar] [CrossRef]
- Pan, K.; Zhu, B.; Wang, L.; Guo, Q.; Shu-Chien, A.C.; Wu, X. Expression pattern of AGPATs isoforms indicate different functions during the triacylglyceride synthesis in Chinese mitten crab, Eriocheir sinensis. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2024, 287, 111535. [Google Scholar] [CrossRef]
- Bargut, T.C.L.; Souza-Mello, V.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Browning of white adipose tissue: Lessons from experimental models. Horm. Mol. Biol. Clin. Investig. 2017, 31, 20160051. [Google Scholar] [CrossRef]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008417. [Google Scholar] [CrossRef] [PubMed]
- Betz, M.J.; Enerbäck, S. Human Brown Adipose Tissue: What We Have Learned So Far. Diabetes 2015, 64, 2352–2360. [Google Scholar] [CrossRef]
- Bargut, T.C.L.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Brown adipose tissue: Updates in cellular and molecular biology. Tissue Cell 2016, 48, 452–460. [Google Scholar] [CrossRef]
- Sakers, A.; De Siqueira, M.K.; Seale, P.; Villanueva, C.J. Adipose-tissue plasticity in health and disease. Cell 2022, 185, 419–446. [Google Scholar] [CrossRef]
- Farmer, S.R. Molecular determinants of brown adipocyte formation and function: Figure 1. Genes. Dev. 2008, 22, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Pinckard, K.M.; Stanford, K.I. The Heartwarming Effect of Brown Adipose Tissue. Mol. Pharmacol. 2022, 102, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Peres Valgas da Silva, C.; Shettigar, V.K.; Baer, L.A.; Abay, E.; Madaris, K.L.; Mehling, M.R.; Hernandez-Saavedra, D.; Pinckard, K.M.; Seculov, N.P.; Ziolo, M.T.; et al. Brown adipose tissue prevents glucose intolerance and cardiac remodeling in high-fat-fed mice after a mild myocardial infarction. Int. J. Obes. 2022, 46, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Werner, C.D.; Kebebew, E.; Celi, F.S. Functional thermogenic beige adipogenesis is inducible in human neck fat. Int. J. Obes. 2014, 38, 170–176. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Tunison, K.; Vale, G.; McDonald, J.G.; Li, X.; Scherer, P.E.; Horton, J.D.; Garg, A. Regulated adipose tissue-specific expression of human AGPAT2 in lipodystrophic Agpat2-null mice results in regeneration of adipose tissue. iScience 2023, 26, 107806. [Google Scholar] [CrossRef]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef]
- Distel, R.J.; Ro, H.-S.; Rosen, B.S.; Groves, D.L.; Spiegelman, B.M. Nucleoprotein complexes that regulate gene expression in adipocyte differentiation: Direct participation of c-fos. Cell 1987, 49, 835–844. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef]
- Bose, G.S.; Kalakoti, G.; Kulkarni, A.P.; Mittal, S. AP-1/C-FOS and AP-1/FRA2 differentially regulate early and late adipogenic differentiation of mesenchymal stem cells. J. Cell. Biochem. 2024, 125, e30543. [Google Scholar] [CrossRef]
- Stephens, J.M.; Morrison, R.F.; Pilch, P.F. The Expression and Regulation of STATs during 3T3-L1 Adipocyte Differentiation. J. Biol. Chem. 1996, 271, 10441–10444. [Google Scholar] [CrossRef]
- Deng, J.; Hua, K.; Lesser, S.S.; Harp, J.B. Activation of Signal Transducer and Activator of Transcription-3 during Proliferative Phases of 3T3-L1 Adipogenesis*. Endocrinology 2000, 141, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Harp, J.B.; Franklin, D.; Vanderpuije, A.A.; Gimble, J.M. Differential Expression of Signal Transducers and Activators of Transcription during Human Adipogenesis. Biochem. Biophys. Res. Commun. 2001, 281, 907–912. [Google Scholar] [CrossRef]
- Ambele, M.A.; Dhanraj, P.; Giles, R.; Pepper, M.S. Adipogenesis: A Complex Interplay of Multiple Molecular Determinants and Pathways. Int. J. Mol. Sci. 2020, 21, 4283. [Google Scholar] [CrossRef] [PubMed]
- Christodoulides, C.; Lagathu, C.; Sethi, J.K.; Vidal-Puig, A. Adipogenesis and WNT signalling. Trends Endocrinol. Metab. 2009, 20, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-N.; Wu, J.-F. TGF-β/SMAD signaling regulation of mesenchymal stem cells in adipocyte commitment. Stem. Cell. Res. Ther. 2020, 11, 41. [Google Scholar] [CrossRef]
- Jiao, Y.; Ahmed, U.; Sim, M.F.M.; Bejar, A.; Zhang, X.; Talukder, M.M.U.; Rice, R.; Flannick, J.; Podgornaia, A.I.; Reilly, D.F.; et al. Discovering metabolic disease gene interactions by correlated effects on cellular morphology. Mol. Metab. 2019, 24, 108–119. [Google Scholar] [CrossRef]
- Subauste, A.R.; Das, A.K.; Li, X.; Elliott, B.G.; Evans, C.; El Azzouny, M.; Treutelaar, M.; Oral, E.; Leff, T.; Burant, C.F. Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes 2012, 61, 2922–2931. [Google Scholar] [CrossRef]
- Talukder Md, M.U.; Sim, M.F.M.; O’Rahilly, S.; Edwardson, J.M.; Rochford, J.J. Seipin oligomers can interact directly with AGPAT2 and lipin 1, physically scaffolding critical regulators of adipogenesis. Mol. Metab. 2015, 4, 199–209. [Google Scholar] [CrossRef]
- Sim, M.F.M.; Persiani, E.; Talukder, M.M.U.; Mcilroy, G.D.; Roumane, A.; Edwardson, J.M.; Rochford, J.J. Oligomers of the lipodystrophy protein seipin may co-ordinate GPAT3 and AGPAT2 enzymes to facilitate adipocyte differentiation. Sci. Rep. 2020, 10, 3259. [Google Scholar] [CrossRef]
- Qi, Y.; Sun, L.; Yang, H. Lipid droplet growth and adipocyte development: Mechanistically distinct processes connected by phospholipids. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 1273–1283. [Google Scholar] [CrossRef]
- Fernández-Galilea, M.; Tapia, P.; Cautivo, K.; Morselli, E.; Cortés, V.A. AGPAT2 deficiency impairs adipogenic differentiation in primary cultured preadipocytes in a non-autophagy or apoptosis dependent mechanism. Biochem. Biophys. Res. Commun. 2015, 467, 39–45. [Google Scholar] [CrossRef]
- Tapia, P.J.; Tapia, P.J.; Figueroa, A.M.; Eisner, V.; González-Hódar, L.; Robledo, F.; Agarwal, A.K.; Garg, A.; Cortés, V. Absence of AGPAT2 impairs brown adipogenesis, increases IFN stimulated gene expression and alters mitochondrial morphology. Metabolism 2020, 111, 154341. [Google Scholar] [CrossRef] [PubMed]
- González-Hódar, L.; McDonald, J.G.; Vale, G.; Thompson, B.M.; Figueroa, A.M.; Tapia, P.J.; Robledo, F.; Agarwal, A.K.; Garg, A.; Horton, J.D.; et al. Decreased caveolae in AGPAT2 lacking adipocytes is independent of changes in cholesterol or sphingolipid levels: A whole cell and plasma membrane lipidomic analysis of adipogenesis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 166167. [Google Scholar] [CrossRef] [PubMed]
- Berardinelli, W. An Undiagnosed Endocrinometabolic Syndrome: Report of 2 Cases. J. Clin. Endocrinol. Metab. 1954, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Seip, M.; Trygstad, O. Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr. 1996, 85, 2–28. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, O.; Marcus-Samuels, B.; Graham, D.; Kim, J.K.; Shulman, G.I.; Castle, A.L.; Vinson, C.; Eckhaus, M.; Reitman, M.L. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J. Clin. Investig. 2000, 105, 271–278. [Google Scholar] [CrossRef]
- Shimomura, I.; Hammer, R.E.; Richardson, J.A.; Ikemoto, S.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: Model for congenital generalized lipodystrophy. Genes. Dev. 1998, 12, 3182–3194. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Gavrilova, O.; Chen, Y.; Reitman, M.L.; Shulman, G.I. Mechanism of Insulin Resistance in A-ZIP/F-1 Fatless Mice. J. Biol. Chem. 2000, 275, 8456–8460. [Google Scholar] [CrossRef]
- Ebihara, K.; Ogawa, Y.; Masuzaki, H.; Shintani, M.; Miyanaga, F.; Aizawa-Abe, M.; Hayashi, T.; Hosoda, K.; Inoue, G.; Yoshimasa, Y.; et al. Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes 2001, 50, 1440–1448. [Google Scholar] [CrossRef]
- Cortés, V.A.; Cautivo, K.M.; Rong, S.; Garg, A.; Horton, J.D.; Agarwal, A.K. Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2 lipodystrophic mice independent of hepatocyte leptin receptors. J. Lipid Res. 2014, 55, 276–288. [Google Scholar] [CrossRef]
- Chong, A.Y.; Lupsa, B.C.; Cochran, E.K.; Gorden, P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 2010, 53, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Simsir, I.; Yurekli, B.; Polat, I.; Saygili, F.; Akinci, B. Metreleptin replacement treatment improves quality of life and psychological well-being in congenital generalized lipodystrophy. Natl. Med. J. India 2020, 33, 278. [Google Scholar] [CrossRef] [PubMed]
- Cortés, V.A.; Curtis, D.E.; Sukumaran, S.; Shao, X.; Parameswara, V.; Rashid, S.; Smith, A.R.; Ren, J.; Esser, V.; Hammer, R.E.; et al. Molecular Mechanisms of Hepatic Steatosis and Insulin Resistance in the AGPAT2-Deficient Mouse Model of Congenital Generalized Lipodystrophy. Cell Metab. 2009, 9, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Chen, X.; Pei, K.; Wang, X.; Ma, X.; Liang, C.; Dong, Q.; Liu, Z.; Han, M.; Liu, G.; et al. Lipodystrophic gene Agpat2 deficiency aggravates hyperlipidemia and atherosclerosis in Ldlr mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2024, 1870, 166850. [Google Scholar] [CrossRef]
- Feng, T.; Tao, Y.; Yan, Y.; Lu, S.; Li, Y.; Zhang, X.; Qiang, J. Transcriptional Inhibition of AGPAT2 Induces Abnormal Lipid Metabolism and Oxidative Stress in the Liver of Nile Tilapia Oreochromis niloticus. Antioxidants 2023, 12, 700. [Google Scholar] [CrossRef]
- Debray, F.-G.; Baguette, C.; Colinet, S.; Van Maldergem, L.; Verellen-Dumouin, C. Early infantile cardiomyopathy and liver disease: A multisystemic disorder caused by congenital lipodystrophy. Mol. Genet. Metab. 2013, 109, 227–229. [Google Scholar] [CrossRef]
- Garg, A.; Chandalia, M.; Vuitch, F. Severe Islet Amyloidosis in Congenital Generalized Lipodystrophy. Diabetes Care 1996, 19, 28–31. [Google Scholar] [CrossRef]
- Garg, A. Dyslipidemias: Pathophysiology, Evaluation and Management; Humana Press: Humana Totowa, NJ, USA, 2015. [Google Scholar]
- Simha, V.; Garg, A. Inherited lipodystrophies and hypertriglyceridemia. Curr. Opin. Lipidol. 2009, 20, 300–308. [Google Scholar] [CrossRef]
- do Rêgo, A.G.; Mesquita, E.T.; Faria, C.A.; Rêgo, M.A.; Baracho, M.d.F.; Santos, M.G.; Egito, E.S.; Brandão Neto, J. Anormalidades cardiovasculares e metabólicas em pacientes com a síndrome de Berardinelli-Seip. Arq. Bras. Cardiol. 2010, 94, 109–118. [Google Scholar] [CrossRef]
- Hummadi, A.; Nahari, A.A.; Alhagawy, A.J.; Zakri, I.; Abutaleb, R.; Yafei, S. Congenital generalized lipodystrophy in two siblings from Saudi Arabia: A case report. Clin. Case Rep. 2022, 10, e05720. [Google Scholar] [CrossRef]
- Mann, J.P.; Savage, D.B. What lipodystrophies teach us about the metabolic syndrome. J. Clin. Investig. 2019, 129, 4009–4021. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, V.; Bernardes, I.; Pereira, J.; Silva, R.P.; Fernandes, S.; Carvalho, D.; Freitas, P. Acromegaly with congenital generalized lipodystrophy—Two rare insulin resistance conditions in one patient: A case report. J. Med. Case Rep. 2020, 14, 34. [Google Scholar] [CrossRef]
- Semple, R.K.; Savage, D.B.; Cochran, E.K.; Gorden, P.; O’Rahilly, S. Genetic Syndromes of Severe Insulin Resistance. Endocr. Rev. 2011, 32, 498–514. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.L.; Oral, E.A. Clinical Classification and Treatment of Congenital and Acquired Lipodystrophy. Endocr. Pract. 2010, 16, 310–323. [Google Scholar] [CrossRef]
- Van Maldergem, L. Berardinelli-Seip Congenital Lipodystrophy. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Agarwal, A.K.; Simha, V.; Oral, E.A.; Moran, S.A.; Gorden, P.; O’Rahilly, S.; Zaidi, Z.; Gurakan, F.; Arslanian, S.A.; Klar, A.; et al. Phenotypic and Genetic Heterogeneity in Congenital Generalized Lipodystrophy. J. Clin. Endocrinol. Metab. 2003, 88, 4840–4847. [Google Scholar] [CrossRef]
- Ferraria, N.; Pedrosa, C.; Amaral, D.; Lopes, L. Berardinelli–Seip syndrome: Highlight of treatment challenge. BMJ Case Rep. 2013, bcr2012007734. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, H.; Panigrihi, I.; Bhatia, P. Oil Red-O Positive lipid blobs on peripheral blood film examination in a muscular infant with the diagnosis of Berardinelli-Seip syndrome. Oxf. Med. Case Rep. 2019, 2019, omz062. [Google Scholar]
- Rahman, O.U.; Khawar, N.; Khan, M.A.; Ahmed, J.; Khattak, K.; Al-Aama, J.Y.; Naeem, M.; Jelani, M. Deletion mutation in BSCL2 gene underlies congenital generalized lipodystrophy in a Pakistani family. Diagn. Pathol. 2013, 8, 78. [Google Scholar] [CrossRef]
- Garg, A.; Misra, A. Lipodystrophies: Rare disorders causing metabolic syndrome. Endocrinol. Metab. Clin. N. Am. 2004, 33, 305–331. [Google Scholar] [CrossRef]
- Miranda, D.M.; Wajchenberg, B.L.; Calsolari, M.R.; Aguiar, M.J.; Silva, J.M.; Ribeiro, M.G.; Fonseca, C.; Amaral, D.; Boson, W.L.; Resende, B.A.; et al. Novel mutations of the BSCL2 and AGPAT2 genes in 10 families with Berardinelli–Seip congenital generalized lipodystrophy syndrome. Clin. Endocrinol. 2009, 71, 512–517. [Google Scholar] [CrossRef]
- Fu, M.; Kazlauskaite, R.; de Fátima Paiva Baracho, M.; Santos, M.G.; Brandão-Neto, J.; Villares, S.; Celi, F.S.; Wajchenberg, B.L.; Shuldiner, A.R. Mutations in Gng3lg and AGPAT2 in Berardinelli-Seip Congenital Lipodystrophy and Brunzell Syndrome: Phenotype Variability Suggests Important Modifier Effects. J. Clin. Endocrinol. Metab. 2004, 89, 2916–2922. [Google Scholar] [CrossRef]
- Magré, J.; Delépine, M.; Van Maldergem, L.; Robert, J.J.; Maassen, J.A.; Meier, M.; Panz, V.R.; Kim, C.A.; Tubiana-Rufi, N.; Czernichow, P.; et al. Prevalence of Mutations in AGPAT2 Among Human Lipodystrophies. Diabetes 2003, 52, 1573–1578. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Sukumaran, S.; Cortés, V.A.; Tunison, K.; Mizrachi, D.; Sankella, S.; Gerard, R.D.; Horton, J.D.; Garg, A. Human 1-Acylglycerol-3-phosphate O-Acyltransferase Isoforms 1 and 2. J. Biol. Chem. 2011, 286, 37676–37691. [Google Scholar] [CrossRef] [PubMed]
- Sankella, S.; Garg, A.; Agarwal, A.K. Activation of Sphingolipid Pathway in the Livers of Lipodystrophic Agpat2−/− Mice. J. Endocr. Soc. 2017, 1, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.B.; Pardini, V.C.; Ferreira ACde, S.; Fernandes, A.P. Phenotypic heterogeneity in biochemical parameters correlates with mutations in AGPAT2 or Seipin genes among Berardinelli–Seip congenital lipodystrophy patients. J. Inherit. Metab. Dis. 2005, 28, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Antuna-Puente, B.; Boutet, E.; Vigouroux, C.; Lascols, O.; Slama, L.; Caron-Debarle, M.; Khallouf, E.; Lévy-Marchal, C.; Capeau, J.; Bastard, J.P.; et al. Higher Adiponectin Levels in Patients with Berardinelli-Seip Congenital Lipodystrophy due to Seipin as compared with 1-Acylglycerol-3-Phosphate- O.-Acyltransferase-2 Deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 1463–1468. [Google Scholar] [CrossRef]
- Akinci, B.; Onay, H.; Demir, T.; Ozen, S.; Kayserili, H.; Akinci, G.; Nur, B.; Tuysuz, B.; Nuri Ozbek, M.; Gungor, A.; et al. Natural History of Congenital Generalized Lipodystrophy: A Nationwide Study From Turkey. J. Clin. Endocrinol. Metab. 2016, 101, 2759–2767. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Tunison, K.; Dalal, J.S.; Nagamma, S.S.; Hamra, F.K.; Sankella, S.; Shao, X.; Auchus, R.J.; Garg, A. Metabolic, Reproductive, and Neurologic Abnormalities in Agpat1-Null Mice. Endocrinology 2017, 158, 3954–3973. [Google Scholar] [CrossRef]
- Simha, V.; Garg, A. Phenotypic Heterogeneity in Body Fat Distribution in Patients with Congenital Generalized Lipodystrophy Caused by Mutations in the AGPAT2 or Seipin Genes. J. Clin. Endocrinol. Metab. 2003, 88, 5433–5437. [Google Scholar] [CrossRef]
- Patni, N.; Garg, A. Congenital generalized lipodystrophies—New insights into metabolic dysfunction. Nat. Rev. Endocrinol. 2015, 11, 522–534. [Google Scholar] [CrossRef]
- Costa, S.; Sampaio, L.; Berta Sousa, A.; Xing, C.; Agarwal, A.K.; Garg, A. Face-sparing Congenital Generalized Lipodystrophy Type 1 Associated With Nonclassical Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2022, 107, 2433–2438. [Google Scholar] [CrossRef] [PubMed]
- Altay, C.; Seçil, M.; Demir, T.; Atik, T.; Akıncı, G.; Özdemir Kutbay, N.; Keskin Temeloğlu, E.; Yıldırım Şimşir, I.; Özışık, S.; Demir, L.; et al. Determining residual adipose tissue characteristics with MRI in patients with various subtypes of lipodystrophy. Diagn. Interv. Radiol. 2017, 23, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Ben Turkia, H.; Tebib, N.; Azzouz, H.; Abdelmoula, M.S.; Ben Chehida, A.; Hubert, P.; Douira, W.; Ben Dridi, M.F. Lipodystrophie congénitale généralisée de type 1 avec atteinte neurologique. Arch. Pédiatrie 2009, 16, 27–31. [Google Scholar] [CrossRef]
- Haghighi, A.; Kavehmanesh, Z.; Haghighi, A.; Salehzadeh, F.; Santos-Simarro, F.; Van Maldergem, L.; Cimbalistiene, L.; Collins, F.; Chopra, M.; Al-Sinani, S.; et al. Congenital generalized lipodystrophy: Identification of novel variants and expansion of clinical spectrum. Clin. Genet. 2016, 89, 434–441. [Google Scholar] [CrossRef]
- Ren, M.; Shi, J.; Jia, J.; Guo, Y.; Ni, X.; Shi, T. Genotype-phenotype correlations of Berardinelli-Seip congenital lipodystrophy and novel candidate genes prediction. Orphanet J. Rare Dis. 2020, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Teboul-Coré, S.; Rey-Jouvin, C.; Miquel, A.; Vatier, C.; Capeau, J.; Robert, J.J.; Pham, T.; Lascols, O.; Berenbaum, F.; Laredo, J.D.; et al. Bone imaging findings in genetic and acquired lipodystrophic syndromes: An imaging study of 24 cases. Skelet. Radiol. 2016, 45, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Brener, A.; Zeitlin, L.; Wilnai, Y.; Birk, O.S.; Rosenfeld, T.; Chorna, E.; Lebenthal, Y. Looking for the skeleton in the closet—Rare genetic diagnoses in patients with diabetes and skeletal manifestations. Acta Diabetol. 2022, 59, 711–719. [Google Scholar] [CrossRef]
- Oswiecimska, J.; Dawidziuk, M.; Gambin, T.; Ziora, K.; Marek, M.; Rzonca, S.; Guilbride, D.L.; Jhangiani, S.N.; Obuchowicz, A.; Sikora, A.; et al. A Patient with Berardinelli-Seip Syndrome, Novel AGPAT2 Splicesite Mutation and Concomitant Development of Non-diabetic Polyneuropathy. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 319–326. [Google Scholar] [CrossRef]
- Yamamoto, A.; Kusakabe, T.; Sato, K.; Tokizaki, T.; Sakurai, K.; Abe, S. Seipin-linked congenital generalized lipodystrophy type 2: A rare case with multiple lytic and pseudo-osteopoikilosis lesions. Acta Radiol. Open 2018, 8, 2058460119892407. [Google Scholar] [CrossRef]
- Guo, D.-D.; Liu, X.-F.; Duan, Y.-D. Multiple subcutaneous nodules for 46 days in an infant aged 66 days. Zhongguo Dang Dai Er Ke Za Zhi 2020, 22, 903–908. [Google Scholar]
- Lima, J.G.; Nobrega, L.H.C.; Lima, N.N.; Dos Santos, M.C.F.; Baracho, M.F.P.; Bandeira, F.; Capistrano, L.; Freire Neto, F.P.; Jeronimo, S.M.B. Bone Density in Patients With Berardinelli-Seip Congenital Lipodystrophy Is Higher in Trabecular Sites and in Type 2 Patients. J. Clin. Densitom. 2018, 21, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Magré, J.; Delépine, M.; Khallouf, E.; Gedde-Dahl, T., Jr.; Van Maldergem, L.; Sobel, E.; Papp, J.; Meier, M.; Mégarbané, A.; Bachy, A.; et al. Identification of the gene altered in Berardinelli–Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 2001, 28, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Craveiro Sarmento, A.S.; Gomes Lima, J.; de Souza Timoteo, A.R.; Galvão Ururahy, M.A.; Antunes de Araújo, A.; Carvalho Vasconcelos, R.; Cândido Dantas, V.K.; Fassarella Agnez-Lima, L.; Araújo de Melo Campos, J.T. Changes in redox and endoplasmic reticulum homeostasis are related to congenital generalized lipodystrophy type 2. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158610. [Google Scholar] [CrossRef]
- Araújo de Melo Campos, J.T.; Dantas de Medeiros, J.L.; Cardoso de Melo, M.E.; Alvares da Silva, M.; Oliveira de Sena, M.; Sales Craveiro Sarmento, A.; Fassarella Agnez Lima, L.; de Freitas Fregonezi, G.A.; Gomes Lima, J. Endoplasmic reticulum stress and muscle dysfunction in congenital lipodystrophies. Biochim. Biophys. Acta—Mol. Basis Disease 2021, 1867, 166120. [Google Scholar] [CrossRef]
- Akinci, G.; Topaloglu, H.; Demir, T.; Danyeli, A.E.; Talim, B.; Keskin, F.E.; Kadioglu, P.; Talip, E.; Altay, C.; Yaylali, G.F.; et al. Clinical spectra of neuromuscular manifestations in patients with lipodystrophy: A multicenter study. Neuromuscul. Disord. 2017, 27, 923–930. [Google Scholar] [CrossRef]
- Dantas de Medeiros, J.L.; Carneiro Bezerra, B.; Brito de Araújo, T.A.; Craveiro Sarmento, A.S.; de Azevedo Medeiros, L.B.; Peroni Gualdi, L.; Luna Cruz, M.D.S.; Xavier Nobre, T.T.; Gomes Lima, J.; Araújo de Melo Campos, J.T. Impairment of respiratory muscle strength in Berardinelli-Seip congenital lipodystrophy subjects. Respir. Res. 2018, 19, 173. [Google Scholar] [CrossRef]
- Vogel, P.; Read, R.; Hansen, G.; Wingert, J.; Dacosta, C.M.; Buhring, L.M.; Shadoan, M. Pathology of Congenital Generalized Lipodystrophy in Agpat2 –/– Mice. Vet. Pathol. 2011, 48, 642–654. [Google Scholar] [CrossRef]
- Craveiro Sarmento, A.S.; de Azevedo Medeiros, L.B.; Agnez-Lima, L.F.; Lima, J.G.; de Melo Campos, J.T.A. Exploring Seipin: From Biochemistry to Bioinformatics Predictions. Int. J. Cell Biol. 2018, 2018, 1–21. [Google Scholar] [CrossRef]
- Payne, V.A.; Grimsey, N.; Tuthill, A.; Virtue, S.; Gray, S.L.; Dalla Nora, E.; Semple, R.K.; O’Rahilly, S.; Rochford, J.J. The Human Lipodystrophy Gene BSCL2/Seipin May Be Essential for Normal Adipocyte Differentiation. Diabetes 2008, 57, 2055–2060. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E.; El Mourabit, H.; Prot, M.; Nemani, M.; Khallouf, E.; Colard, O.; Maurice, M.; Durand-Schneider, A.M.; Chrétien, Y.; Grès, S.; et al. Seipin deficiency alters fatty acid Δ9 desaturation and lipid droplet formation in Berardinelli-Seip congenital lipodystrophy. Biochimie 2009, 91, 796–803. [Google Scholar] [CrossRef]
- Sołtysik, K.; Ohsaki, Y.; Tatematsu, T.; Cheng, J.; Maeda, A.; Morita, S.Y.; Fujimoto, T. Nuclear lipid droplets form in the inner nuclear membrane in a seipin-independent manner. J. Cell Biol. 2021, 220, e202005026. [Google Scholar] [CrossRef]
- Gomes, K.B.; Fernandes, A.P.; Ferreira, A.C.; Pardini, H.; Garg, A.; Magré, J.; Pardini, V.C. Mutations in the Seipin and AGPAT2 Genes Clustering in Consanguineous Families with Berardinelli-Seip Congenital Lipodystrophy from Two Separate Geographical Regions of Brazil. J. Clin. Endocrinol. Metab. 2004, 89, 357–361. [Google Scholar] [CrossRef]
- Haghighi, A.; Razzaghy-Azar, M.; Talea, A.; Sadeghian, M.; Ellard, S.; Haghighi, A. Identification of a novel nonsense mutation and a missense substitution in the AGPAT2 gene causing congenital generalized lipodystrophy type 1. Eur. J. Med. Genet. 2012, 55, 620–624. [Google Scholar] [CrossRef]
- Craveiro Sarmento, A.S.; Ferreira, L.C.; Lima, J.G.; de Azevedo Medeiros, L.B.; Barbosa Cunha, P.T.; Agnez-Lima, L.F.; Galvão Ururahy, M.A.; de Melo Campos, J.T.A. The worldwide mutational landscape of Berardinelli-Seip congenital lipodystrophy. Mutat. Res. /Rev. Mutat. Res. 2019, 781, 30–52. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Montenegro Junior, R.M.; Lima, G.E.D.C.P.; Fernandes, V.O.; Montenegro, A.P.D.R.; Ponte, C.M.M.; Martins, L.V.; Pinheiro, D.P.; de Moraes, M.E.A.; de Moraes Filho, M.O.; d’Alva, C.B. Leu124Serfs*26, a novel AGPAT2 mutation in congenital generalized lipodystrophy with early cardiovascular complications. Diabetol. Metab. Syndr. 2020, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Cortés, V.A.; Smalley, S.V.; Goldenberg, D.; Lagos, C.F.; Hodgson, M.I.; Santos, J.L. Divergent Metabolic Phenotype between Two Sisters with Congenital Generalized Lipodystrophy Due to Double AGPAT2 Homozygous Mutations. A Clinical, Genetic and In Silico Study. PLoS ONE 2014, 9, e87173. [Google Scholar] [CrossRef] [PubMed]
- Les Laboratoires Servier. Servier Medical. Available online: https://smart.servier.com/ (accessed on 12 April 2024).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Melo, M.E.C.; da Silva, L.M.G.; Cavalcante, A.C.C.; Lima, J.G.; Campos, J.T.A.d.M. The Role of the AGPAT2 Gene in Adipose Tissue Biology and Congenital Generalized Lipodystrophy Pathophysiology. Int. J. Mol. Sci. 2025, 26, 5416. https://doi.org/10.3390/ijms26115416
de Melo MEC, da Silva LMG, Cavalcante ACC, Lima JG, Campos JTAdM. The Role of the AGPAT2 Gene in Adipose Tissue Biology and Congenital Generalized Lipodystrophy Pathophysiology. International Journal of Molecular Sciences. 2025; 26(11):5416. https://doi.org/10.3390/ijms26115416
Chicago/Turabian Stylede Melo, Maria Eduarda Cardoso, Letícia Marques Gomes da Silva, Ana Carolina Costa Cavalcante, Josivan Gomes Lima, and Julliane Tamara Araújo de Melo Campos. 2025. "The Role of the AGPAT2 Gene in Adipose Tissue Biology and Congenital Generalized Lipodystrophy Pathophysiology" International Journal of Molecular Sciences 26, no. 11: 5416. https://doi.org/10.3390/ijms26115416
APA Stylede Melo, M. E. C., da Silva, L. M. G., Cavalcante, A. C. C., Lima, J. G., & Campos, J. T. A. d. M. (2025). The Role of the AGPAT2 Gene in Adipose Tissue Biology and Congenital Generalized Lipodystrophy Pathophysiology. International Journal of Molecular Sciences, 26(11), 5416. https://doi.org/10.3390/ijms26115416